
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of 68Ga-labelled ultrasound microbubbles for whole-body PET imaging†
Javier
Hernández-Gil
 *a,
Marta
Braga
b,
Bethany I.
Harriss
a,
Laurence S.
Carroll
b,
Chee Hau
Leow
c,
Meng-Xing
Tang
c,
Eric O.
Aboagye
*b and
Nicholas J.
Long
*a
*a,
Marta
Braga
b,
Bethany I.
Harriss
a,
Laurence S.
Carroll
b,
Chee Hau
Leow
c,
Meng-Xing
Tang
c,
Eric O.
Aboagye
*b and
Nicholas J.
Long
*a
aDepartment of Chemistry, Imperial College London, UK. E-mail: j.hernandez-gil@imperial.ac.uk; n.long@imperial.ac.uk
bDepartment of Surgery & Cancer, Imperial College London, UK. E-mail: eric.aboagye@imperial.ac.uk
cDepartment of Bioengineering, Imperial College London, UK
First published on 1st May 2019
Abstract
Microbubble (MB) contrast agents have revolutionalised the way ultrasound (US) imaging can be used clinically and pre-clinically. Contrast-enhanced US offers improvements in soft-tissue contrast, as well as the ability to visualise disease processes at the molecular level. However, its inability to provide in vivo whole-body imaging can hamper the development of new MB formulations. Herein, we describe a fast and efficient method for achieving 68Ga-labelling of MBs after a direct comparison of two different strategies. The optimised approach produces 68Ga-labelled MBs in good yields through the bioorthogonal inverse-electron-demand Diel–Alder reaction between a trans-cyclooctene-modified phospholipid and a new tetrazine-bearing HBED-CC chelator. The ability to noninvasively study the whole-body distribution of 68Ga-labelled MBs was demonstrated in vivo using positron emission tomography (PET). This method could be broadly applicable to other phospholipid-based formulations, providing accessible solutions for in vivo tracking of MBs.
Introduction
The development and further refinement of contrast agents plays a central role in the fields of anatomical and molecular imaging, acting as a driving force to overcome limitations inherent to existing imaging modalities.1 In ultrasound (US), the use of microbubbles (MBs, Scheme 1), which consist of a gas core stabilised by a phospholipid or polymer shell, has improved the poor ability of this technique to distinguish between tissues with similar acoustic responses.2 In the presence of an acoustic field, these compressible MBs produce strong nonlinear signals, which can be differentiated from the surrounding tissue, enhancing soft-tissue contrast and signal-to-noise ratios in regions of interest. The fact that MBs typically present sizes of 1–5 μm, and therefore are restricted to intravascular targets, alongside their ability to incorporate disease-targeted ligands, has recently oriented the use of US imaging towards detecting and monitoring vascular pathologies at the molecular level.2c In this regard, different preclinical studies have successfully demonstrated the capability of MBs to visualise receptors overexpressed in inflammation, angiogenesis and/or early tumour formation,3a–c and a phospholipid-based formulation has even recently entered first in-human clinical trials in various cancer types.3d,e These examples, together with the proven capacity of MBs to load drugs3f will further support the development of alternative MB agents for new diagnostic and therapeutic US applications in patients. | ||
| Scheme 1 Schematic structure/composition of the developed microbubbles (MBs), showing two surface conjugation sites (not to scale). | ||
Clinical translation, however, is always a long, high-risk and expensive endeavour, and therefore, early evaluation of the biodistribution and/or pharmacokinetics of MBs could facilitate their (pre)clinical development.4a Whole-body imaging has long demonstrated utility in the evaluation of in vivo fates and/or possible off-target effects.4 Unfortunately, contrast-enhanced US is limited to local imaging. This limitation has created a demand for new MB formulations that can provide whole-body imaging and therefore, offer a better understanding of diagnostic and/or drug activity during the initial stages of MB development.
Nuclear imaging techniques, namely positron emission tomography (PET) and single-photon-emission tomography (SPECT), provide non-invasive and whole-body imaging with high sensitivity and unlimited penetration depth.5 Hence, a few groups have attempted to develop dual nuclear/US MB agents.6 Most of these studies, however, have used streptavidin-biotin interactions to radiolabel MBs and/or employed SPECT tracers (iodine-125, technetium-99m or indium-111).6a–d Although elegant and encouraging, these approaches face disadvantages: (i) immunogenic streptavidin-coated MBs are not recommended for human use and (ii) SPECT cannot provide quantitative analysis in vivo. PET, unlike SPECT, allows determination of tracer concentration in tissue with higher sensitivity and image quality. Despite these advantages, only one group has incorporated a 18F-labelled phospholipid onto MBs, allowing non-invasive assessment of the whole-body MB fates.6e However, 18F-labelling usually requires time-consuming multi-step synthesis, and more importantly, depends on expensive cyclotron facilities. So, there is an unmet need for a rapid and robust method to generate radiolabelled MBs using broadly accessible PET isotopes.
Gallium-68 (hereafter, 68Ga) is a radiometal that offers aqueous single-step and high-yielding complexations with high positron emission (89%) and on-site availability, allowing hospitals economical production of a high-quality and generator-produced PET isotope.7 In addition, its short half-life (67.7 min) enables the perfect monitoring of MBs, which have circulation times of around 10–20 minutes.2 However, the development of 68Ga-labelled MBs (hereafter, 68Ga-MBs) has never been attempted, which inspired us to focus on this particular problem in the field of US imaging. We reasoned that the optimised method should offer a fast, accessible and versatile way to radiolabel the shell components of MBs, as well as to provide reliable validation checkpoints with conventional To test this hypothesis, two 68Ga labelling strategies were initially devised: (i) direct conjugation of a metal chelator to the phospholipid head-groups and, (ii) bioorthogonal ‘click’ ligation between a chelator-based prosthetic group and a complementary functionalised-phospholipid (Scheme 2A and B). Through a direct comparison between both approaches, we have developed a rapid and efficient method for labelling ultrasound MB agents with a generator-produced PET isotope, offering accessible solutions for in vivo tracking of MBs.
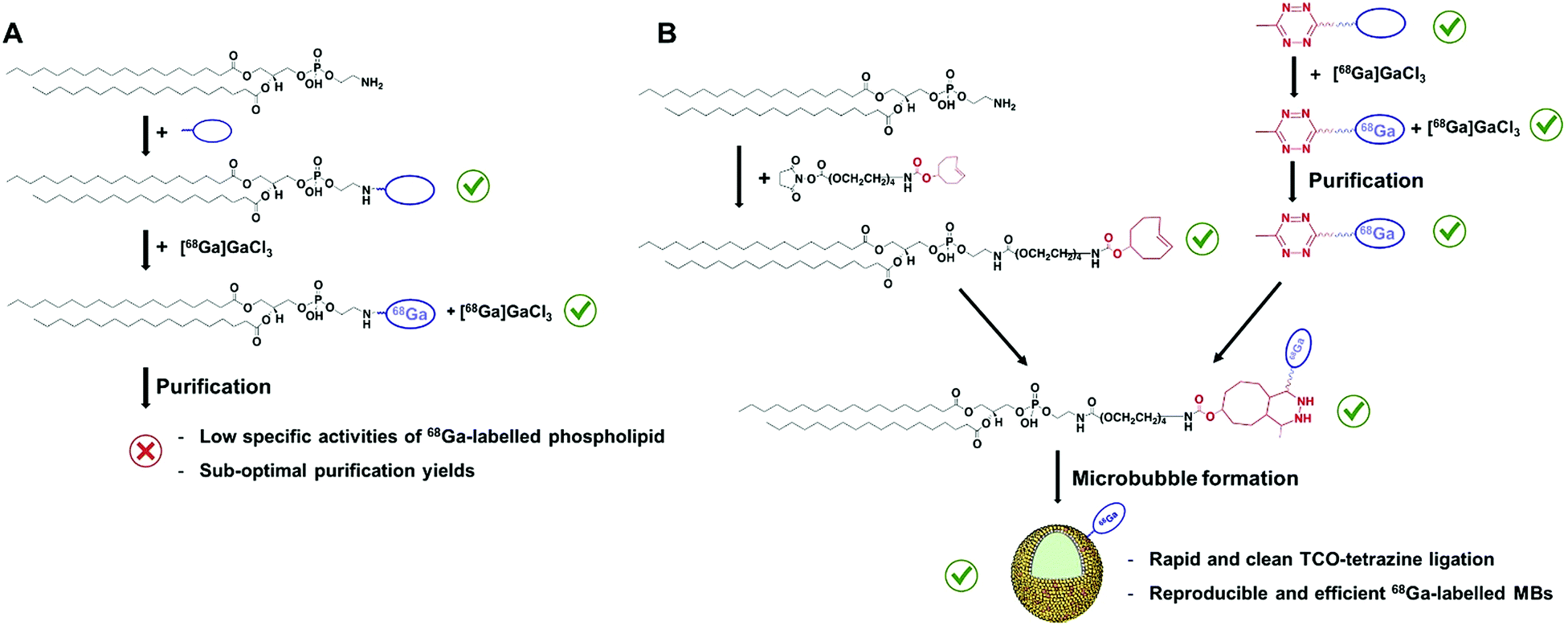 | ||
| Scheme 2 (A) “Direct labelling approach”: conjugation of a metal chelator to the headgroup of PE and subsequent labelling and purification. (B) “Click labelling approach”: development of bioorthogonal components for ‘click’ ligation of a 68Ga-chelator-tetrazine and a trans-cyclooctene-modified phospholipid to yield 68Ga-labelled MBs. | ||
Results and discussion
Design and preparation of microbubbles
Here, we focused on phospholipid-shelled MBs, which represent the most common and versatile US contrast agent.2,3 We recently demonstrated that a cyanine7.5-modified phospholipid could be successfully incorporated into a MB formulation of commercially available 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (poly-ethylene glycol)-2000] (DSPE-PEG(2000)) phospholipids.8a We reasoned that 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PE), which shares DSPE-PEG(2000) backbone without its PEGylated moiety, could therefore, be combined with DPPC and DSPE-PEG(2000), and thus offer an alternative anchor point for the PET tracer. The absence of a bulky PEG45 polymer on PE also facilitates the purification and characterisation of resulting products, while maintaining DSPE-PEG(2000) available for subsequent conjugation of targeting ligands (Scheme 1). With such a rationale in mind, we prepared a new formulation containing DPPC, DSPE-PEG(2000) and PE at 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:10 molar ratios, following standard protocols (Table S1†).8 Optical microscopy was used to visualise the size and concentration of the new MB formulation and, as shown in Table S2,† no significant difference was observed between PE-containing MBs and our previous formulation without PE. The stock MB solution containing 10% of PE yielded a mean bubble diameter of 2.10 ± 1.07 μm and a concentration of (1.22 ± 0.68) × 1010 MBs mL−1 while the unmodified formulation generated bubbles with a mean size of 2.18 ± 1.06 μm and a concentration of (8.02 ± 0.03) × 109 MB mL−1. The acoustic response of both MBs was also compared. The two formulations were diluted to a concentration of approximately 106 MBs mL−1 in a 2 L water tank and their US contrast enhancements quantified (Table S2†). Both MBs produced a similar US contrast at 4.5 MHz, indicating that the new formulation can also be used for US imaging.
:5:10 molar ratios, following standard protocols (Table S1†).8 Optical microscopy was used to visualise the size and concentration of the new MB formulation and, as shown in Table S2,† no significant difference was observed between PE-containing MBs and our previous formulation without PE. The stock MB solution containing 10% of PE yielded a mean bubble diameter of 2.10 ± 1.07 μm and a concentration of (1.22 ± 0.68) × 1010 MBs mL−1 while the unmodified formulation generated bubbles with a mean size of 2.18 ± 1.06 μm and a concentration of (8.02 ± 0.03) × 109 MB mL−1. The acoustic response of both MBs was also compared. The two formulations were diluted to a concentration of approximately 106 MBs mL−1 in a 2 L water tank and their US contrast enhancements quantified (Table S2†). Both MBs produced a similar US contrast at 4.5 MHz, indicating that the new formulation can also be used for US imaging.
Both MBs produced a similar US contrast at 4.5 MHz, indicating that the new formulation can also be used for US imaging.
Evaluation of the first approach: direct conjugation of a metal chelator to the phospholipid head-groups
To test the feasibility of our first approach (Scheme 2A), we functionalised PE with three chelators of known affinity for 68Ga:7a 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA), 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) and 2-(4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecan-1-yl)- pentanedioic acid (DOTA-GA). PE-NOTA (1) and PE-DOTA (2) were prepared by conjugation of the primary amine headgroup of PE with a NHS-activated form of each chelator, whereas PE-DOTAGA (3) was obtained via reaction of a p-isothiocyanatobenzyl PE intermediate with an ethylendiamine-modified DOTAGA reagent. 1–3 were purified by preparative thin-layer chromatography (TLC) and obtained in high yields (Fig. 1A and ESI†).To examine whether 1–3 can efficiently chelate 68Ga, a fixed amount of generator-produced 68Ga3+ (∼2 MBq) was separately reacted with 1–3 over a range of phospholipid concentrations and two pH values (3.5 and 4.5). All reactions were incubated for 10 min at 90 °C and the radiochemical yields (RCYs) were measured using radioTLC. 1 demonstrated the highest labelling efficiency, with RCYs greater than 80% at moderately low chelator concentrations (10 μM) for both pH conditions, corresponding to molar activities of 1–2 MBq nmol−1 (Fig. 1B).
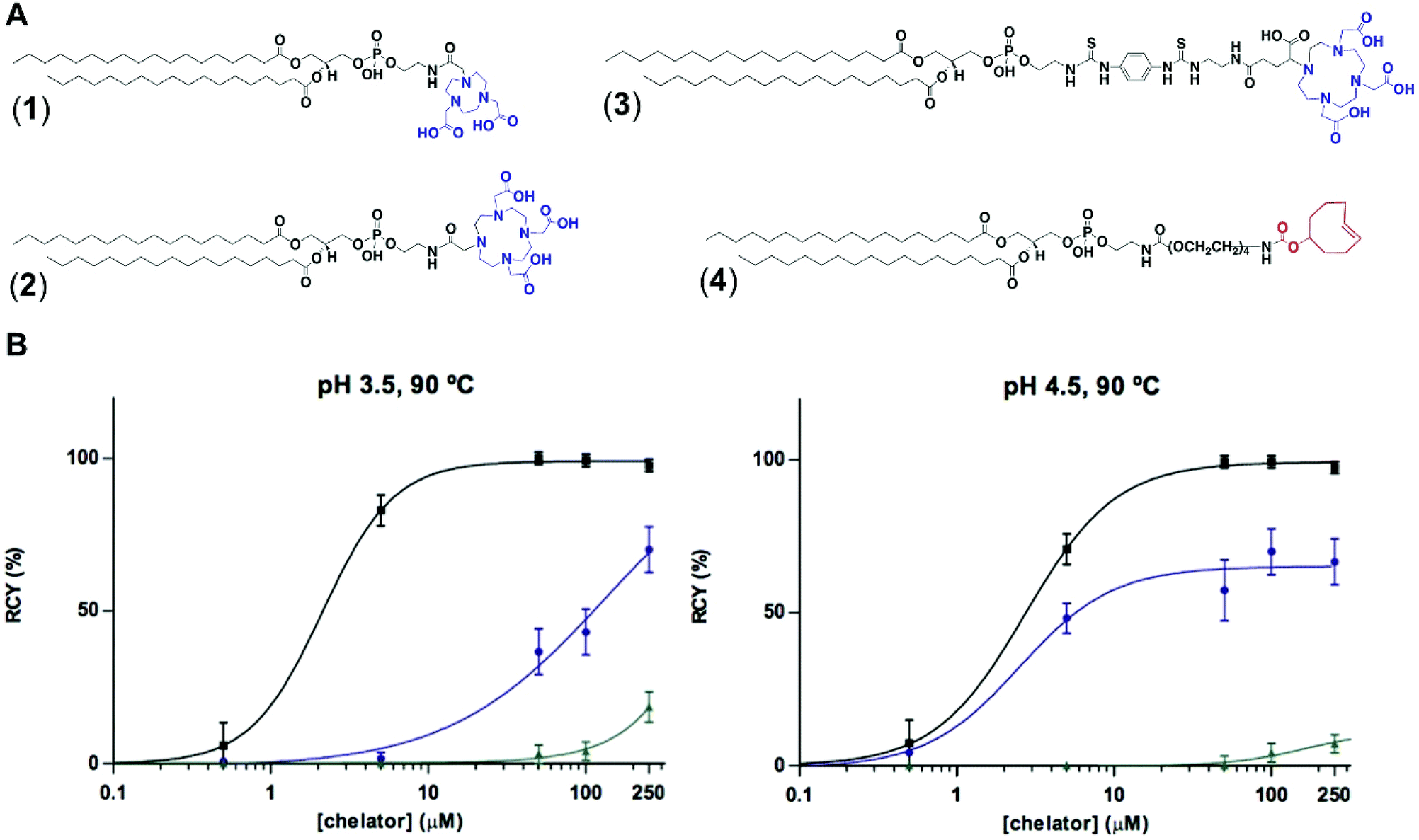 | ||
| Fig. 1 (A) Structure of PE-NOTA (1), PE-DOTA (2), PE-DOTAGA (3) and PE-PEG4-TCO (4). (B) Radiochemical yields for the reaction of 68Ga3+ with 1 (black), 2 (green) and 3 (blue) under different phospholipid concentrations (0.05 to 250 μM) and pH values (3.5 and 4.5), after 10 min at 90 °C. | ||
As previously reported, NOTA derivatives show better affinity for 68Ga3+ than DOTA conjugates as a consequence of the close fit of the relatively small Ga(III) metal ion in the pseudo octahedral cavity of the potentially six-coordinate 9-aneN3, with attached carbonyl and amide groups.9 We then selected 1 for optimising a 68Ga-labelling method under in vivo conditions. Purified 68GaCl3 (70–75 MBq) was reacted with 1 (∼20 nmol) in acetate buffer (0.1 M, pH 4.0) for 15 min at 90 °C. The RCY of [68Ga][Ga(1)], under these conditions, was >85% (non-isolated, estimated by radio-HPLC). Next, different solid-phase extraction (SPE) cartridges were used for [68Ga][Ga(1)] purification but, unfortunately all sorbents tested presented low recovery yields (Table S3†). The best results were obtained with Oasis® HLB cartridges with an average eluted activity of 27 ± 5%. These all-purpose cartridges, with a strongly hydrophilic and reversed-phase polymer, allow acidic, basic and neutral analyte purifications from pH 0 to 14. Under our labelling conditions, pH = 3.5–4.0, [68Ga][Ga(1)] behaves as a zwitterionic phospholipid with a positively charged Ga-chelating moiety and a negatively charged phosphate group (pKa ∼1.5–2.5), meeting a priori the manufacturer requirements for high recovery yields. Phospholipids, however, are a major concern for LC-MS analysis of small molecules in biological matrices and many SPE cartridges (in our case, HybridSPE®, Oasis® Prime and Oasis® HLB cartridges) contain sorbents that can retain phospholipids. This effect was clearly observed when using Oasis® Prime cartridges, retaining 62 ± 15% of the initial loaded activity and, also noticeable with HybridSPE® and Oasis® HLB cartridges trapping 32 ± 7 and 17 ± 3%, respectively. Therefore, the sub-optimal purification yields obtained because of the lipid nature of 1 alongside its low molar activities prompted us to focus on the second approach.
Evaluation of the second approach: bioorthogonal ‘click’ ligation between a chelator-based prosthetic group and a complementary functionalised-phospholipid
For the second strategy (Scheme 2B), we hypothesised that a tetrazine-bearing radioconjugate could combine high molar activities and straightforward work-up procedures from small-sized chelators with high-yielding and fast reactivities from ‘click’ ligations. In this regard, our group and others have been designing new probes suited for bioorthogonal radiolabelling via the inverse-electron-demand Diels–Alder (iEDDA) cycloadditions between tetrazines and olefins.10 iEDDA reactions avoid the use of metal catalysts, which could compete with the far less abundant 68Ga3+ radiometal, as well as offer rapid kinetics at low concentrations, essential when working with short-lived radioisotopes.11 Thus, a trans-cyclooctene (TCO)-modified phospholipid was synthesised using a commercially available TCO-PEG4-NHS ester and PE. TCO was chosen because of its higher reactivity compared to other strained olefins.12 PE-PEG4-TCO (4) was purified by preparative-TLC and characterised by 1H NMR spectroscopy and MALDI-TOF mass spectrometry (Fig. 1a and ESI†).Compound 5, containing N,N′-bis[2-hydroxy-5-(carboxyethyl)- benzyl]ethylenediamine-N,N-diacetic acid (HBED-CC) and a tetrazine, was also prepared (Fig. 2 and ESI†). In a six-step sequence, the desired product was ultimately obtained by in situ coupling HATU-activated [Fe(HBED-CC)]− with (4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanamine. Here, we used HATU, which required a 15 minute activation, instead of the most common 2 day tetrafluorophenol/dicyclohexylcarbodiimide process.13 The formation of the iron complex assures the protection of coordinating phenolic and carboxylate groups while exposing the two propionic acid arms for amine conjugation. HBED-CC-tetrazine derivative 5 was purified by flash chromatography and its monosubstituted nature confirmed using 1H, 13C NMR spectroscopy and ESI mass spectrometry (see ESI†). As a reference, a tetrazine-bearing DOTA-GA tracer (6), previously designed by our group,10b was prepared (Fig. 2 and ESI†).
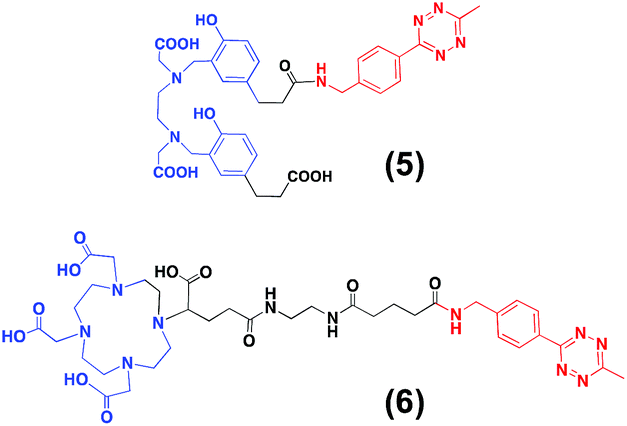 | ||
| Fig. 2 (A) Structure of HBED-CC-tetrazine (5) and DOTA-GA-tetrazine (6). | ||
A head-to-head comparison of the 68Ga-chelating efficiency between 5 and 6 was performed (Fig. 3). At room temperature, the difference between both chelators was remarkable. Whilst 5 performed extremely well, 6 presented poor ability to coordinate 68Ga3+. This suggested that the labelling efficiency of 6 was limited by the higher kinetic barriers that macrocyclic-based chelators generally present compared to acyclic ligands.7a,145 also showed little pH-dependence, yielding near-quantitative RCYs at the low concentration of 5 μM for all pH values tested (based on radioTLC analysis of the reaction solution). At high temperature, both chelators exhibited a similar efficiency, with conversions higher than 90% at 5 μM. At lower concentrations, however, 5 again demonstrated better performance over 6. At 1 μM and pH values of 3.5 and 4.5, RCYs of [68Ga][Ga(5)] were near quantitative while RCYs of [68Ga][Ga(6)] were lower than 30%. This corresponded to maximum molar activities for 5 of 15–20 MBq nmol−1. Next, we selected 5 to evaluate its 68Ga-labelling efficiency under in vivo conditions. This time, [68Ga][Ga(5)], obtained after 10 min incubation at room temperature, could be successfully purified with conventional reverse-phase cartridges. The entire procedure was performed in less than 20 min, yielding purities >98% and RCYs >95% (determined by radio-HPLC analysis of the pure product).
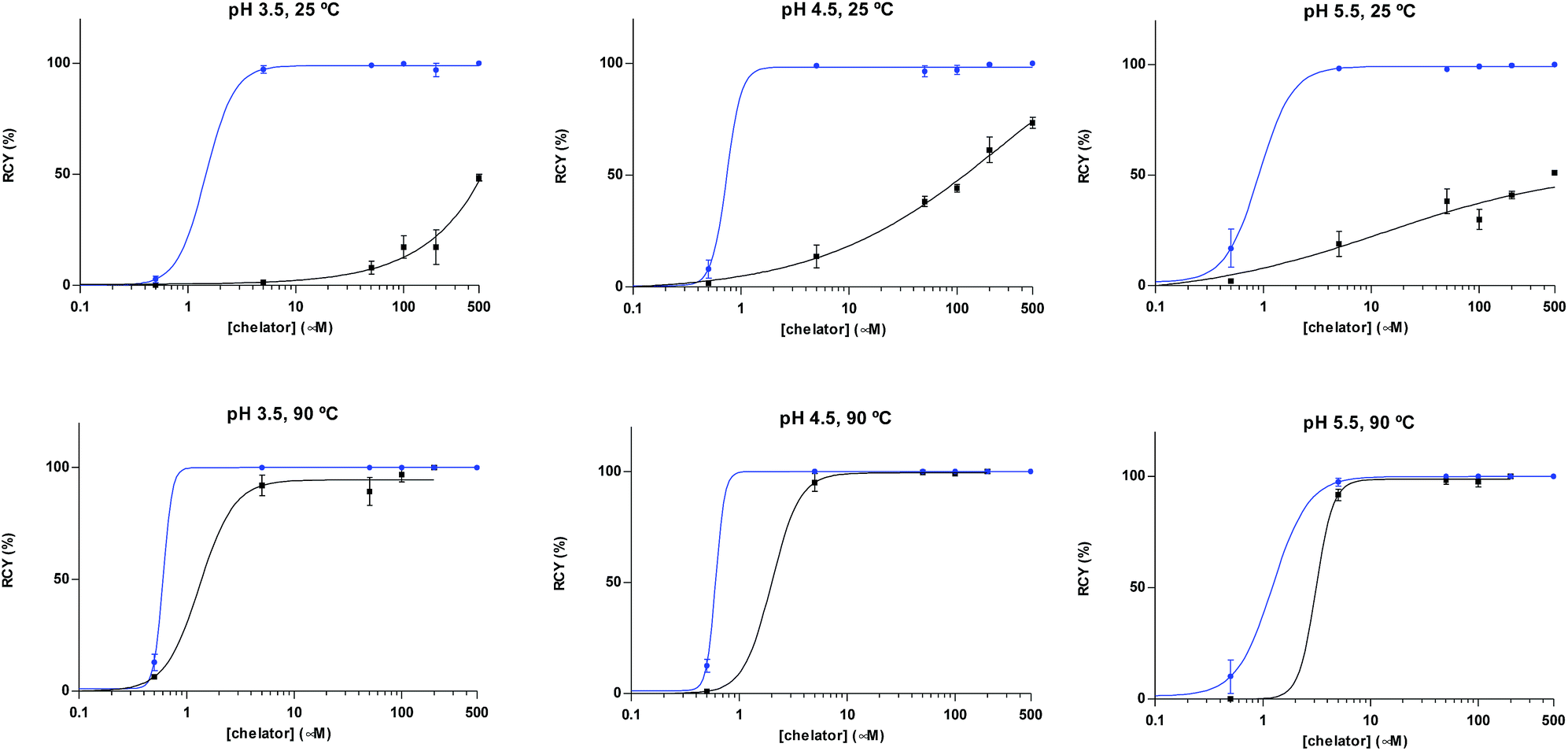 | ||
| Fig. 3 Radiochemical yields for the reaction of 68Ga3+ with 5 (blue) and 6 (black) under different concentrations of chelator (0.05 to 500 μM), pH values (3.5, 4.5 and 5.5) and temperatures (25 and 90 °C), after 10 min. | ||
Preparation of 68Ga-labelled MBs
Fig. 4A shows the chromatogram of purified [68Ga][Ga(5)] labelled at room temperature and pH 4.2 ± 0.3. As for other HBED-based chelators, 5 forms different geometric isomers when complexed to Ga3+.15 Although a mixture of species can potentially present different pharmaceutical profiles, a recent study measuring the cell binding and internalisation properties of [68Ga]Ga-PSMA-HBED-CC labelled at room temperature (containing a mixture of species) and at 95 °C (containing the thermodynamically most stable isomer) on PSMA-expressing LNCaP cells showed comparable results for both fractions, indicating a stereochemical independent receptor-interaction of the radioligand.16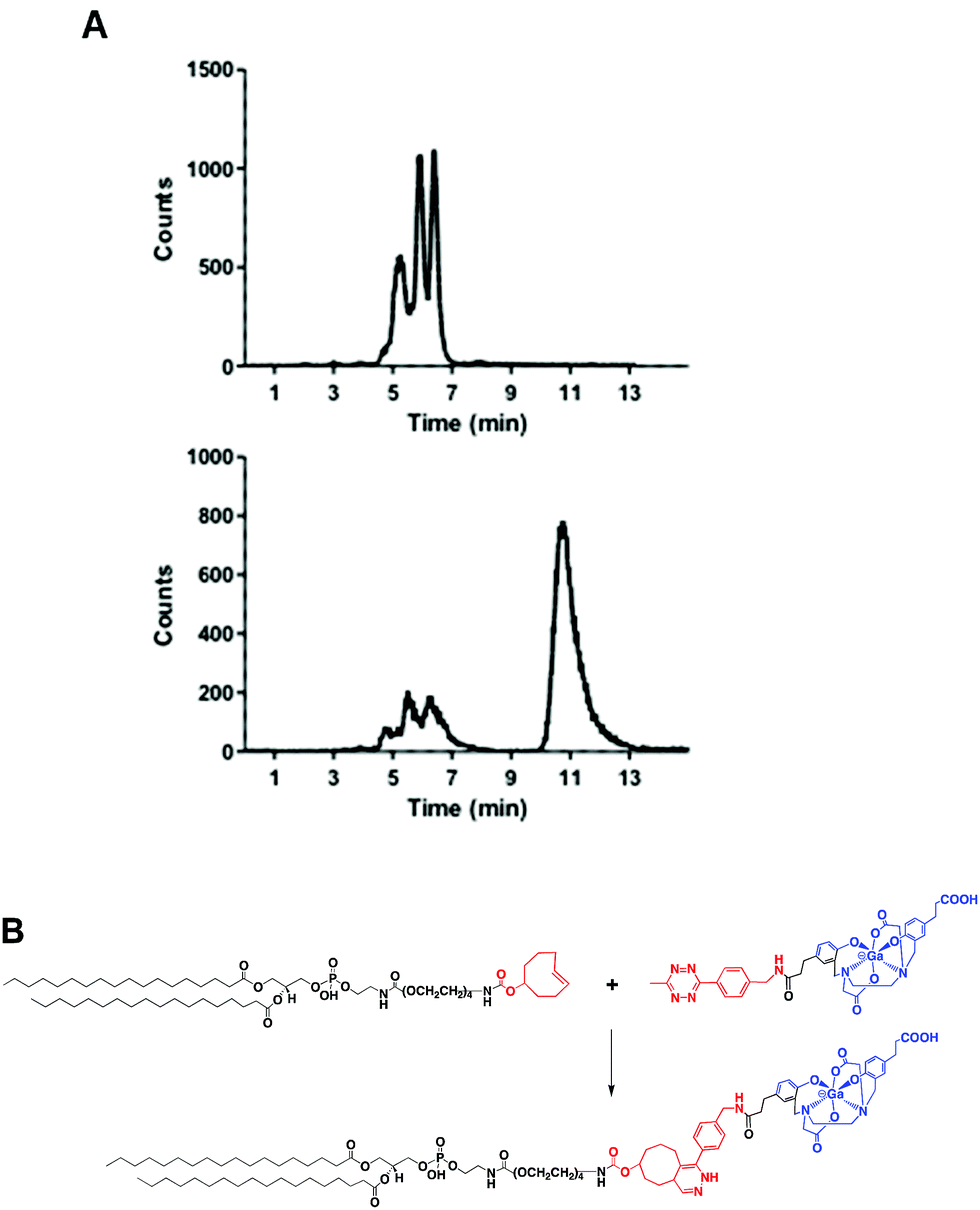 | ||
| Fig. 4 (A) Radio-HPLC chromatograms of (top) [68Ga]Ga-HBED-CC-tetrazine precursor and (bottom) reaction between 4 and 5, after 15 min at 60 °C. (B) Scheme of the reaction between TCO-modified PE (4) and [68Ga]Ga-HBED-CC-tetrazine (5). | ||
Next, we reacted a purified fraction of [68Ga][Ga(5)] (∼75 MBq) with PE-PEG4-TCO (0.107 μmol) at 60 °C (Fig. 4A). After 15 minutes, the radio-HPLC chromatogram showed the appearance of a new major peak at a slower retention time (τr = 10.8 min) than the labelled precursor (τr = 5.3–6.4 min), which corresponded to radiochemical conversions of the dihydropyridazine products of ∼80–85% (non-isolated, estimated by radio-HPLC) (Fig. 4A and B). Next, 68Ga-labelled PE (hereafter, 68Ga-PE) was combined with DPPC and DSPE-PEG(2000) and the resulting formulation was activated to form gas-filled MBs. Excess of unreacted [68Ga][Ga(5)] and free lipids were removed via a centrifugation/washing methodology.17 The RCY of purified 68Ga-MBs was around 40–50%, generating samples with ∼30–35 MBq and concentrations of (1.28 ± 0.68)·109 MB mL−1, in 40–50 minutes after 68Ga elution from the generator.
In vivo study of 68Ga-labelled MBs
Encouraged by the successful 68Ga-labelling of MBs, comparative dynamic PET imaging and biodistribution studies of [68Ga][Ga(5)], 68Ga-PE and 68Ga-MBs were undertaken on Balb/c nude mice (n = 5–6). The optimised labelling approach yielded MB stock solutions with high activity and concentration, allowing us to use concentrations of approximately 5 × 107 MB per injection with activities between 0.37–0.74 MBq. These MB concentration values are in the same order of typical diagnostic doses used in US imaging, suggesting that this strategy generates 68Ga-MBs that can also be exploited for complementary US studies.Kinetic analysis showed that the MBs had a pharmacokinetic profile different from that of freely-injected PE and 5 (Fig. 5 and S6–S8†). Following the injection of free [68Ga][Ga(5)], activity accumulated in the bladder, gallbladder and intestines within the first 5 minutes, which confirmed that [68Ga][Ga(5)] was characterised by a rapid tissue distribution and subsequent clearance through the urinary route. 68Ga-PE and 68Ga-MBs, on the contrary, presented longer circulation times in the animals, with high blood pool mean values persisting for the entire imaging period. Both formulations, however, presented differences in their biodistribution, which became more evident at later time points. Between 0 to 2 min post-injection (PI), 68Ga-PE and 68Ga-MBs were mainly present in the heart and liver, whereas at 5–20 min time interval, 68Ga-PE was mainly accumulated in the bladder and gallbladder, and 68Ga-MBs were present in the liver, gallbladder and spleen. The uptake in the liver and spleen reached equilibrium around 3 min PI at approximately 66 and 40% ID g−1, respectively. Previous studies have also indicated the liver and the spleen as the major organs for the accumulation of MBs with sizes around 1–10 μm.6a,b In addition, 68Ga-MBs showed an initial peak of activity in the lungs of 43 ± 12% ID g−1, which decreased to 20 ± 5% ID g−1 at 20 min, indicating that 68Ga-MBs were gradually redistributed into the systemic circulation after an initial accumulation in the lungs. Biodistribution data confirmed the PET results. At 20 min PI, both [68Ga][Ga(5)] and 68Ga-PE were mostly present in the urine (689 ± 64 and 146 ± 26% ID g−1, respectively), consistent with an efficient urinary excretion route. 68Ga-MBs, on the other hand, showed a more distributed uptake within the animal models, with accumulation of radioactivity mainly observed in the liver (47 ± 4% ID g−1), spleen (40 ± 2%ID g−1) and urine (57 ± 11% ID g−1).
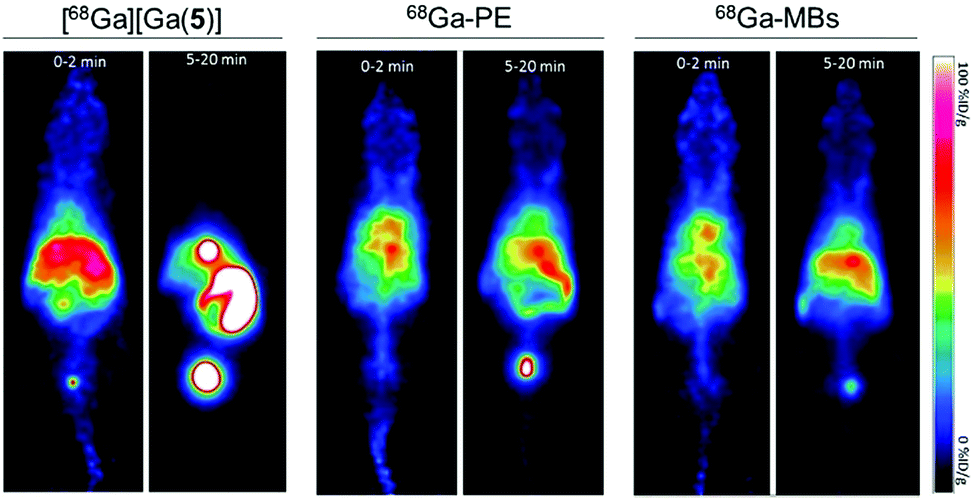 | ||
| Fig. 5 Small-animal PET imaging of Balb/c nude mice after intravenous injection of [68Ga][Ga(5)] (left), 68Ga-labelled PE (middle) and, 68Ga-labelled MBs (right) at early (0–2 min) and late (5–20 min) time intervals. | ||
Conclusions
In conclusion, we have developed an efficient method for labelling MBs with a generator-produced PET isotope using a rapid and clean ligation between a TCO-modified phospholipid and a 68Ga-HBED-CC-tetrazine tracer. Bioconjugation of TCO to phospholipids is simple and reproducible, and the new HBED-CC-tetrazine chelator provides efficient and high-yielding 68Ga-labelling, affording reproducible synthesis of 68Ga-MBs under mild conditions. This method offers real-time monitoring and the possibility of easily customising alternative phospholipid-based formulations. We also confirmed that the resulting 68Ga-MBs allow non-invasive study of the whole-body distribution of MBs in mice. As contrast-enhanced US has emerged as a promising imaging modality for clinical applications, we believe that this strategy is a favourable way of non-invasively evaluating the pharmacokinetics and off-target accumulation of new MB formulations in their initial stages of (pre)clinical development. Therefore, this strategy can be easily implemented across multiple centres and hospitals, accelerating MB evaluation and development. Future work will explore the incorporation of targeted-modified phospholipids into the MB formulation and compare 68Ga-labelled MB containing targeted and non-targeted vectors.Experimental section
Materials
All the syntheses were carried out with the following commercially available reagents used without further purification: 4-(aminomethyl)benzonitrile hydrochloride (97%, Sigma Aldrich), di-tert-butyl dicarbonate (≥99%, Sigma Aldrich), nickel(II) trifluoromethanesulfonate (96%, Sigma Aldrich), hydrazine hydrate (50–60%, Sigma Aldrich), sodium nitrite (NaNO2, ≥97%, Sigma Aldrich), 3-(4-hydroxyphenyl)propionic acid (98%, Sigma Aldrich), sodium hydroxide (98.5–100.5%, VWR International), ethylenediamine-N,N′-diacetic acid (≥98%, Sigma Aldrich), formaldehyde (37 wt% in H2O, Sigma Aldrich), hydrochloric acid (37%, VWR International), trifluoroacetic acid (≥99.5%, Tokyo Chemical Industry Co., Ltd.), N,N-diisopropylethylamine (≥99%, Sigma Aldrich), iron(III) chloride (97%, Sigma Aldrich). 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate,N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU, 97%, Sigma Aldrich), glutaric anhydride (95%, Sigma Aldrich). Triethylamine (≥99%, Sigma Aldrich), N-hydroxysuccinimide (98%, Sigma Aldrich). N,N′-Dicyclohexylcarbodiimide (DCC, 99%, Sigma Aldrich), trans-cyclooctene-PEG4-NHS ester (TCO-PEG4-NHS, >95%, Jena Bioscience), trans-cyclooctene-NHS ester (E)-cyclooct-4-enyl-2,5-dioxo-1-pyrrolidinyl carbonate (TCO-NHS ester, >95%, Jena Bioscience), 4-nitroaniline (≥99%, Sigma Aldrich), p-phenylene diisothiocyanate (98%, Sigma Aldrich), 2,2′,2′′-(10-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (NH2-DOTA-GA, ChemMtech mdt), 2,2′,2′′-(10-(2-((2,5-dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid (DOTA-NHS ester, ChemMtech mdt), 2,2′-(7-(2-((2,5-dioxopyrrolidin-1-yl)oxy)-2-oxoethyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (NOTA-NHS ester, ChemMtech mdt), sodium acetate (≥99%, Sigma Aldrich), acetic acid (≥99%, Sigma Aldrich), ammonium acetate (≥98%, Sigma Aldrich), decafluorobutane (99%, FluorMed, L.P.), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC, ≥99%, Avanti Polar Lipids), 1-palmitoyl-2-hydroxy-sn-glycero-3 phosphocholine (LPC, ≥99%, Avanti Polar Lipids), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (18:0 PE-NH2, ≥99%, Avanti Polar Lipids), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG(2000)-NH2, ≥99%, Avanti Polar Lipids), primuline (50%, Sigma Aldrich), propylene glycol (≥99.5%, Sigma Aldrich), glycerol (99%, Sigma Aldrich), Dulbecco's phosphate-buffered saline (10×, Sigma Aldrich). Solvents were HPLC grade and obtained from VWR Chemicals. Anhydrous chloroform and methanol were purified and dried according to standard methods prior to use and, anhydrous dimethyl sulfoxide was obtained from Sigma Aldrich (≥99.5%).For experiments working with radioactive substances, water (Ultrapur, VWR) was used for preparation of buffers, HCl and NaCl solutions. 5.5 M HCl prepared by dilution of hydrochloric acid 37% (Emprove, VWR).
Methods
1D 1H and 13C NMR spectra were recorded on either a Bruker AV-500 or on a Bruker AV-400 spectrometer at room temperature using standard pulse programs. Chemical shifts (δ) are quoted in parts per million (ppm) and referenced to the appropriate residual solvent signal. Coupling constants (J) are reported to the nearest 0.5 Hz. Mass spectra (m/z) of phospholipids were run on a MALDI micro MX – TOF mass spectrometer from Waters. Samples were spotted 1:1 v/v with a matrix solution (4-nitroaniline, 10 mg mL−1 in methanol) and measured in reflector mode. High resolution mass spectra (m/z) were recorded on either a VG platform II or VG AutoSpec spectrometers, with only molecular ions (M+, MH+, MNa+, MK+, MNH4+) and major peaks being reported. Flash chromatography (FC) was performed on silica gel (Merck Kieselgel 60 F254 320–400 mesh). Thin Layer Chromatography (TLC) was performed using TLC Silica Gel 60 F24 (aluminium sheets 20 × 20 cm for analytical runs and glass plates 20 × 20 cm for preparative TLC purifications of phospholipids) from Merck. Plates were visualised either by quenching of ultraviolet fluorescence (λ = 254 and 366 nm) or by charring with 10% KMnO4 in 1 M H2SO4. Phospholipids were additionally visualised by charring with 5% primuline in acetone:water (8:2 v/v), lipids appearing as.yellow spots under 365 nm wavelength. Flash purifications were carried out in a Isolera™ Spektra System using Biotage® SNAP Ultra C18 for reverse phase conditions (12, 30 and 60 g sizes) and, Biotage® SNAP Ultra C18 for normal phase conditions (25, 50 and 100 g sizes). Gradients are indicated throughout the text. Analytical HPLC chromatograms were run in a Waters AutoPurification™ System equipped with a 2489 UV/vis Dectector and a 3100 Mass Detector, using a Phenomenex Gemini C18 column (150 mm × 4.6 mm) at a flow rate of 1.2 mL min−1 and gradient A: (5–98% B over 12 min then 98% B for 3 min) buffer A = H2O (0.1% TFA), buffer B = MeCN (0.1% TFA). Supelco Iso-Disc™ Filters PFTE-4-4 (4 mm × 0.45 μm) from Sigma Aldrich were used for sample filtration prior injection.
The lipid-coated decafluorobutane-filled microbubbles (MBs) were produced using a modified formulation described previously.18 1 mL of the resulting lipid solution (refer to Section 4 in this document for further details) was sealed in a 2 mL glass vial and the headspace was then purged with decafluorobutane at room temperature. The microbubbles were produced via mechanical agitation using a dental HL-AH High Speed Digital Amalgamator Amalgam Capsule Blend Mixer HL-AH (4000 rpm for 30 s, two cycles). Particle size of non-radioactive MBs was measured on a bright-field microscope (Nikon Eclipse 50×i, 40× objective), 10 μL diluted samples (1:100 dilution factor from stock sample) were first introduced into a haemocytometer and then sized and counted according to our reported protocol.19 ζ-potential analyses of microbubbles in solution (1:1000 dilution factor from stock sample) were measured with a NanoSizer (Malvern Nano-Zs, UK) and using DTS1070 cuvettes. Alternatively, 68Ga-labelled MBs were counted and sized using a Countess II FL Automated Cell Counter from Life Technologies. Non-radioactive controls were run to confirm size and concentration of MBs presented non-significant differences from both methods used to count and size MBs. MBs were purified by centrifugation adapting a reported method using a ROTINA 35 R centrifuge from Hettich Zentrifugen.17
68Ga was eluted from an Eckert & Ziegler Radiopharma GmbH GalliaPharm® 68Ge/68Ga generator (Berlin, Germany) using a fully automated Modular-Lab system. Aqueous HCl solutions (0.1 M, 5 mL) was passed through the generator and the eluate was collected in a single fraction and used directly for 68Ga-labeling reactions (refer to radiochemistry section below for further details).
Different purification cartridges were used for radioactive experiments: silica-based strong cation exchange cartridge (Bond Elut-SCX, 100 mg, 1 mL) purchased from Agilent (further referred as SCX cartridge); Sep-Pack® Light tC18 cartridge (145 mg sorbent per cartridge), Sep-Pack® Plus Light C8 cartridge (145 mg sorbent per cartridge), Oasis® HLB cartridge (1cc/30 mg) and Oasis® Prime HLB cartridge Plus Light (100 mg) acquired from Waters. HybridSPE® – Phospholipid (30 mg/1 mL SPE Tubes) acquired from SUPELCO. iTLC experiments were measured on a Scan-RAM™ PET/SPECT radio-TLC Scanner from LabLogic using iTLC-SG – Glass microfibre chromatography paper impregnated with silica gel from Agilent Technologies. Analysis data was performed with Laura 3 software (LabLogic, Sheffield, UK). Radio-HPLC was carried out on an Agilent 1100 series HPLC system (Agilent Technologies, Stockport, UK) equipped with a γ-RAM Model 3 gamma-detector (IN/US Systems Inc., Florida, USA) and Laura 3 software (LabLogic, Sheffield, UK) was used for processing all analytical HPLC chromatograms. For tetrazine chelators, a Phenomenex Germini 5 u C18 (150 × 4.6 mm) was used and an acidic gradient: 5–95% B over 15 min, then 95% B for 5 min, where A = water (0.1% TFA) and B = acetonitrile (0.1% TFA). For phospholipids a Phenomenex Bondclone 10 C18 (150 × 3.90 mm) was used, as well as a neutral gradient: 5–95% B over 7 min, then 95% B for 8 min, where A = 100 mM ammonium formate (H2O) and B = methanol.
68GaCl3 was eluted from a GalliaPharm® 68Ge/68Ga Generator from Eckert & Ziegler Radiopharma GmbH (0.74 GBq) using a fully automated Modular-Lab system and an adapted NaCl based method reported previously.20 Briefly, 68GaCl3 was eluted in 5 mL of a 0.1 M HCl solution, which was subsequently trapped on a SCX cartridge (pre-conditioned with 1 mL 5.5 M HCl and 10 mL water) and the activity was then eluted, with minimal loss (2–5%), using a mixture of 12.5 μL of 5.5 M HCl and 500 μL of 5 M NaCl solution. This eluate was subsequently buffered for direct use in 68Ga-labelling experiments.
Organic syntheses
All the syntheses and experiments containing NHS ester compounds were performed under anhydrous conditions and an atmosphere of nitrogen in flame-dried glassware. All purified products were kept in the freezer and found to be stable for more than six months.Synthesis of PE-NOTA (1) and PE-DOTA (2). 18:0 PE-NH2 (7.5 mg, 10 μmol) was dissolved in 3 mL of a chloroform:methanol mixture (0.6
:0.4 v/v) and triethylamine (50 μL). To this solution, either DOTA-NHS (10.0 mg, 13.1 μmol) or NOTA-NHS (10.0 mg, 15.2 μmol) was added and the mixture was left to stir for 5 h. After that, the crude was concentrated in rotavapour and purified by preparative-TLC. Rf for PE-DOTA = 0.10–0.19 (CHCl3:MeOH:H2O 6:4:0.2 v/v) and, Rf for PE-NOTA = 0.08–0.16 (CHCl3:MeOH:H2O 6:4:0.2 v/v).
PE-DOTA. 9.2 mg, 8.2 μmol, 82%.1H NMR (400 MHz, CDCl3): δ (ppm) 0.87 (t, CH3, 6H, 3JH–H = 6.5 Hz), 1.25 (s, CH2, 56H), 1.57 (m, CH2CH2CO, 4H), 2.27 (m, CH2CH2CO, 4H), 2.41–3.18 (m, NCH2CH2N + NCH2CH2N + NCH2COOH, 22H), 3.64–4.43 (m, CH2CH2N + CH2CH2N + PO4CH2CH + COOCH2COH + COCH2N, 10H), 5.17 (s, PO4CH2CH, 1H). MALDI (matrix: 4-nitroaniline); m/z for [C59H118N6O16PNa]+ ([M − H + NH4 + Na + CH3CH2OH]+) expected = 1220.8, found 1220.7 and [C59H117N6O16PNa2]+ ([M − 2H + NH4 + 2Na + CH3CH2OH]+) expected = 1242.8, found 1242.6.
PE-NOTA. 8.3 mg, 8.0 μmol, 80%. 1H NMR (400 MHz, CDCl3): δ (ppm) 0.87 (t, CH3, 6H, 3JH–H = 6.6 Hz), 1.25 (s, CH2, 56H), 1.56–1.90 (m, CH2CH2CO + NCH2CH2N + NCH2CH2N, 16H), 2.26 (m, CH2CH2CO, 4H), 2.64–2–92 (m, NCH2COOH, 6H), 3.25–3.45 (m, CH2CH2N, 2H), 3.56–4.41 (m, CH2CH2N + PO4CH2CH + COOCH2COH + 6H), 5.19 (s, PO4CH2CH, 1H). MALDI (matrix: 4-nitroaniline); m/z for [C55H109N5O14PNa]− ([M − 3H + NH4 + Na + CH3CH2OH]−) expected = 1117.8, found 1117.5 and [C55H110N5O14P]− ([M − 2H + NH4 + CH3CH2OH]−) expected = 1095.7, found 1095.4.
Synthesis of PE-isothiocyanate
18:0 PE-NH2 (50 mg, 66.9 μmol) was dissolved in 15 mL of a chloroform:methanol mixture (0.8:0.2 v/v) and triethylamine (10 μL). To this solution, a solution of p-phenylene diisothiocyanate (130 mg, 690.0 μmol, dissolved in 3 mL of chloroform) was dropwise added and the mixture was left to stir for 5 h. After that, the crude was concentrated in rotavapour and purified by flash column chromatography. Rf = 0.5 (CHCl3:MeOH:H2O 8:2:0.2 v/v). (57 mg, 60.2 μmol, 90%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.88 (t, CH3, 6H, 3JH–H = 6.5 Hz), 1.25 (s, CH2, 56H), 1.53 (m, CH2CH2CO, 4H), 2.26 (m, CH2CH2CO, 4H), 3.70–4.20 (m, CH2CH2N + CH2CH2N + trans-PO4CH2CH + COOCH2COH, 7H), 4.36 (s, cis-PO4CH2CH, 1H), 5.19 (s, PO4CH2CH, 1H), 7.09 (d, CH, 2H, 3JH–H = 8.3 Hz), 7.47 (m, CH, 2H). MALDI (matrix: 4-nitroaniline); m/z for [C51H93N3O9PS2]+ ([M + H + CH3CH2OH]+) expected = 986.6, found 986.6.
Synthesis of PE-iso-DOTA-GA (3)
PE-iso (7.0 mg, 7.45 μmol) was dissolved in 1 mL of a chloroform:methanol mixture (0.5:0.5 v/v) and triethylamine (50 μL). To this solution, 2,2′,2′′-(10-(4-((2-aminoethyl)amino)-1-carboxy-4-oxobutyl)-1,4,7,10 tetraazacyclododecane-1,4,7-triyl)triacetic acid (NH2-DOTA-GA, 10.0 mg, 19.28 μmol) was added and the mixture was left to stir for 5 h. After that, the crude was concentrated in rotavapour and purified by preparative TLC. Rf = 0.15–0.25 (CHCl3:MeOH:H2O 5:5:0.2 v/v). (6.8 mg, 4.65 μmol, 62%). 1H NMR (400 MHz, CDCl3): δ (ppm) 0.71 (t, CH3, 6H, 3JH–H = 6.5 Hz), 1.09 (s, CH2, 56H), 1.44 (m, CH2CH2CO, 4H), 1.69 (m, CH2CH2, 2H), 1.93–2.55 (m, NCH2CH2N + NCH2CH2N + CH2CH2CO, 20H), 2.58–2.73 (m, NCH2COOH, 6H), 2.88 (m, CH2CH2, 2H), 3.62–3.88 (m, CH2CH2N + CH2CH2N + PO4CH2CH + COOCH2COH + CH2CH2, 8H), 4.17–4.27 (m, CH2CH2, 2H), 5.06 (s, PO4CH2CH, 1H), 7.21 (s, CH, 4H). MALDI (matrix: 4-nitroaniline); m/z for [C72H132N10O18PS2Na2]+ ([M − 2H + NH4 + 2Na + CH3CH2OH]+) expected = 1565.9, found 1566.0; [C72H133N10O18PS2Na]+ ([M − H + NH4 + Na + CH3CH2OH]+) expected = 1543.9, found 1544.0.
Synthesis of PE-PEG4-TCO (4) and PE-TCO (7)
18:0 PE-NH2 (10 mg, 13.3 μmol) was dissolved in 7.5 mL of a chloroform:methanol mixture (0.56:0.44 v/v) and triethylamine (50 μL). To this solution, either a solution of TCO-PEG4-NHS (10 mg, 19.43 μmol dissolved in 0.3 mL chloroform) or TCO-NHS (5 mg, 18.71 μmol) was added and the mixture was left to stir for 5 h. After that, the crude was concentrated in vacuo and purified by preparative TLC. Rf for PE-PEG4-TCO = 0.45 (CHCl3:MeOH:H2O 8:2:0.2 v/v) and, Rf for PE-TCO = 0.50 (CHCl3:MeOH:H2O 8:2:0.2 v/v).
Synthesis of bifunctional chelators
3-(4-Hydroxyphenyl)propionic acid (3.39 g, 20.8 mmol) was dissolved in 21 mL of a solution of methanol: water: 4.0 N NaOH (0.71:0.19:0.1 v:v:v). To this solution, ethylenediamine-N,N′-diacetic acid (1.80 g, 10.4 mmol) suspended in 5 mL of NaOH (2 N) was added. The pH of the mixture was adjusted to 6.7 and the solution was cooled in an ice bath. Then, formaldehyde (aqueous solution at 37%, 20.8 mmol, 1.5 mL) was added dropwise while keeping the pH of the solution at 6.7 by intermittent addition of NaOH (4 N) and, the suspension was refluxed for 4 h at 70 °C. After that, the solution was cooled to room temperature and methanol was removed on a rotary evaporator. The remaining aqueous solution was diluted with water (15 mL), acidified with HCl (4 N) to pH 5.5 and extracted with diethyl ether (15 mL × 3) to remove unreacted 3-(4-hydroxyphenyl)propionic acid. The aqueous phase was then adjusted to pH 3 with HCl (6 N) and unreacted ethylenediamine-N,N′-diacetic acid was filtered off. The filtrate was then brought to neutrality with NaOH (4 N) and concentrated in vacuo. The crude was allowed to flash chromatography using an Isolera™ Spektra System (Biotage® SNAP Ultra C18 60 g cartridge, A: H2O with 0.1% TFA, B: CH3CN with 0.1% TFA. Gradient: 2–40% B, 55 CV). (277 mg, 0.52 mmol, 6%). 1H NMR (400 MHz, dmso d6): δ (ppm) 2.45 (t, CH2CH2COOH, 3JH–H = 7.8 Hz, 4H), 2.70 (t, CH2CH2COOH, 3JH–H = 7.8 Hz, 4H), 3.20 (s, CH2N, 4H), 3.65 (s, CH2COOH, 4H), 4.02 (s, CH2N, 4H), 6.79 (d, CHarom, 3JH–H = 8.2 Hz, 2H), 7.07 (m, CHarom, 4H). 13C NMR (100 MHz, dmso- d6): δ (ppm) 29.5 (2C), 35.6 (2C), 49.5 (CH2N, 2C), 52.2 (2C), 52.7 (2C), 115.4 (2C), 118.7 (2C), 129.8 (2C), 131.4 (2C), 131.9 (2C), 154.5 (2C), 170.2 (2C), 173.8 (2C). HPLC/MS τr = 7.70 min (gradient A); m/z for [C26H33N2O10]+ ([M + H]+) expected = 533.2135, found 533.2142.
HBED-CC (5.3 mg, 9.4 μmol) was dissolved in 0.7 mL of a mixture of H2O:CH3CN (50:50 v/v) containing 20 μL in N,N-diisopropylethylamine. To this solution, 190 μL of FeCl3 (0.1 M in water) was added and the reaction mixture was stirred for 1 h and then evaporated under reduced pressure to dryness. The resulting solid was dissolved in CH3CN and evaporated to dryness again to remove free water. 1.25 mL of anhydrous dmso was then added. To this solution, a solution of HATU (3.5 mg, 9.5 μmol) in 0.25 mL of anhydrous dmso and 0.050 mL N,N-diisopropylethylamine was added dropwise. After 15 min, (4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)methanamine (2.3 mg, 11.3 μmol) dissolved in 0.5 mL of anhydrous dmso was added dropwise and the reaction was left to react for 24 h. The reaction was then quenched with 2 mL of H2O and the solution was lyophilized. The remaining solid was dissolved in a mixture of H2O with 10–15% of dmso and purified by flash chromatography (Isolera™ Spektra System). Briefly, the crude was loaded in a Biotage® SNAP Ultra C18 30 g cartridge and flushed with 25 mL of HCl (1 M) and washed with 5 mL of H2O. The disappearance of the violet colour indicated the removal of iron(III) species. The remaining mixture was eluted with the following gradient: A: H2O with 0.1% TFA, B: CH3CN with 0.1% TFA. A to B: 2 to 30%, 30 CV, 30 to 70%, 5 CV and 70 to 98%, 5 CV. The purified product was lyophilized and the residue dried in vacuo to give a pink solid as 7 (1.5 mg, 2.1 μmol, 22%). The product was stable in the freezer for more than six months. 1H NMR (400 MHz, methanol-d4): δ (ppm) 6.82 (dd, CH, 3JH–H = 22.3, 8.4 Hz, 2H), 7.18–7.11 (m, CH, 4H), 7.23 (d, CH, 3JH–H = 8.2 Hz, 2H), 8.42 (d, CH, 3JH–H = 8.2 Hz, 2H), 4.42 (s, NHCH2C, 2H), 4.21 (s, NCH2C, 2H), 4.11 (s, NCH2C, 2H), 3.78 (s, NCH2COOH, 2H), 3.64 (s, NCH2COOH, 2H), 3.41 (s, NCH2CH2N, 2H), 3.02 (s, CH3, 3H), 2.91 (t, CH2CH2CONH, 3JH–H = 6.8 Hz, 2H), 2.81 (t, CH2CH2CONH, 3JH–H = 7.5 Hz, 2H), 2.59 (t, CH2CH2CONH, 3JH–H = 6.8 Hz, 2H), 2.59 (t, CH2CH2CONH, 3JH–H = 7.5 Hz, 2H). 13C NMR (100 MHz, methanol-d4): δ (ppm) 21.1, 31.0, 31.8, 36.9, 38.8, 43.5, 48.4, 48.6, 48.8, 49.0, 49.2, 49.4, 49.6, 51.1, 51.4, 54.2, 116.6, 119.9, 120.5, 128.9, 129.0, 131.7, 131.8, 132.2, 133.2, 133.3, 133.6, 133.7, 144.9, 156.0, 165.2, 168.7, 171.6, 172.0, 175.2, 176.7. HPLC: τr = 8.85 min (gradient A); ESI (negative mode): m/z for [C36H40N7O9]− ([M − H]−) expected = 714.2888, found 714.2893.
Synthesis of DOTA-GA-tetrazine (2,2′,2′′-(10-(1-carboxy-4-((2-(5-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-5-oxopentanamido)ethyl)amino)-4-oxobutyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetic acid) (5) was carried out according to a method reported by our group, with some minor modifications:10b
:0 v/v to 96:4 v/v) to give tert-butyl (4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)carbamate (0.37 g, 62% yield) as a bright pink solid. Rf = 0.39 (2% Et2O/DCM). 1H NMR (400 MHz, CDCl3): δ (ppm) 1.46 (s, COOC(CH3)3, 9H), 3.07 (s, CH3, 3H), 4.41 (d, CH2, 3JH–H = 5.6 Hz, 2H), 5.09 (sbr, NH, 1H), 7.47 (d, CHarom, 3JH–H = 8.4 Hz, 2H), 8.51 (d, CHarom, 3JH–H = 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ (ppm) 21.2 (1C), 28.5 (3C), 44.5 (1C), 79.9 (1C), 128.1 (2C), 128.3 (2C), 130.9 (1C), 144.1 (1C), 156.1 (1C) 164.0 (1C), 167.3 (1C). ESI (positive mode): m/z for [C15H20N5O2] ([M + H]+) expected = 302.1617, found 302.1621.
Radiochemistry
68GaCl3 was eluted from a GalliaPharm® 68Ge/68Ga Generator from Eckert & Ziegler Radiopharma GmbH (0.74 GBq) using a fully automated Modular-Lab system and an adapted NaCl-based method previously reported.20 Briefly, 68GaCl3 was eluted in 5 mL of a 0.1 M HCl solution, which was subsequently trapped on a SCX cartridge (preconditioned with 1 mL 5.5 M HCl and 10 mL water) and the activity was then eluted, with minimal loss (2–5%), using a mixture of 12.5 μL of 5.5 M HCl and 500 μL of 5 M NaCl solution. This eluate was subsequently buffered for direct use in 68Ga-labelling experiments.:water 80:20 v/v) for all chelators. [68Ga(PE-NOTA)] Rf = 0.66–0.76; [68Ga(PE-DOTA)] Rf = 0.67–0.78; [68Ga(PE-iso-DOTAGA)] Rf = 0.62–0.76; [68Ga(DOTAGA-Tetrazine)] Rf = 0.67–0.77 [68Ga(HBED-CC-Tetrazine)] Rf = 0.68–0.78; non-chelated 68Ga3+Rf < 0.3. iTLC plates were imaged and quantified by digital autoradiography using instruments and software described above.
:5:80, v/v/v)). The vial was sealed and activated to form microbubbles (see in the next section the experimental details for this process).
In order to remove unreacted [68Ga][Ga(5)], microbubbles were purified by centrifugation adapting a reported method.17 The vial containing the 68Ga-labelled microbubbles was put upside down and centrifuged at 350 g during 2 min using a ROTINA 35 R centrifuge. Then, the microbubbles were collected into a concentrated cake on the top of the vial and the remaining suspension (infranatant), which contained unreacted precursor and residual lipids and vesicles, which did not form part of the microbubble shell, was discarded. The microbubble cake was re-dispersed to a 1 mL volume of 20 vol% glycerol solution in PBS and stored in the same vial with PFB headspace until further use. The washing step was repeated three times or until no activity was measured in the infranatant solution. 68Ga-labelled MBs were counted and sized on a Countess II FL Automated Cell Counter and stored in the same vial with PFB headspace until further use.
(*) An alternative dienophile-modified phospholipid without the PEG4 spacer, named PE-TCO (7), was also tested for reaction with [68Ga][Ga(5)]. Under the same conditions, 7 showed low conversions (approx. 10%), probably as a consequence of its poor solubility in ethanol (data not shown).
Microbubble production
The lipid-coated, decafluorobutane-filled microbubbles (MBs) were manufactured using a modified formulation.18 Matsunaga et al. prepared MBs containing a lipid mixture of 85 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 10 mol% 1-palmitoyl-2-hydroxy-sn-glycero-3 phosphocholine (LPC), and 5 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N [methoxy(polyethylene glycol)-2000] (DSPE-PEG2000-NH2). In this paper, we studied the possibility of incorporating 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (PE-NH2) lipid into the MB shell. For that reason, we prepared MBs having a final lipid mixture of 85 mol% DPPC, 10 mol%, (either PE-NH2 or PE-PEG4-TCO) and 5 mol% DSPE-PEG2000-NH2. In all cases, the total lipid concentration was between 0.86 and 0.88 mg mL−1. The mixture of lipids was first dissolved in chloroform, dried over nitrogen gas, and then further dried in vacuo overnight to remove residual solvent from the lipid films. Then, 1 mL of a solution of propylene glycol, glycerol, and phosphate-buffered saline (PBS) (15:5:80, v/v/v) was used to rehydrate the films. This created a lipid suspension, which was immediately stirred for 10 min at 50–60 °C and later, decanted in a sealed 2 mL glass vial. Prior to activation, the headspace was purged with decafluorobutane at room temperature and, MBs were produced via mechanical agitation.
Biology experimental procedures
All animal experiments were conducted by licensed investigators in accordance with the United Kingdom Home Office Guidance on the Operation of the Animals (Scientific Procedures) Act 1986 (HSMO, London, UK, 1990) and with the published guidelines for the Welfare of Use of Animals in Cancer Research Institute Committee on Welfare of Animals in Cancer Research.22Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This work was supported by the Cancer Research UK (CRUK Multidisciplinary Award – C53470/A22353).References
- (a) The Chemistry of Molecular Imaging, ed.N. J. Long and W. T. Wong, Wiley-VCH, Weinheim, 2014 Search PubMed; (b) R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS.
- (a) L. Abou-Elkacem, S. V. Bachawal and J. K. Willmann, Eur. J. Radiol., 2015, 84, 1685–1693 CrossRef PubMed; (b) J. R. Lindner, Nat. Rev. Drug Discovery, 2004, 3, 527–532 CrossRef CAS PubMed; (c) F. Kiessling, S. Fokong, P. Koczera, W. Lederle and T. Lammers, J. Nucl. Med., 2012, 53, 345–348 CrossRef CAS PubMed.
- (a) B. A. Kaufmann, J. M. Sanders, C. Davis, A. Xie, P. Aldred, I. J. Sarembock and J. R. Lindner, Circulation, 2007, 116, 276–284 CrossRef CAS PubMed; (b) G. Korpanty, J. G. Carbon, P. A. Grayburn, J. B. Fleming and R. A. Brekken, Clin. Cancer Res., 2007, 13, 323–330 CrossRef CAS PubMed; (c) G. E. R. Weller, M. K. K. Wong, R. A. Modzelewski, E. Lu, A. L. Klibanov, W. R. Wagner and F. S. Villanueva, Cancer Res., 2005, 65, 533–539 CAS; (d) M. Smeenge, F. Tranquart, C. K. Mannaerts, T. M. de Reijke, M. J. van de Vijver, M. P. Laguna, S. Pochon, J. J. M. C. H. de la Rosette and H. Wijkstra, Invest. Radiol., 2017, 52, 419–427 CrossRef CAS PubMed; (e) J. K. Willmann, L. Bonomo, A. C. Testa, P. Rinaldi, G. Rindi, K. S. Valluru, G. Petrone, M. Martini, A. M. Lutz and S. S. Gambhir, J. Clin. Oncol., 2017, 35, 2133–2140 CrossRef CAS PubMed; (f) K. Ferrara, R. Pollard and M. Borden, Annu. Rev. Biomed. Eng., 2007, 9, 415–447 CrossRef CAS PubMed.
- (a) J. K. Willmann, N. van Bruggen, L. M. Dinkelborg and S. S. Gambhir, Nat. Rev. Drug Discovery, 2008, 7, 591–607 CrossRef CAS PubMed; (b) R. Weissleder and M. J. Pittet, Nature, 2008, 452, 580–589 CrossRef CAS PubMed.
- S. S. Gambhir, Nat. Rev. Cancer, 2002, 2, 683–693 CrossRef CAS PubMed.
- (a) N. Lazarova, P. W. Causey, J. A. Lemon, S. K. Czorny, J. R. Forbes, A. Zlitni, A. Genady, F. Stuart Foster and J. F. Valliant, Nucl. Med. Biol., 2011, 38, 1111–1118 CrossRef CAS PubMed; (b) J. K. Willmann, Z. Cheng, C. Davis, A. M. Lutz, M. L. Schipper, C. H. Nielsen and S. S. Gambhir, Radiology, 2008, 249, 212–219 CrossRef PubMed; (c) M. Palmowski, B. Morgenstern, P. Hauff, M. Reinhardt, J. Huppert, M. Maurer, E. C. Woenne, S. Doerk, G. Ladewig, J. W. Jenne, S. Delorme, L. Grenacher, P. Hallscheidt, G. W. Kauffmann, W. Semmler and F. Kiessling, Invest. Radiol., 2008, 43, 162–169 CrossRef CAS PubMed; (d) P. Walday, H. Tolleshaug, T. Gjoen, G. M. Kindberg, T. Berg, T. Skotland and E. Holtz, Biochem. J., 1994, 299, 437–443 CrossRef CAS PubMed; (e) M. S. Tartis, D. E. Kruse, H. Zheng, H. Zhang, A. Kheirolomoom, J. Marik and K. W. Ferrara, J. Controlled Release, 2008, 131, 160–166 CrossRef CAS PubMed.
- (a) P. Spang, C. Herrmann and F. Roesch, Semin. Nucl. Med., 2016, 46, 373–394 CrossRef PubMed; (b) M. Fani, J. P. André and H. R. Maecke, Contrast Media Mol. Imaging, 2008, 3, 53–63 CrossRef CAS PubMed.
- (a) S. Lin, A. Shah, J. Hernández-Gil, A. Stanziola, B. I. Harriss, T. O. Matsunaga, N. Long, J. Bamber and M.-X. Tang, J. Photoacoust., 2017, 6, 26–36 CrossRef PubMed; (b) P. S. Sheeran, S. Luois, P. A. Dayton and T. O. Matsunaga, Langmuir, 2011, 27, 10412–10420 CrossRef CAS PubMed.
- (a) A. Schmidtke, T. Lappchen, C. Weinmann, L. Bier-Schorr, M. Keller, Y. Kiefer, J. P. Holland and M. D. Bartholoma, Inorg. Chem., 2017, 56, 9097–9110 CrossRef CAS PubMed; (b) E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 181, 273–280 CrossRef CAS; (c) E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 190, 37–46 CrossRef CAS; (d) R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- (a) H. L. Evans, L. Carroll, E. O. Aboagye and A. C. Spivey, J. Labelled Compd. Radiopharm., 2014, 57, 291–297 CrossRef CAS PubMed; (b) H. L. Evans, Q.-D. Nguyen, L. S. Carroll, M. Kaliszczak, F. J. Twyman, A. C. Spivey and E. O. Aboagye, Chem. Commun., 2014, 50, 9557–9560 RSC; (c) J.-P. Meyer, P. Adumeau, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 2791–2807 CrossRef CAS PubMed.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- R. Selvaraj and J. M. Fox, Curr. Opin. Chem. Biol., 2013, 17, 753–760 CrossRef CAS PubMed.
- M. Eder, B. Wängler, S. Knackmuss, F. LeGall, M. Little, U. Haberkorn, W. Mier and M. Eisenhut, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 1878–1886 CrossRef CAS PubMed.
- S. R. Banerjee, Z. Chen, M. Pullambhatla, A. Lisok, J. Chen, R. C. Mease and M. G. Pomper, Bioconjugate Chem., 2016, 27, 1447–1455 CrossRef PubMed.
- (a) J. Schuhmacher, G. Klivenyi, R. Matys, M. Stadler, T. Regiert, H. Hauser, J. Doll, W. Maier-Borst and M. Zoller, Cancer Res., 1995, 55, 115–123 CAS; (b) J. Schuhmacher, G. Klivenyi, W. E. Hull, R. Matys, H. Hauser, H. Kalthoff, W. H. Schmiegel, W. Maier-Borst and S. A. Matzku, Int. J. Radiat. Appl. Instrum., Part B, 1992, 19, 809–824 CrossRef CAS.
- M. Eder, O. Neels, M. Müller, U. Bauder-Wüst, Y. Remde, M. Schäfer, U. Hennrich, M. Eisenhut, A. Afshar-Oromieh, U. Haberkor and K. Kopka, Pharmaceuticals, 2014, 7, 779–796 CrossRef CAS PubMed.
- J. A. Feshitan, C. C. Chen, J. J. Kwan and M. A. Borden, J. Colloid Interface Sci., 2009, 329, 316–324 CrossRef CAS PubMed.
- (a) S. Lin, A. Shah, J. Hernández-Gil, A. Stanziola, B. I. Harriss, T. O. Matsunaga, N. Long, J. Bamber and M.-X. Tang, J. Photoacoust., 2017, 6, 26–36 CrossRef PubMed; (b) P. S. Sheeran, S. Luois, P. A. Dayton and T. O. Matsunaga, Langmuir, 2011, 27, 10412–10420 CrossRef CAS PubMed.
- C. A. Sennoga, V. Mahue, J. Loughran, J. Casey, J. M. Seddon, M. Tang and R. J. Eckersley, Ultrasound Med. Biol., 2010, 36, 2093–2096 CrossRef PubMed.
- M. K. Schultza, D. Muellerc, R. P. Baumc, G. L. Watkinsa and W. A. P. Breeman, Appl. Radiat. Isot., 2013, 76, 46–54 CrossRef PubMed.
- M. Zöller, J. Schuhmacher, J. Reed, W. Maier-Borst and S. Matzku, J. Nucl. Med., 1992, 33, 1366–1372 Search PubMed.
- P. Workman, E. O. Aboagye, F. Balkwill, A. Balmain, G. Bruder, D. J. Chaplin, J. A. Double, J. Everitt, D. A. H. Farningham, M. J. Glennie, L. R. Kelland, V. Robinson, I. J. Stratford, G. M. Tozer, S. Watson, S. R. Wedge, S. A. Eccles, V. Navaratnam and S. Ryder, Br. J. Cancer, 2010, 102, 1555–1577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00684b |
| This journal is © The Royal Society of Chemistry 2019 |