
Heterologous production of asperipin-2a: proposal for sequential oxidative macrocyclization by a fungi-specific DUF3328 oxidase†
Ying
Ye
a,
Taro
Ozaki
 *a,
Myco
Umemura
bc,
Chengwei
Liu
a,
Atsushi
Minami
a and
Hideaki
Oikawa
*a
*a,
Myco
Umemura
bc,
Chengwei
Liu
a,
Atsushi
Minami
a and
Hideaki
Oikawa
*a
aDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Hokkaido, Japan. E-mail: ozaki@sci.hokudai.ac.jp; hoik@sci.hokudai.ac.jp
bBioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba 305-8566, Ibaraki, Japan
cComputational Bio Big Data Open Innovation Laboratory (CBBD-OIL), AIST, Tsukuba 305-8566, Ibaraki, Japan
First published on 28th November 2018
Abstract
Asperipin-2a is a ribosomally synthesized and post-translationally modified peptide isolated from Asperigillus flavus. Herein, we report the heterologous production of asperipin-2a and determination of its absolute structure. Notably, the characteristic bicyclic structure was likely constructed by a single oxidase containing the DUF3328 domain.
RiPPs (ribosomally synthesized and post-translationally modified peptides) are a diverse and rapidly expanding class of natural products with potent biological activities as well as unique structures.1 Although RiPPs can be found in all three domains of life (archaea, bacteria, and eukaryotes), they have rarely been identified in fungi. In Basidiomycetes, α-amanitin, phallacidin, and omphalotin were demonstrated to be RiPPs.2–4 In contrast, in Ascomycetes, there were no such reports until the biosynthetic gene cluster for ustiloxin B was discovered (Fig. 1).5–7 Recently, it was also reported that phomopsin A from Phomopsis leptostromformis and epichloëcyclins from the genus Epichloë belong to the RiPP class.8,9 Led by the identification and characterization of the biosynthetic genes for ustiloxin B, Nagano and co-workers extensively searched similar gene clusters in fungal genomes and classified them into more than 40 types.10 They also identified asperipin-2a (1) as a product derived from one of the gene clusters. The co-occurrence of genes encoding precursor peptides and enzymes with the DUF3328 domain is a unique feature of these gene clusters. By employing an Aspergillus oryzae expression system, we revealed that two oxidases UstYa and UstYb, both of which contain the DUF3328 domain, and tyrosinase UstQ were involved in the oxidation and macrocyclization of the precursor peptide that furnishes the 13-membered ring of ustiloxins.11 The details of the macrocyclization process as well as the function of DUF3328 proteins in the other gene clusters are yet to be elucidated.
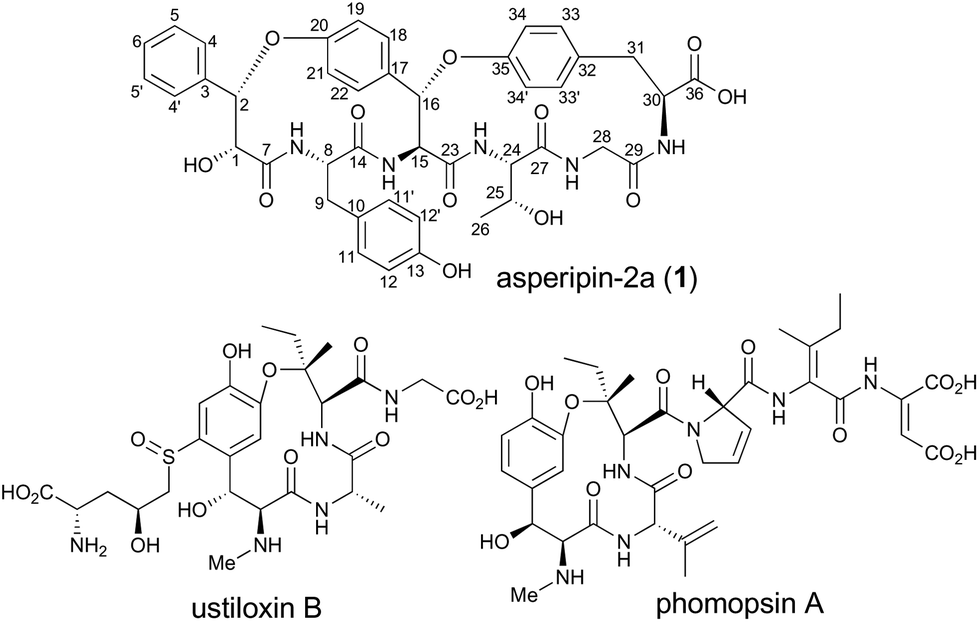 | ||
| Fig. 1 Asperipin-2a (1) and related cyclic peptides. | ||
1 is a bicyclic peptide that possesses two macrocyclic ether rings consisting of 14- and 17-membered paracyclophans. The putative biosynthetic gene cluster for 1 is composed of four genes encoding the precursor peptide (AFLA_041400, referred to aprA in this study), UstYa/Yb homolog (AFLA_041390, aprY), a transporter (AFLA_041380, aprT), and isoflavone reductase-related enzyme (AFLA_041370, aprR) (Fig. S1†). As proposed in the biosynthesis of ustiloxin B, AprY containing the functionally unknown domain DUF3328 likely catalyzes oxidative macrocyclization during the biosynthesis of 1. However, in the previous study, only aprA and aprY gene deletions were investigated and the biosynthetic pathway of 1 remains unclear.10 In the present study, we report the heterologous expression of the four genes involved in the production of 1, showing that DUF3328 oxidase is involved in the biosynthesis of this compound. The improved production yield of 1 also allowed us to determine its absolute configuration by chemical degradation, chiral HPLC, and NMR analyses.
To reconstitute the biosynthetic pathway, four genes, aprA, aprY, aprR, and aprT, were amplified from genomic DNA of Aspergillus flavus. The aprA gene was cloned into the plasmid pUSA212 to construct pUSA2-aprA, while the aprY gene was inserted into pUARA212 to generate pUARA2-aprY (Fig. S2†). The two other genes, aprR and aprT, were cloned into pAdeA213 to construct pAdeA2-aprRT (Fig. S2†). Sequencing the aprA gene revealed that the precursor peptide contains eight repeats of the core sequence “FYYTGY” while the AprA sequence deposited in NCBI contains eleven repeats (see ESI, page S17†). The two plasmids, pUARA2-aprY and pAdeA2-aprRT, were introduced into A. oryzae NSAR1 (AO-WT) to generate AO-aprYRT. The resulting transformant did not produce 1 as the precursor peptide was absent in this strain. We then introduced pUSA2-aprA into this transformant to generate AO-aprAYRT. As we expected, the resulting transformant successfully produced 1 (Fig. 2). The 1D- and 2D-NMR spectra of the isolated compound as well as its HR-ESI-MS were in good agreement with previously reported spectra, confirming that the above four genes were sufficient for the biosynthesis of 1 (Table S3†). We also prepared the transformants AO-aprAYR and AO-aprAYT to test whether AprR and/or AprT were essential for biosynthesis. Although these showed significantly decreased levels of production, both transformants still produced trace amounts of 1, suggesting that AprR and AprT were required for biosynthesis and that adventitious proteins in the host strain complement their function. Considering that deletion of aprY in A. flavus abolished the production of 1 in the previous study, this observation suggests that AprY is involved in the formation of the bicyclic structure (Scheme 1).
 | ||
| Fig. 2 LC-MS profiles of the metabolites extracted from transformants. Chromatograms were extracted at m/z 810. | ||
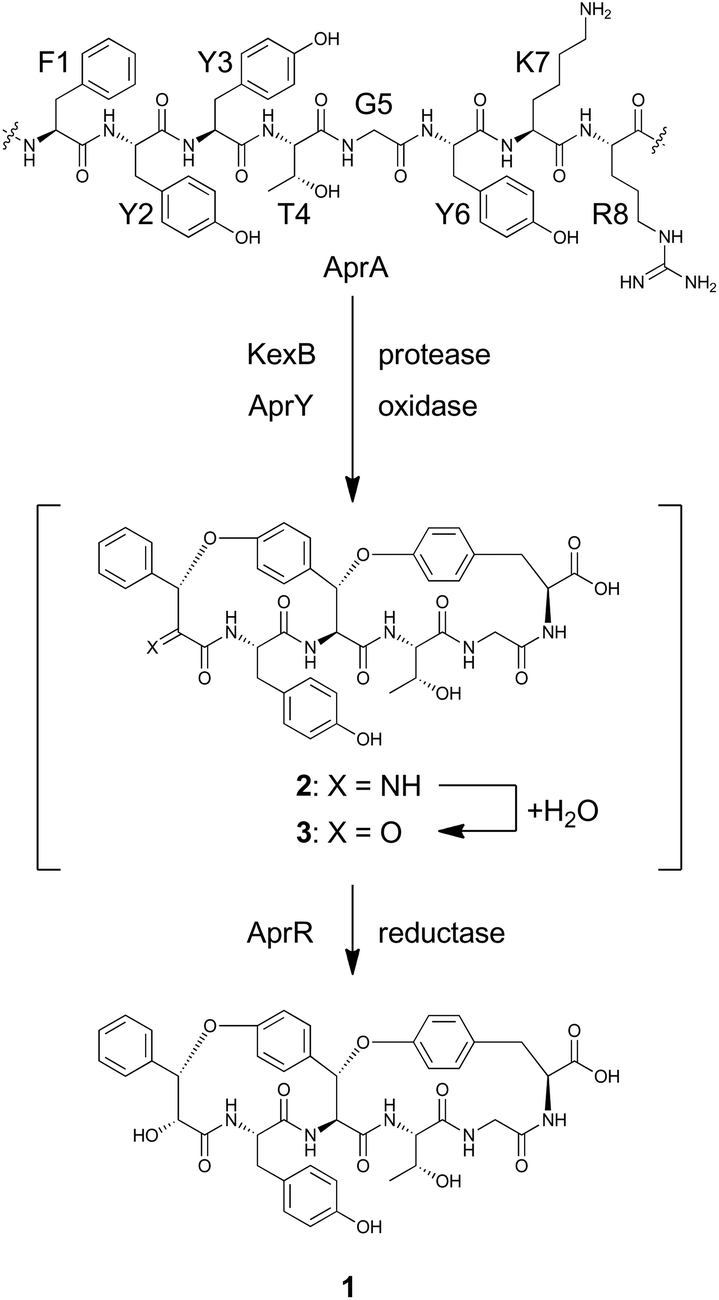 | ||
| Scheme 1 Putative biosynthetic pathway for 1. | ||
Using AO-aprYRT as a host strain, we also investigated the conversion of two AprA analogs, AprA/Y3F and AprA/Y6F. As either the third or the sixth tyrosine of the core peptide is substituted with phenylalanine, we expected that these mutants could not undergo cyclization and that monocyclic products might be obtained. We constructed the plasmids pUSA2-aprA/Y3F and pUSA2-aprA/Y6F and introduced them into AO-aprYRT. Neither AO-aprAYRT/Y3F nor AO-aprAYRT/Y6F generated the expected products, thus suggesting the importance of both the third and the sixth tyrosine for cyclization (Fig. S3†). As no related metabolites such as monocyclic compounds, were observed, the cyclization might be a sequential process. However, further experimental verification is necessary to address this hypothesis.
A BLAST search using AprA as a query retrieved orthologous precursor proteins from at least three strains including Aspergillus oryzae, Aspergillus parasiticus, and Aspergillus arachidicola (Table S4 and Fig. S4†). Notably, each gene was flanked by aprY, aprR, and aprT homologs, suggesting that they are involved in the biosynthesis of 1 or its congeners. The number of core peptide repeats of each AprA ortholog varies from one to eight. The third and the sixth tyrosine residues were strictly conserved among each repeat (Fig. S5†). The first phenylalanine was also conserved, indicating the importance of these three residues in the cyclization process. On the other hand, in some cases, the second, the forth, and the fifth residues were substituted with phenylalanine, histidine, and asparagine, respectively (Fig. S5†). This observation indicated that these positions did not affect cyclization and could be substituted for structural diversification.
The highly-strained 14-membered paracyclophane ring of 1 has limited conformational flexibility and is rotationally restricted,14 thus allowing us to elucidate its relative stereochemistry by 1D- and 2D-NMR analyses. Both H1 and H2 were observed as broad singlets, suggesting a small 3JHH value between these protons (Fig. 3A). On the basis of this observation, the relative stereochemistry at C1 and C2 can be deduced as (1R,2S) or (2R,1S). In addition, the NOEs between H16 and 15-NH, H16 and H18, and H15 and H22 clearly indicated that the relative configuration at C15 and C16 was either all-S or all-R (Fig. 3B).
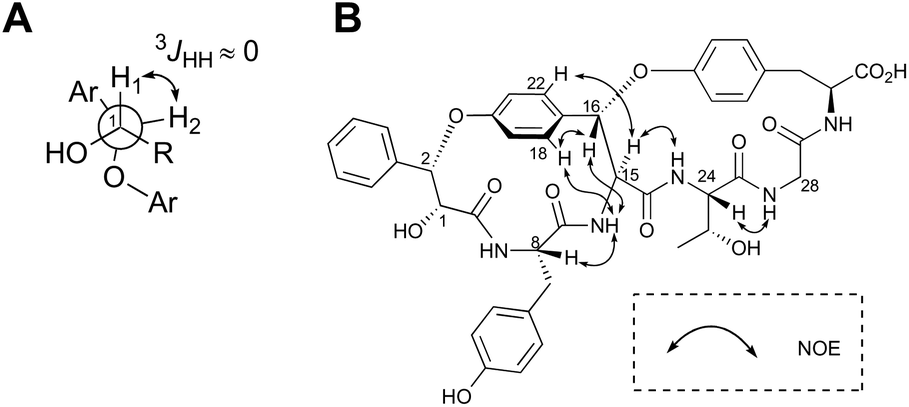 | ||
| Fig. 3 Stereochemical analysis of 1. (A) Newman projection of 3-phenyllactic acid moiety of 1. (B) Key ROESY correlations. | ||
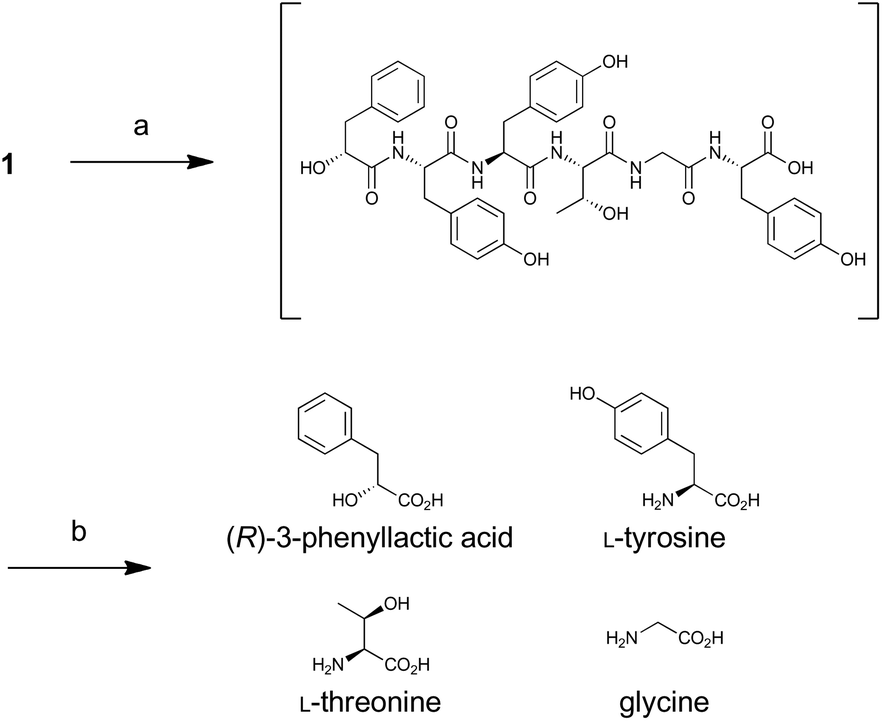
For determination of the absolute configuration, 1 was subjected to a Pd-mediated hydrogenolysis, followed by hydrolysis with 6 M HCl to give a mixture of 3-phenyllactic acid and amino acids (Scheme 2). A mixture of amino acids were then converted with Nα-(5-fluoro-2,4-dinitrophenyl)-L-leucinamide (L-FDLA) to yield L-FDLA derivatives.15–17 LC-MS analysis revealed that the retention times of the two constituent amino acids Tyr and Thr from 1 were identical to those of the L-Tyr- and L-Thr-derivatives, respectively (Fig. 4A and B). L-FDLA-Gly was also observed (Fig. 4C). The molar ratio of L-Tyr and Gly was roughly estimated to be 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by comparing the peak areas of FDLA derivatives (Fig. S6†). These results are consistent with the ribosomal synthesis of the precursor peptide AprA. The hydrolysate was also analyzed by chiral HPLC without derivatization. The retention time of 3-phenyllactic acid from 1 was identical to that of commercial (R)-3-phenyllactic acid, thus confirming the stereochemistry at C1 to be R (Fig. 4D). Together with the relative configuration as described above, the absolute structure of 1 was determined as shown in Fig. 1.
:1 by comparing the peak areas of FDLA derivatives (Fig. S6†). These results are consistent with the ribosomal synthesis of the precursor peptide AprA. The hydrolysate was also analyzed by chiral HPLC without derivatization. The retention time of 3-phenyllactic acid from 1 was identical to that of commercial (R)-3-phenyllactic acid, thus confirming the stereochemistry at C1 to be R (Fig. 4D). Together with the relative configuration as described above, the absolute structure of 1 was determined as shown in Fig. 1.
 | ||
| Scheme 2 (a) HCOONH4, 10% Pd/C, MeOH, 70 °C; (b) 6 M HCl, 110 °C. | ||
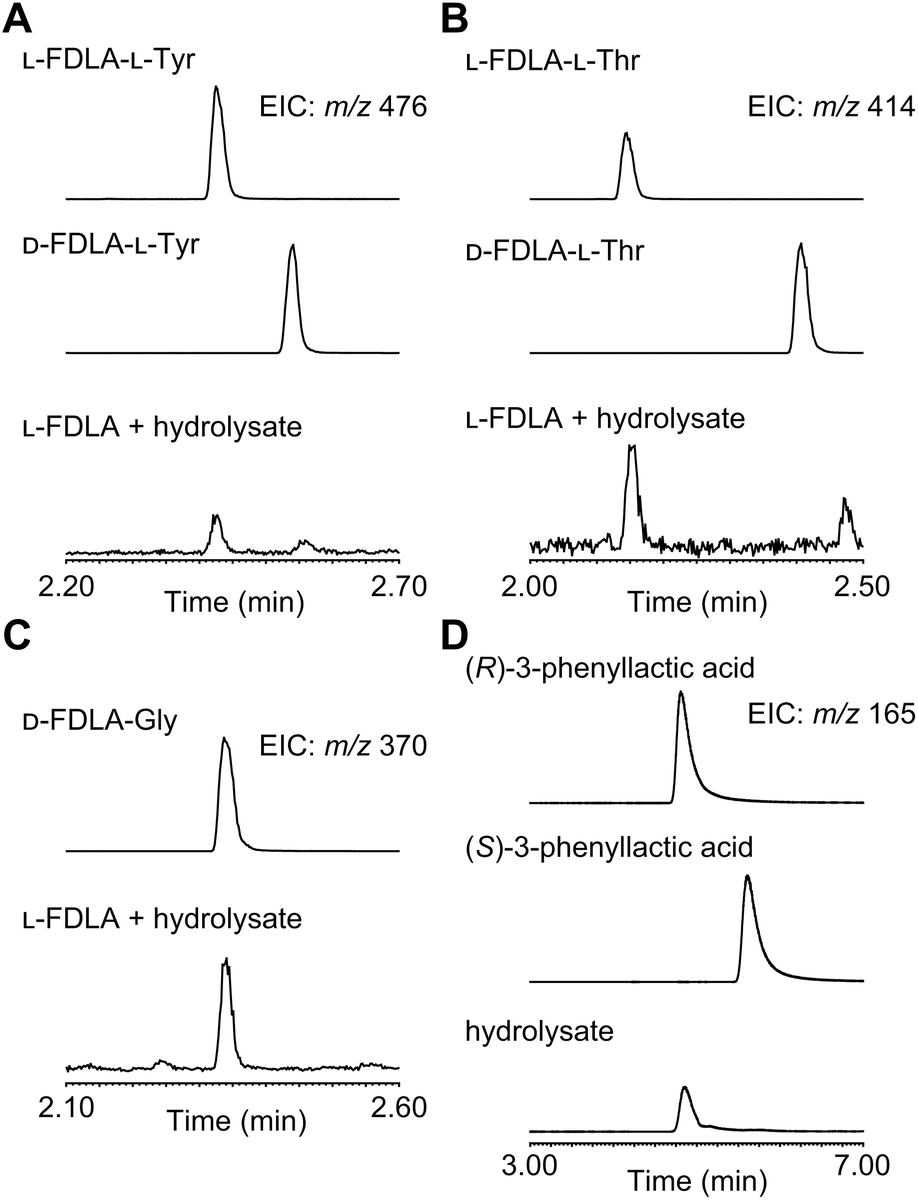 | ||
| Fig. 4 LC-MS profiles of the constituents of 1. (A) L-FDLA derivatized Tyr, (B) Thr, (C) Gly, and (D) chiral LC-MS analysis with authentic (R)- and (S)-phenyllactic acid. Chromatograms were extracted at m/z 476 for FDLA-Tyr, m/z 414 for FDLA-Thr, m/z 370 for FDLA-Gly, and m/z 165 for phenyllactic acid. | ||
The biosynthesis of 1 starts with transcription and translation of the structural gene aprA. The synthesized precursor peptide AprA contains eight repeats of the core sequence FYYTGY. Each repeat is flanked by KR, which is a target site of Golgi protease, KexB, suggesting that AprA undergoes a similar proteolytic process to that known in the biosynthesis of ustiloxin B.18 AprA is also modified by AprY and AprR to furnish 1. However, there is no information concerning the order of these post-translational modifications. As discussed above, the bicyclic structure of 1 is likely synthesized by the single enzyme AprY. Considering the relatively small size of AprY and that two C–O bonds were generated in the same stereochemical course, formation of these two bonds might be successively catalyzed in the same active site of the enzyme. In ustiloxin biosynthesis, initial hydroxylation and subsequent cyclization gave a macrocyclic system.11 This transformation accompanied hydroxylation at the Cβ position of L-Tyr. Similarly, macrocyclization of 1 may accompany an α-hydroxylation–dehydration sequence to give imine 2, which is readily hydrolyzed to yield putative ketone intermediate 3. We tempted to explain that the reductase AprR may be required for the final reduction to yield 1 (Scheme 1). As AprT shows homology to major facilitator superfamily protein (MFS_1, Pfam ID: PF07690), it is likely that this protein is not involved in biosynthesis but rather exports the product.
Besides RiPPs produced by Ascomycetes such as ustiloxin B and phomopsin A, plants also produce cyclopeptides that are structurally related to 1. Cyclopeptides, such as mauritine A,14 sanjoinin A,14,19,20 and ophiorrhisine A,21 possess 14-membered paracyclophane rings similar to that of 1 (Fig. S7†). Although DUF3328 proteins do not exist in plants, other oxidases that operate with a similar mechanism might be involved in the biosynthesis of these compounds. Future studies focusing on the mechanism of DUF3328 proteins may contribute to studies of similar cyclopeptides from other genera.
Conclusions
In this study, we achieved the heterologous production of 1 and determined its absolute structure, showing that DUF3328 protein AprY was involved in formation of the characteristic bicyclic structure. Together with our previous results on ustiloxin B, the present findings established the common strategy for macrocyclization of RiPPs produced by filamentous fungi. In both cases, oxidative cyclization catalyzed by UstY homologs accompanies a C–O bond formation between the phenol and Cβ of an amino acid moiety. Compared with the macrocyclization of ustiloxins, which requires three enzymes (two DUF3328s and one tyrosinase), macrocyclization of 1 would be more feasible for biochemical characterization, as it requires only one or two enzymes (AprY and/or AprR). However, preliminary attempts to reconstitute this enigmatic reaction were unsuccessful. Further studies are currently underway in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (JSPS KAKENHI Grants (15H01835 (H. O.), 16H06446 (A. M.), 17K15263 & 17H05425 (T. O.), 17H05456 (M. U.)). We are grateful to DAICEL Corp. for the use of chiral column.References
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- H. E. Hallen, H. Luo, J. S. Scott-Craig and J. D. Walton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 19097–19101 CrossRef CAS.
- S. Ramm, B. Krawczyk, A. Mühlenweg, A. Poch, E. Mösker and R. D. Süssmuth, Angew. Chem., Int. Ed., 2017, 56, 9994–9997 CrossRef CAS.
- N. S. van der Velden, N. Kälin, M. J. Helf, J. Piel, M. F. Freeman and M. Künzler, Nat. Chem. Biol., 2017, 13, 833–835 CrossRef CAS.
- M. Umemura, N. Nagano, H. Koike, J. Kawano, T. Ishii, Y. Miyamura, M. Kikuchi, K. Tamano, J. Yu, K. Shin-ya and M. Machida, Fungal Genet. Biol., 2014, 68, 23–30 CrossRef CAS.
- M. Umemura, H. Koike, N. Nagano, T. Ishii, J. Kawano, N. Yamane, I. Kozone, K. Horimoto, K. Shin-ya, K. Asai, J. Yu, J. W. Bennett and M. Machida, PLoS One, 2013, 8, e84028 CrossRef.
- T. Tsukui, N. Nagano, M. Umemura, T. Kumagai, G. Terai, M. Machida and K. Asai, Bioinformatics, 2015, 31, 981–985 CrossRef CAS.
- W. Ding, W. Q. Liu, Y. Jia, Y. Li, W. A. van der Donk and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3521–3526 CrossRef CAS.
- R. D. Johnson, G. A. Lane, A. Koulman, M. Cao, K. Fraser, D. J. Fleetwood, C. R. Voisey, J. M. Dyer, J. Pratt, M. Christensen, W. R. Simpson, G. T. Bryan and L. J. Johnson, Fungal Genet. Biol., 2015, 85, 14–24 CrossRef CAS.
- N. Nagano, M. Umemura, M. Izumikawa, J. Kawano, T. Ishii, M. Kikuchi, K. Tomii, T. Kumagai, A. Yoshimi, M. Machida, K. Abe, K. Shin-Ya and K. Asai, Fungal Genet. Biol., 2016, 86, 58–70 CrossRef CAS.
- Y. Ye, A. Minami, Y. Igarashi, M. Izumikawa, M. Umemura, N. Nagano, M. Machida, T. Kawahara, K. Shin-Ya, K. Gomi and H. Oikawa, Angew. Chem., Int. Ed., 2016, 55, 8072–8075 CrossRef CAS.
- K. Tagami, A. Minami, R. Fujii, C. Liu, M. Tanaka, K. Gomi, T. Dairi and H. Oikawa, ChemBioChem, 2014, 15, 2076–2080 CrossRef CAS.
- T. Ugai, A. Minami, R. Fujii, M. Tanaka, H. Oguri, K. Gomi and H. Oikawa, Chem. Commun., 2015, 51, 1878–1881 RSC.
- T. Gulder and P. S. Baran, Nat. Prod. Rep., 2012, 29, 899–934 RSC.
- P. Marfey, Carlsberg Res. Commun., 1984, 49, 591–596 CrossRef CAS.
- K. Fujii, Y. Ikai, T. Mayumi, H. Oka, M. Suzuki and K. Harada, Anal. Chem., 1997, 69, 3346–3352 CrossRef CAS.
- K. Fujii, Y. Ikai, H. Oka, M. Suzuki and K. Harada, Anal. Chem., 1997, 69, 5146–5151 CrossRef CAS.
- A. Yoshimi, M. Umemura, N. Nagano, H. Koike, M. Machida and K. Abe, AMB Express, 2016, 6, 9 CrossRef.
- R. Tschesche and I. Reutel, Tetrahedron Lett., 1968, 3817–3818 CrossRef CAS.
- R. Tschesche and H. Last, Tetrahedron Lett., 1968, 2993–2998 CrossRef CAS.
- T. Onozawa, M. Kitajima, N. Kogure, N. Peerakam, D. Santiarworn and H. Takayama, J. Nat. Prod., 2017, 80, 2156–2160 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob02824a |
| This journal is © The Royal Society of Chemistry 2019 |