
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A K+-promoted Diels–Alder reaction by using a self-assembled macrocyclic boronic ester containing two crown ether moieties†
Kosuke
Ono
 ,
Morikazu
Niibe
and
Nobuharu
Iwasawa
*
,
Morikazu
Niibe
and
Nobuharu
Iwasawa
*
Department of Chemistry, Tokyo Institute of Technology, O-okayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: niwasawa@chem.titech.ac.jp
First published on 3rd July 2019
Abstract
A K+-promoted Diels–Alder reaction of 1,4,9,10-anthradiquinone with various dienes is achieved in the presence of a self-assembled macrocyclic boronic ester [2+2]crown containing two crown ether moieties. The reaction rate is remarkably accelerated (up to 206-fold) compared to that in the absence of the promoter. Furthermore, the reaction proceeds regioselectively to yield an internal adduct. The self-assembly protocol was also demonstrated.
Introduction
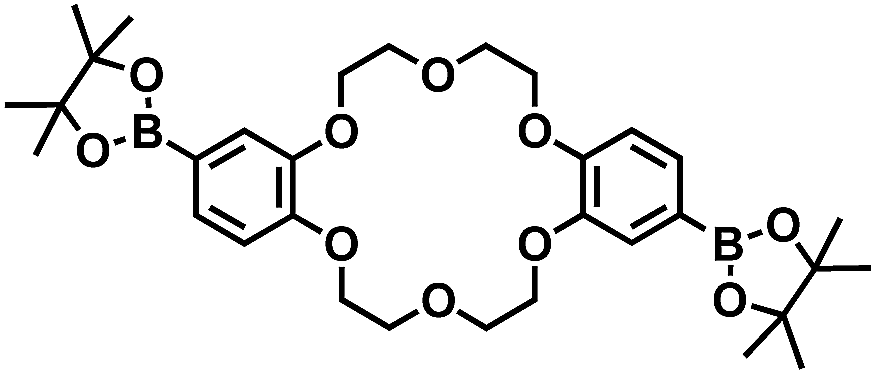
One of the major goals for utilization of synthetic host molecules is their application as catalysts.1 Among various organic reactions, the Diels–Alder reaction, which is one of the most useful reactions for the construction of 6-membered carbocycles, has been studied using synthetic host molecules, such as hydrogen-bonded capsules,2 cyclic metalloporphyrin trimers,3 coordination cages,4 and so on.5 Through these studies, reaction characteristics such as acceleration of the reaction rate,1–5 formation of products with unique regio- or stereo-selectivity,4b–d catalytic turnover,2c,d,4b,h or asymmetric induction4f were demonstrated depending on the host molecules. These reaction characteristics were mostly achieved by a strategy in which both a diene and a dienophile were pre-organized inside the host molecules. However, this strategy often led to a problem known as product inhibition, because the host usually binds the product more strongly than the substrates.1–5 To solve this problem, we devised a new type of supramolecular catalyst using a macrocyclic boronic ester containing two crown ether moieties. The macrocyclic host binds and activates the dienophile through M+-crown ether moieties and promotes the Diels–Alder reaction with various dienes. After the reaction, the bent-shaped product that showed weaker binding affinity could be replaced by the dienophile easily through the open framework. In this strategy, the entropic disadvantage arising from the need to bind both substrates is also reduced. Thus, the exchange process of the product with the dienophile is thought to be energetically neutral. Recently, the group of Lusby introduced this simple dienophile-binding strategy and realized the efficient catalytic turnover by using the coordination cage that activated the dienophile by hydrogen bonding.4h,6Research is being carried out on dynamic self-assembly utilizing boronic ester formation of diboronic acids with an indacene-type tetrol.7 During the examination of the possibility of utilizing our hosts as catalysts, we observed a concise self-assembly of the macrocyclic boronic ester [2+2]crown, containing two dibenzo-18-crown-6 moieties, and a hitherto unknown K+-accelerated Diels–Alder reaction, which showed not only acceleration of the reaction rate but also enhancement of the internal regioselectivity in the reaction of 1,4,9,10-anthradiquinone and various dienes (Fig. 1).
 | ||
| Fig. 1 (a) Schematic representation of the Diels–Alder reaction of 1,4,9,10-anthradiquinone 1 and various dienes accelerated using [2+2]crown·2K+. (b) Self-assembly of [2+2]crown. (c) X-ray structure of [2+2]crown. | ||
Results and discussion
The macrocyclic boronic ester [2+2]crown was quite easily prepared in high yield by simply mixing a diboronic acid of dibenzo-18-crown-6 2 and optically pure tetrol (+)-3 (ref. 8) in MeOH–CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), followed by GPC purification (Fig. 1b).9,10 Formation of the desired [2+2]crown was fully confirmed by 1H NMR and FAB-MS, and the structure was confirmed by X-ray crystallographic analysis (Fig. 1c), which suggested that its cavity size (16.9 × 12.7 Å) was suitable for inclusion of quinone type compounds (Fig. 1c).11
:1), followed by GPC purification (Fig. 1b).9,10 Formation of the desired [2+2]crown was fully confirmed by 1H NMR and FAB-MS, and the structure was confirmed by X-ray crystallographic analysis (Fig. 1c), which suggested that its cavity size (16.9 × 12.7 Å) was suitable for inclusion of quinone type compounds (Fig. 1c).11
With the desired macrocyclic boronic ester [2+2]crown in hand, the binding behavior of [2+2]crown containing alkaline metal salts with several quinonoid compounds was examined. From 1H NMR and isothermal titration calorimetry (ITC) studies, 1:1 complexation behavior was revealed for 1,4,9,10-anthradiquinone 1 and [2+2]crown·2K+ with a high association constant (Ka = 3.54 × 103 M−1; CHCl3/CH3CN (1:1))12 (Fig. 2)9,13 (see the ESI†). It should be noted that the use of other metal cations (Li+ or Na+) instead of K+ or a mixture of monomeric pinacol ester of dibenzo-18-crown-6 diboronic acid 2pin,14 KOTf, and 1 did not show an obvious shift of the signals of 1 by 1H NMR. These results suggested that the macrocyclic structure of [2+2]crown with K+ ions was essential for the efficient binding of 1.9,13
 | ||
| Fig. 2 Complexation of [2+2]crown with diquinone 1 in the presence of K+. | ||
As it is known that the Diels–Alder reaction of 1 with dienes occurs at both the C-2 and C-4a double bonds giving an internal and a terminal regioisomeric adduct and sometimes a diadduct,15 the reaction of 1 with anthracene 4 in the presence or absence of [2+2]crown and K+ was examined with the expectation that high regioselectivity owing to the restriction of the host framework would be achieved.16 When a mixture of 1 and 4 was kept at room temperature in a mixed solution of CHCl3 and CH3CN (1:1) for 1.5 hours, the Diels–Alder reaction proceeded very slowly (2% conversion) and a mixture of the internal adduct and terminal adduct was obtained in about a 4:1 ratio (Table 1, entry 1). Addition of [2+2]crown without K+ did not affect the result of the reaction (Table 1, entry 2). On the other hand, when the same reaction was carried out in the presence of a stoichiometric amount of [2+2]crown and 2 equivalents of KOTf, the reaction was dramatically accelerated to give the product in 95% yield under the same reaction conditions, and the rate constant k was 206 times larger than that of the reaction without [2+2]crown and KOTf. Importantly, only the adduct with the internal alkene was obtained selectively (entry 3). Control experiments were carried out using monomeric 2pin with KOTf, and almost no accelerating effect was observed (entry 4). Thus, the macrocyclic host structure of [2+2]crown was essential for the acceleration. Potassium cations were much more effective than sodium cations, which only had a small accelerating effect (entry 3 vs. 5).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Crown ether | M+OTf− | Conversionb at 90 min |
5int:5terb |
k (M−1 min−1) | k/kno catc |
| a Reaction conditions: 1 (6.5 mM), 4 (9.8 mM), M+OTf− (13 mM), [2+2]crown (6.5 mM). b Conversion at 1.5 h and ratio of 5int/5ter were determined by 1H NMR. c Reaction rate k was estimated by using a second-order kinetic model. The value of kno cat is taken from the reaction in the absence of crown ether and M+OTf−. d 2pin (13 mM). | ||||||
| 1 | None | None | 2% | 4:1 |
0.033 | — |
| 2 | [2+2]crown | None | 2% | 4:1 |
0.030 | 0.9 |
| 3 | [2+2]crown | KOTf | 95% | 5int only | 6.87 | 206 |
| 4d | 2pin | KOTf | 2% | 4:1 |
0.038 | 1.1 |
| 5 | [2+2]crown | NaTOf | 5% | 13:1 |
0.175 | 5.2 |

It is well known that Lewis acids accelerate the Diels–Alder reaction by coordinating with the lone pair of the carbonyl group resulting in lowering of the LUMO level of the dienophile.17 TiCl4, SnCl4, BF3·OEt2, etc. are employed as typical Lewis acids, and the use of alkaline metals for the acceleration of the Diels–Alder reaction has been mostly limited to the reaction using LiClO4 in ether.18 In fact, Na+ or K+ has not been employed for this purpose due to their very low ability to accept electron pairs in the vacant orbital of the metal to activate dienophiles.19,20 To our knowledge, this is the first example that the Diels–Adler reaction was effectively promoted by potassium ions19 and this unique effect was specific to the combination of [2+2]crown and potassium ions.
The catalytic version of this reaction was also examined, and even 5 mol% of [2+2]crown·2K+ was sufficient to promote the reaction (Scheme 1). After 3 hours, 45% of 1 was transformed into the Diels–Alder adduct 5 (5int/5ter = 30:1), while only 5% conversion was observed without the catalyst. From the second-order plot, after 90 minutes, the reaction rate became slightly slower than that in the initial period, suggesting moderate product inhibition (Fig. 3).21 However, reasonable catalytic activity was maintained under the conditions even where the amount of the product was considerably larger than that of the catalyst.
 | ||
| Scheme 1 Catalytic conditions of the Diels–Alder reaction. | ||
 | ||
| Fig. 3 Second-order plot (1/([4]0 − [1]0)ln([4][1]0/[1][4]0)/M−1vs. t/min) for the catalytic conditions of the Diels–Alder reaction. [4]0 = initial concentration of 4, [1]0 = initial concentration of 1. | ||
The acceleration of the reaction was observed with various 2-mono and 2,3-di-substituted 1,3-butadienes (Table 2). When dienes with small substituents 6–8 were used, the reactions were remarkably accelerated and the reaction rate constants kcat were approximately 20–50 times larger than those of the reaction without [2+2]crown·2K+ (entry 1–6). On the other hand, the reactions with dienes with bulky substituents 9–12 were less accelerated (kcat/kno cat ∼ 10) as shown in entries 7–14. Steric repulsion between the [2+2]crown framework and the bulky substituent has made it difficult to promote the reaction smoothly. However, in all cases, enhancement of the regioselectivity of the product was observed in the presence of [2+2]crown·2K+. In particular, in the case of diene 12 (entry 13 and 14), the ratio of the internal adduct/terminal adduct was dramatically increased from 1:1 to 45:1 by the addition of [2+2]crown·2K+. Furthermore, chirality induction was observed when 2-mono-substituted 1,3-dienes were used (see the ESI, Table S1†). This also suggested that the [2+2]crown framework recognized the substituent of dienes and the reaction proceeded inside the host, although the enantioselectivities were low (up to 19% ee). 1-Mono- and 1,4-di-substituted 1,3-butadienes were not applicable to this reaction probably because these dienes could not approach the quinone inside [2+2]crown·2K+ due to the steric hindrance of the [2+2]crown framework (see the ESI, Table S2†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Diene | [2+2]crown·2K+ | k (M−1 min−1) | k cat/kno cate | Internal:terminalf |
Conversionf at 30 min |
| a Reaction conditions: 1 (6.5 mM) in the presence of the catalyst; 1 (15 mM) in the absence of the catalyst. b 3 equiv. of diene were used. c 1 (13 mM). d reaction rate k was estimated by using a second-order kinetic model with the assumption that 1 is completely complexed with [2+2]crown·2K+, although the estimated value of complexation based on the association constant is about 80% in the beginning. e The value of kcat or kno cat is taken from the reaction in the presence or absence of the catalyst. f Ratio of internal adduct/terminal adduct and conversion were determined by 1H NMR. | ||||||
| 1b |

|
1.0 equiv. | 1.43 | 19 | 19:1 |
60% |
| 2b | None | 0.077 | — | 1.6:1 |
8% | |
| 3 |

|
1.0 equiv. | 10.90 | 51 | Internal only | 84% |
| 4 | None | 0.21 | — | 2.4:1 |
11% | |
| 5 |

|
1.0 equiv. | 18.27 | 43 | Internal only | 93% |
| 6 | None | 0.43 | — | 9:1 |
21% | |
| 7 |

|
1.0 equiv. | 0.74 | 9.1 | 10:1 |
13% (40% at 90 min) |
| 8c | None | 0.081 | — | 1.5:1 |
4% (13% at 90 min) | |
| 9 |
|
1.0 equiv. | 4.64 | 10 | 13:1 |
60% |
| 10 | None | 0.46 | — | 2:1 |
25% | |
| 11 |

|
1.0 equiv. | 0.64 | 8.1 | Terminal trace | 15% (37% at 90 min) |
| 12 | None | 0.078 | — | Terminal trace | 7% (17% at 90 min) | |
| 13 |
|
1.0 equiv. | 3.30 | 11 | 45:1 |
51% |
| 14 | None | 0.32 | — | 1:1 |
38% | |

The reason why anthracene 4 shows excellent activity compared to the other dienes 6–12 was also investigated. In the reaction of anthracene (Table 1, entry 3), the signal of the anthracene shifted slightly up-field compared to that of free anthracene in 1H NMR spectra, suggesting the formation of a weak Michaelis complex 1·4@[2+2]crown·2K+ (Fig. S10†), while no obvious shift was observed without [2+2]crown·2K+ (Fig. S11†).22 The formation of a substrate pair inside the host was thought to contribute to the acceleration of the reaction of anthracene as a result of the high local concentration of substrates. Subsequently, the possibility of whether the stabilization of the transition state (TS) was involved in the high activity of anthracene was also investigated. The adducts were used as TS analogues and their association constants with the host [2+2]crown·2K+ were measured by ITC study.4h Interestingly, the association constant of the anthracene adduct 5int was 1.08 × 103 M−1 in CHCl3/CH3CN (1:1) (Fig. S12†),21 while the association constants of other internal adducts derived from acyclic dienes such as 6, 9 and 11 that show lower activity were almost 0 M−1 (Fig. S25†). These results suggested that the TS stabilization effect of [2+2]crown·2K+ could also contribute to the high activity of anthracene.
Finally, we examined the possibility of the self-assembly protocol for this reaction. All the components, 1, 2, (+)-3, and 4, were mixed together and the process of the formation of the Diels–Alder adduct was monitored by 1H NMR (Scheme 2). The internal adduct 5int was obtained as the exclusive product in 83% yield after 90 min. From the second-order plot shown in Fig. 4, the gradual increase of the reaction rate was observed and the reaction rate reached 5.95 M−1 min−1 from 60 min later. This value is comparable with that obtained by the reaction using preformed [2+2]crown·2K+ (6.88 M−1 min−1, Table 1 entry 2). Thus, the self-assembly of the active [2+2]crown·2K+ occurred rapidly in the reaction mixture simply by mixing the component molecules to accelerate the reaction.
 | ||
| Scheme 2 Self-assembly protocol for the Diels–Alder reaction of 1 and 4. | ||
 | ||
| Fig. 4 Second-order plot (1/([4]0 − [1]0)ln([4][1]0/[1][4]0)/M−1vs. t/min) for the self-assembled promoter system, where all components were mixed at the same time. [4]0 = initial concentration of 4, [1]0 = initial concentration of 1. | ||
Conclusions
The present study shows that the macrocyclic boronic ester [2+2]crown·2K+ efficiently promotes the Diels–Alder reactions of 1,4,9,10-anthradiquinone and various dienes with high regioselectivity. It is noteworthy that four-point binding of the carbonyl groups with potassium cations in the [2+2]crown framework effectively accelerated the Diels–Alder reaction. Furthermore, the self-assembly protocol was successfully demonstrated by utilizing the dynamic nature of boronic ester linkages, offering the possibility of a novel catalytic system combined with the reversibility of boronic ester formation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks are due to Dr Hidehiro Uekusa and Dr Kohei Johmoto for performing X-ray analysis. We are grateful to a referee for useful comments about the high reactivity of anthracene. This work was supported by a Core Research for Evolutional Science and Technology (CREST) project from the Japan Science and Technology Agency (JST) and by JSPS KAKENHI Grant Number 15H05800.Notes and references
- For representative reviews of supramolecular catalysis, see: (a) F. Hof, S. L. Craig, C. Nuckolls and J. Rebek Jr, Angew. Chem., Int. Ed., 2002, 41, 1488–1508 CrossRef CAS; (b) D. M. Vriezema, M. C. Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed; (c) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (d) B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC; (e) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed; (f) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (g) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (h) M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef CAS; (i) M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed; (j) Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS PubMed; (k) C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS PubMed; (l) Y. Yu and J. Rebek Jr, Acc. Chem. Res., 2018, 51, 3031–3040 CrossRef CAS PubMed.
- (a) J. Kang and J. Rebek Jr, Nature, 1997, 385, 50–52 CrossRef CAS PubMed; (b) J. Kang, G. Hilmersson, J. Santamaría and J. Rebek Jr, J. Am. Chem. Soc., 1998, 120, 3650–3656 CrossRef CAS; (c) J. Kang, J. Santamaría, G. Hilmersson and J. Rebek Jr, J. Am. Chem. Soc., 1998, 120, 7389–7390 CrossRef CAS; (d) R. J. Hooley and J. Rebek Jr, Org. Biomol. Chem., 2007, 5, 3631–3636 RSC.
- (a) C. J. Walter, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Chem. Commun., 1993, 458–460 RSC; (b) Z. Clyde-Watson, A. Vidal-Ferran, L. J. Twyman, C. J. Walter, D. W. J. McCallien, S. Fanni, N. Bampos, R. S. Wylie and J. K. M. Sanders, New J. Chem., 1998, 493–502 RSC; (c) M. Marty, Z. Clyde-Watson, L. J. Twyman, M. Nakash and J. K. M. Sanders, Chem. Commun., 1998, 2265–2266 RSC; (d) M. Nakash, Z. Clyde-Watson, N. Feeder, J. E. Davies, S. J. Teat and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 5286–5293 CrossRef CAS; (e) M. Nakash and J. K. M. Sanders, J. Org. Chem., 2000, 65, 7266–7271 CrossRef CAS PubMed.
- (a) T. Kusukawa, T. Nakai, T. Okano and M. Fujita, Chem. Lett., 2003, 32, 284–285 CrossRef CAS; (b) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (c) Y. Nishioka, T. Yamaguchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2007, 129, 7000–7001 CrossRef CAS PubMed; (d) S. Horiuchi, Y. Nishioka, T. Murase and M. Fujita, Chem. Commun., 2010, 46, 3460–3462 RSC; (e) S. Horiuchi, T. Murase and M. Fujita, Chem.–Asian J., 2011, 6, 1839–1847 CrossRef CAS PubMed; (f) T. Murase, S. Peschard, S. Horiuchi, Y. Nishioka and M. Fujita, Supramol. Chem., 2011, 23, 199–208 CrossRef CAS; (g) D. Samanta, S. Mukherjee, Y. P. Patil and P. S. Mukherjee, Chem.–Eur. J., 2012, 18, 12322–12329 CrossRef CAS PubMed; (h) V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed.
- (a) R. Warmuth, Chem. Commun., 1998, 59–60 RSC; (b) B. Brisig, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2003, 42, 1270–1273 CrossRef CAS PubMed; (c) S. Käss, T. Gregor and B. Kersting, Angew. Chem., Int. Ed., 2006, 45, 101–104 CrossRef PubMed; (d) M. M. J. Smulders and J. R. Nitschke, Chem. Sci., 2012, 3, 785–788 RSC.
- Recently, our group also reported the catalytic acceleration of nucleophilic additions to aromatic carbonyl compounds using a macrocyclic boronic ester. The macrocyclic host binds and activates various aromatic carbonyl compounds by donor–acceptor interactions and promotes the nucleophilic addition of a ketene silyl acetal, see: T. Uchikura, K. Ono, K. Takahashi and N. Iwasawa, Angew. Chem., Int. Ed., 2018, 57, 2130–2133 CrossRef CAS PubMed.
- (a) N. Iwasawa and H. Takahagi, J. Am. Chem. Soc., 2007, 129, 7754–7755 CrossRef CAS PubMed; (b) H. Takahagi, S. Fujibe and N. Iwasawa, Chem.–Eur. J., 2009, 15, 13327–13330 CrossRef CAS PubMed; (c) K. Ono, R. Aizawa, T. Yamano, S. Ito, N. Yasuda, K. Johmoto, H. Uekusa and N. Iwasawa, Chem. Commun., 2014, 50, 13683–13686 RSC; (d) Y. Kikuchi, H. Takahagi, K. Ono and N. Iwasawa, Chem.–Asian J., 2014, 9, 1001–1005 CrossRef CAS PubMed; (e) Y. Kikuchi, K. Ono, K. Johmoto, H. Uekusa and N. Iwasawa, Chem.–Eur. J., 2014, 20, 15737–15741 CrossRef CAS PubMed; (f) S. Ito, K. Ono, K. Johmoto, H. Uekusa and N. Iwasawa, Chem. Sci., 2016, 7, 5765–5769 RSC.
- (a) H. Sakurai, N. Iwasawa and K. Narasaka, Bull. Chem. Soc. Jpn., 1996, 69, 2585–2594 CrossRef CAS; (b) S. Ito, K. Ono and N. Iwasawa, J. Am. Chem. Soc., 2012, 134, 13962–13965 CrossRef CAS PubMed.
- For macrocyclic compounds containing two crown ether moieties, see: (a) C.-F. Lin, Y.-H. Liu, C.-C. Lai, S.-M. Peng and S.-H. Chiu, Chem.–Eur. J., 2006, 12, 4594–4599 CrossRef CAS PubMed; (b) C.-F. Lin, Y.-H. Liu, C.-C. Lai, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2006, 45, 3176–3181 CrossRef CAS PubMed; (c) S.-Y. Hsueh, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2007, 46, 2013–2017 CrossRef CAS PubMed; (d) S.-Y. Hsueh, C.-C. Lai, Y.-H. Liu, Y. Wang, S.-M. Peng and S.-H. Chiu, Org. Lett., 2007, 9, 4523–4526 CrossRef CAS PubMed; (e) J. Li, J. Guo, G. Yang, G. Zhang, C. Chen and D.-Q. Zhang, Asian J. Org. Chem., 2012, 1, 166–172 CrossRef CAS; (f) Y. Han, Z. Meng, Y.-X. Ma and C.-F. Chen, Acc. Chem. Res., 2014, 47, 2026–2040 CrossRef CAS PubMed; (g) W.-B. Hu, W.-J. Hu, Y. A. Liu, J.-S. Li, B. Jiang and K. Wen, Chem. Commun., 2016, 52, 12130–12142 RSC.
- For examples of self-assembly of macrocyclic compounds containing two crown ether moieties, see: M. Kim and G. W. Gokel, J. Chem. Soc., Chem. Commun., 1987, 1686–1688 RSC.
- Single crystals, suitable for X-ray diffraction analysis, were obtained as racemic crystals from a mixture of equimolar amounts of macrocyclic boronic esters prepared by using 2 and (+)-tetrol or (−)-tetrol, respectively.
- This value was based on the postulate that K+ was included in [2+2] quantitatively.
- For examples of alkaline metal cation templated synthesis of pseudorotaxanes, rotaxanes and catenanes by using crown ether hosts, see: (a) G. Kaiser, T. Jarrosson, S. Otto, Y.-F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959–1962 CrossRef CAS PubMed; (b) T. Iijima, S. A. Vignon, H.-R. Tseng, T. Jarrosson, J. K. M. Sanders, F. Marchioni, M. Venturi, E. Apostoli, V. Balzani and J. F. Stoddart, Chem.–Eur. J., 2004, 10, 6375–6392 CrossRef CAS PubMed; (c) S. I. Pascu, T. Jarrosson, C. Naumann, S. Otto, G. Kaiser and J. K. M. Sanders, New J. Chem., 2005, 29, 80–89 RSC; (d) T. Han and C.-F. Chen, J. Org. Chem., 2007, 72, 7287–7293 CrossRef CAS PubMed; (e) T. Han and C.-F. Chen, J. Org. Chem., 2008, 73, 7735–7742 CrossRef CAS PubMed; (f) Z. Meng, J.-F. Xiang and C.-F. Chen, Chem. Sci., 2014, 5, 1520–1525 RSC.
- Chemical structure of 2pin
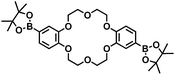 .
. - Diels–Alder reaction of 1 and various dienes has been studied as a direct route to the aglycones of the anthracycline antibiotics daunorubicin and Adriamycin, see: (a) H. H. Inhoffen, H. Muxfeldt, V. Koppe and J. Heimann-Trosien, Chem. Ber., 1957, 90, 1448–1455 CrossRef CAS; (b) W. W. Lee, A. P. Martinez, T. H. Smith and D. W. Henry, J. Org. Chem., 1976, 41, 2296–2303 CrossRef CAS PubMed; (c) T. R. Kelly, R. N. Goerner Jr, J. W. Gillard and B. K. Prazak, Tetrahedron Lett., 1976, 43, 3869–3872 CrossRef.
- For examples of supramolecular catalysts containing crown ether moieties, see: (a) Y. Chao and D. J. Cram, J. Am. Chem. Soc., 1976, 98, 1015–1017 CrossRef CAS; (b) J.-P. Behr and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1978, 143–146 RSC; (c) T. Matui and K. Koga, Tetrahedron Lett., 1978, 19, 1115–1118 CrossRef; (d) D. J. Cram and G. D. Y. Sogah, J. Chem. Soc., Chem. Commun., 1981, 625–628 RSC; (e) R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Org. Chem., 2001, 66, 5926–5928 CrossRef CAS PubMed; (f) R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2003, 125, 2224–2227 CrossRef CAS PubMed; (g) R. Cacciapaglia, A. Casnati, S. Di Stefano, L. Mandolini, D. Paolemili, D. N. Reinhoudt, A. Sartori and R. Ungaro, Chem.–Eur. J., 2004, 10, 4436–4442 CrossRef CAS PubMed.
- For a review of Diels–Alder reaction with Lewis acids, see: Lewis-Acid-Catalyzed Diels–Alder Reaction, in The Diels–Alder Reaction: Selected Practical Methods, ed. F. Fringuelli and A. Taticchi, John Wiley & Sons, Ltd, West Sussex, England, 2002, pp. 99–142 Search PubMed.
- (a) H. Waldmann, Angew. Chem., Int. Ed., 1991, 30, 1306–1308 CrossRef; (b) A. Kumar, Chem. Rev., 2001, 101, 1–19 CrossRef CAS PubMed; (c) T. Imahori, Li(I), Na(I), and K(I) Lewis Acids, in Acid Catalysis in Modern Organic Synthesis, ed. H. Yamamoto and K. Ishihara, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (d) P. A. Grieco, J. J. Nunes and M. D. Gaul, J. Am. Chem. Soc., 1990, 112, 4595–4596 CrossRef CAS; (e) M. A. Forman and W. P. Dailey, J. Am. Chem. Soc., 1991, 113, 2761–2762 CrossRef CAS; (f) G. Desimoni, G. Faita, P. P. Righetti and G. Tacconi, Tetrahedron, 1991, 47, 8399–8406 CrossRef CAS; (g) A. Casaschi, G. Desimoni, G. Faita, A. G. Invernizzi, S. Lanati and P. Righetti, J. Am. Chem. Soc., 1993, 115, 8002–8007 CrossRef CAS; (h) T. Ooi, A. Saito and K. Maruoka, Tetrahedron Lett., 1998, 39, 3745–3748 CrossRef CAS.
- Use of K+ ions for acceleration of the Diels–Alder reaction of benzoquinone with crown ether moieties was reported, although the effect was low (kcat/kno cat < 4), see: (a) A. Tsuda and T. Oshima, New J. Chem., 1998, 1027–1029 RSC; (b) A. Tsuda and T. Oshima, J. Org. Chem., 2002, 67, 1282–1289 CrossRef CAS PubMed; (c) A. Tsuda, C. Fukumoto and T. Oshima, J. Am. Chem. Soc., 2003, 125, 5811–5822 CrossRef CAS PubMed.
- For recent reports of the Diels–Alder reactions in which alkaline salts work as salting-out or in agents, see: (a) C. J. Rizzo, J. Org. Chem., 1992, 57, 6382–6384 CrossRef CAS; (b) C. K. Pai and M. B. Smith, J. Org. Chem., 1995, 60, 3731–3735 CrossRef CAS; (c) S. S. Pawar, U. Phalgune and A. Kumar, J. Org. Chem., 1999, 64, 7055–7060 CrossRef CAS; (d) A. Kumar and S. S. Deshpande, J. Phys. Org. Chem., 2002, 15, 242–245 CrossRef CAS; (e) D. Sarma and A. Kumar, Org. Lett., 2006, 8, 2199–2202 CrossRef CAS PubMed.
- In 1H NMR spectrum of a mixture of product 5int and [2+2]crown·2K+; the proton signals of 5int shifted considerably, suggesting 5int was included in the host (Fig. S12†).
- When 1 (0.65 mM) and 4 (0.65 mM) were mixed in CDCl3/CD3CN (1:1), no shift of the signals was observed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1541693. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc01597c |
| This journal is © The Royal Society of Chemistry 2019 |