
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-in-class ruthenium anticancer drug (KP1339/IT-139) induces an immunogenic cell death signature in colorectal spheroids in vitro†
Debora
Wernitznig
 *a,
Konstantinos
Kiakos
a,
Giorgia
Del Favero
b,
Nathalie
Harrer
c,
Herwig
Machat
c,
Annika
Osswald
c,
Michael A.
Jakupec
a,
Andreas
Wernitznig
c,
Wolfgang
Sommergruber
c and
Bernhard K.
Keppler
a
*a,
Konstantinos
Kiakos
a,
Giorgia
Del Favero
b,
Nathalie
Harrer
c,
Herwig
Machat
c,
Annika
Osswald
c,
Michael A.
Jakupec
a,
Andreas
Wernitznig
c,
Wolfgang
Sommergruber
c and
Bernhard K.
Keppler
a
aDepartment of Inorganic Chemistry and Research Cluster ‘Translational Cancer Therapy Research’, Faculty of Chemistry, University of Vienna, Währinger Straße 42, 1090, Vienna, Austria. E-mail: debora.wernitznig@univie.ac.at
bDepartment of Food Chemistry and Toxicology, Faculty of Chemistry, University of Vienna, Währinger Straße 38, 1090, Vienna, Austria
cBoehringer Ingelheim RCV GmbH & Co KG, 1121, Vienna, Austria
First published on 3rd April 2019
Abstract
The ruthenium complex sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)] (KP1339/IT-139) showed preclinical activity in a variety of in vivo tumor models including a highly predictive colon cancer model. The compound has entered clinical trials, where patients experienced disease stabilization accompanied by mild side effects. KP1339, a GRP78 inhibitor, disrupts endoplasmic reticulum (ER) homeostasis leading to cell death. The PERK/eIF2α-branch of the ER plays an essential role in the cascade of events triggering immunogenic cell death (ICD). ICD makes dying cancer cells ‘visible’ to the immune system, initiating a prolonged immune response against the tumor. As some metal-based chemotherapeutics such as oxaliplatin are able to induce ICD, we investigate whether KP1339 could also trigger induction of the ICD signature. For this, we employ a three-dimensional colon cancer spheroid model and show for the first time that the treatment with KP1339, a ruthenium-based complex, triggers an ICD signature hallmarked by phosphorylation of PERK and eIF2α, exposure of calreticulin on the cell membrane, release of high mobility group box 1 and secretion of ATP.
Significance to metallomicsThe impact of metal-based compounds on the immunological aspects of cancer is now emerging as a critical component of their therapeutic potential. It has been demonstrated based on preclinical and clinical data, that the immunomodulatory effects of platinum complexes provide a promising platform for their combination with immunotherapies. However, further studies are necessary to elucidate the complex interaction between cancer and the immune system and the role of metal-based drugs in modulating their intricate interplay. Our work highlights the importance of studying the immunological aspects of metal-based compounds, and emphasizes the potential of ruthenium-derived complexes in this emerging field. |
The technological advances and better understanding of the biology of cancer have enabled a range of therapeutic interventions involving different cellular targets, compromising the viability of cancer cells and often leading to disease remission. However, due to the suboptimal specificity and efficacy of available therapies and the succeeding adaptive-escaping mechanisms developed by cancer cells, relapses occur, often associated with increased malignancy and chemo-resistance. One important aspect that has gained attention in recent years is the critical role played by the immune system in the complex dynamics of cancer initiation, progression and response to therapies.1–4 The mechanisms underlying the opposing roles played by the different immune cell types (e.g., macrophages that support tumor growth and often contribute to an immunosuppressive milieu) and their interaction with cancer associated fibroblasts and other components of the tumor microenvironment have been shown to interfere with the outcome of treatments and to essentially influence the prognosis of cancer patients.5–8 Major efforts have been made to unveil immunomodulatory mechanisms which shape immune responses during tumorigenesis. In this context, a plethora of studies have reported encouraging results, particularly with checkpoint inhibitors (PD-1/PDL-1). This approach aims at converting ‘cold’ tumors into ‘hot’ tumors, where activated immune cells are able to effectively eliminate cancer cells.9–13
Although classic cytotoxic chemotherapy has been traditionally considered immunosuppressive, various clinical studies demonstrate that it often interacts positively with immunotherapy. Some groups have shown that anticancer agents such as oxaliplatin induce cell death accompanied by the release of crucial signals, rendering cancer cells ‘visible’ to the immune system. These signals initiate a cascade of events that can lead to long-term responses in immunocompetent hosts, a mechanism described as immunogenic cell death (ICD).14–16
Therefore, it is of major therapeutic relevance to explore the immunogenic potential of both clinically applied anti-cancer drugs and currently developed compounds. In this context, it is of utmost interest to characterize the molecular mechanisms of how cells die upon treatment as this subsequently influences the onset of ICD. Chemotherapy-induced ICD depends on the emission of key immunomodulatory damage-associated molecular patterns (DAMPs), such as: pre-apoptotic calreticulin (CRT) surface-exposure, extracellular adenosine triphosphate (ATP) and high mobility group box 1 (HMGB-1).
CRT surface exposure is able to induce dendritic cell (DC) maturation and uptake of tumor antigens.17–19 Release of ATP from dying cells stimulates purinergic P2RX7 receptors on the surface of DCs leading to the activation of the NLRP3 inflammasome. This stimulates IFN-γ production by tumor-specific cytotoxic T cells. ATP also functions as a potent chemoattractant for several cell types involved in immune responses, e.g., monocytes, granulocytes and DCs. Finally, HMGB-1 in the extracellular space binds to toll-like receptor 4 (TLR-4) on the surface of DCs to optimize the presentation of antigens from dying tumor cells. DAMPs are able to induce cross-presentation of processed tumor associated antigens by DCs to CD4+ and CD8+ T cells, resulting in specific immunity against the tumor.17–19
Although only in vivo vaccination studies in mice can furnish the complete proof of induction of ICD,20 we made use of complex 3D models to learn more about the ability of compounds to trigger an ICD-linked signature, a prerequisite for ICD. These 3D models have the advantage of being less experimentally demanding, more high-throughput and of employing cells of human origin. In addition, 3D models much better recapitulate the tumor architecture and biology compared to the widely used two-dimensional cultures (2D). 3D models mimic important characteristics of solid tumors, such as the accumulation of metabolites, the induction of hypoxia and the presence of distinct proliferating, quiescent and necrotic regions.20–23
Platinum-based compounds have been extensively studied and comprise some of the most widely used anticancer drugs in the clinic, although their use is associated with severe side effects, and their efficacy is hampered by the emergence of resistance.24
Ruthenium-derived complexes are an emerging class of anticancer compounds that could complement the existing armory of drugs due to their unique mechanism of action. KP1339 (IT-139, sodium trans-[tetrachloridobis(1H-indazole)ruthenate(III)]) is a clinically investigated drug that induces or enhances endoplasmic reticulum stress (ERS) by downregulating BiP (also known as GRP78).25,26
The endoplasmic reticulum (ER) possesses three different transmembrane receptors: IRE1α, ATF6 and PERK. Normally, these proteins are bound by the ER chaperone BiP. Once unfolded proteins accumulate, BiP releases ATF6, PERK and IRE1α, allowing their activation of the unfolded protein response (UPR). Under prolonged ERS conditions UPR is activated as an attempt to restore homeostasis by shutting down protein synthesis and arresting the cell cycle. If this condition persists, the PERK branch of the UPR via ATF4 induces the activation of CHOP, leading to apoptosis.
Neoplastic cells responding to ICD inducers undergo a phase of intense ERS characterized by the generation of reactive oxygen species (ROS), activation of PERK and increased cytoplasmic Ca2+. The active form of PERK phosphorylates the eukaryotic translation initiation factor 2α (eIF2α), shutting down protein translation. eIF2α activation is essential for CRT translocation to the cell membrane.27–30
Autophagy plays an essential role in the active secretion of ATP in the course of ICD. The activation of the autophagic machinery together with the execution of apoptosis results in the secretion of ATP by dying cells.31–33
HMGB-1 is a protein that is normally localized in the nucleus and under homeostatic conditions is able to shuttle between the nucleus and the cytoplasm. Its functions vary depending on localization and, in the context of ICD, the presence of HMGB-1 in the extracellular space serves as an important DAMP, exerting pro-inflammatory and immunostimulatory effects.34 It was previously shown that the main mode of action of KP1339 relies on the downregulation of BiP, which triggers ERS.26
We hypothesized that KP1339 could induce the hallmarks of ICD, making it the first recognized ruthenium-based complex that is likely able to trigger ICD in vitro. In order to support our hypothesis, 3D tumor spheroids derived from three different colon adenocarcinoma cell lines (HCT-15, HCT-116 and HT-29) were treated with KP1339, oxaliplatin or cisplatin. In response to all treatments, tumor spheroids underwent cell death as indicated by flow cytometry studies with annexin-V fluorescein isothiocyanate and propidium iodide (PI) (SF1, ESI†).
In a first series of experiments we could confirm at the protein level previously published findings using monolayer cultures of colorectal cancer cell lines, which demonstrate ERS upon KP1339 treatment.26 In all three cell lines, KP1339 and oxaliplatin treated spheroids showed enhanced PERK activation compared to untreated spheroids, followed by eIF-2α phosphorylation after 24 hours of treatment (Fig. 1 and SF2, ESI†), in contrast to cisplatin treated spheroids which did not exhibit these effects. The activation of this branch of the ERS resulting in eIF-2α phosphorylation is a prerequisite for CRT translocation to the cell membrane.35,36
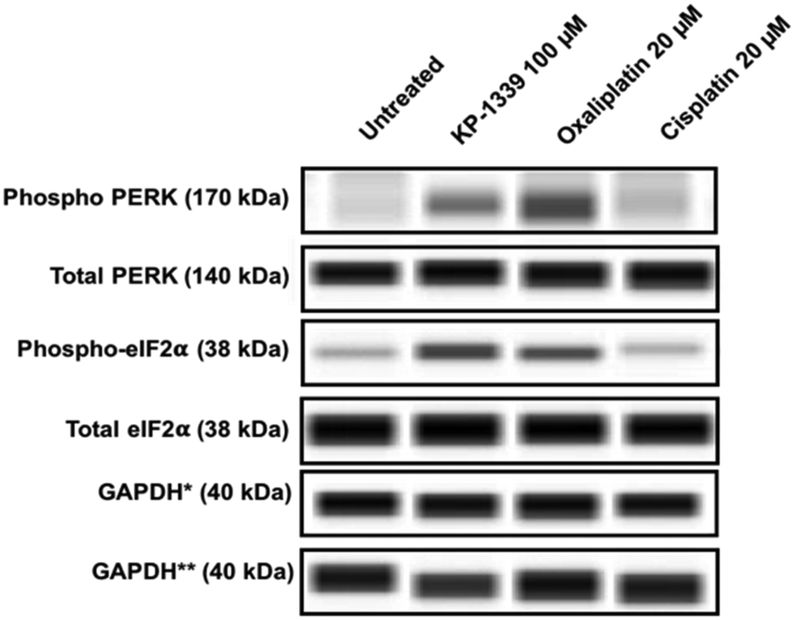 | ||
| Fig. 1 Representative immunoblot images from a capillary Western blot system. The total load of PERK, eIF2α, and GAPDH, respectively, of HCT-116 cells from spheroids is shown. For PERK and eIF2α the phosphorylation status is also analyzed, whereby phosphorylation is only observed upon treatment with KP1339 and oxaliplatin for 24 h. *GAPDH: loading control for pPERK, tPERK, p-eIF-2α. **GAPDH: loading control for t-eIF2α. | ||
The next step was to investigate whether CRT translocates from the cytoplasm to the cell membrane upon treatment. To demonstrate this, we performed flow cytometry studies, where spheroids were treated for 24 hours with oxaliplatin, cisplatin, KP1339 or were left untreated, followed by staining with an anti-CRT antibody and propidium iodide (live/dead staining). In all cell lines, viable (propidium iodide negative) cells isolated from spheroids treated with KP1339 and oxaliplatin showed increased expression of CRT on the cellular membrane compared to cisplatin treated and untreated controls (Fig. 2 and SF3, ESI†). Additionally, cryosections of spheroids treated under the same conditions as described above were subjected to immunofluorescence staining followed by confocal microscopy. Cells were stained with a cell membrane marker (wheat germ agglutinin), anti-CRT antibody and DAPI (for nuclei) under non-cell-permeabilizing conditions. This approach confirmed the results obtained by flow cytometry, with CRT co-localizing with the cell membrane in KP1339 and oxaliplatin treated spheroids (Fig. 3 and SF4–SF6, ESI†).
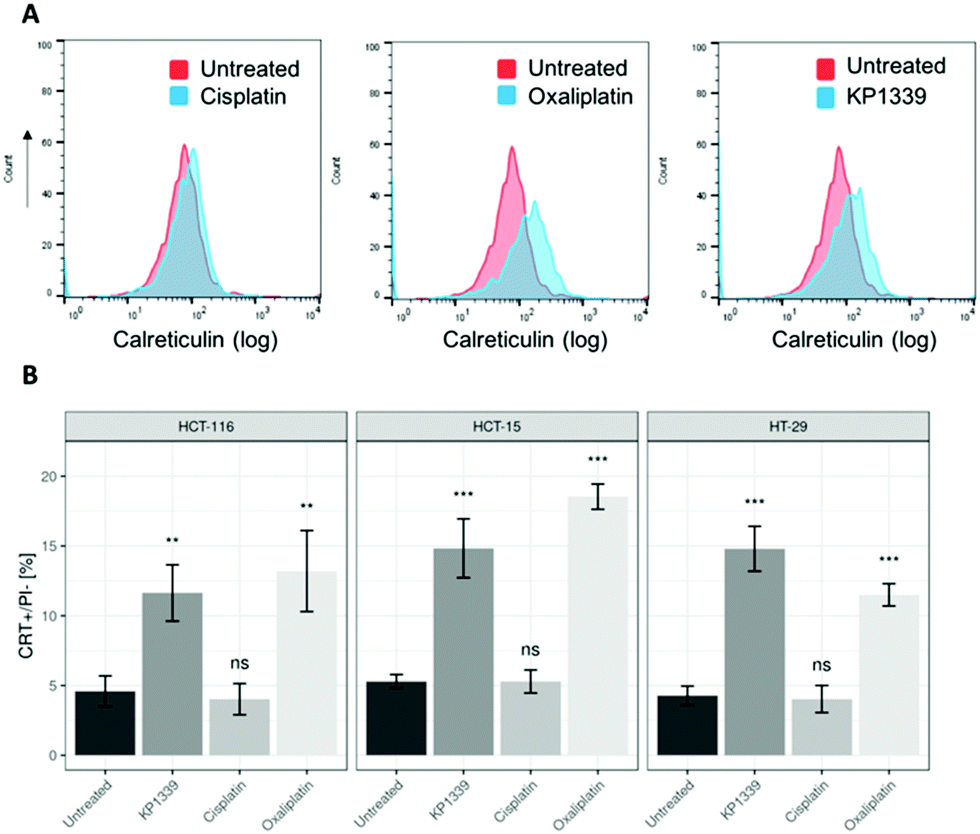 | ||
| Fig. 2 Representative flow cytometry histogram showing increased CRT expression on the surface of (A) cells from HCT-116 spheroids treated with KP1339 and oxaliplatin, compared to untreated (control) and cisplatin-treated spheroids, after 24 h of exposure. (B) Analysis of calreticulin membrane exposure of treated spheroids after drug exposure for 24 h. Mean + STD (ns: not significant; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Tukey's range test). | ||
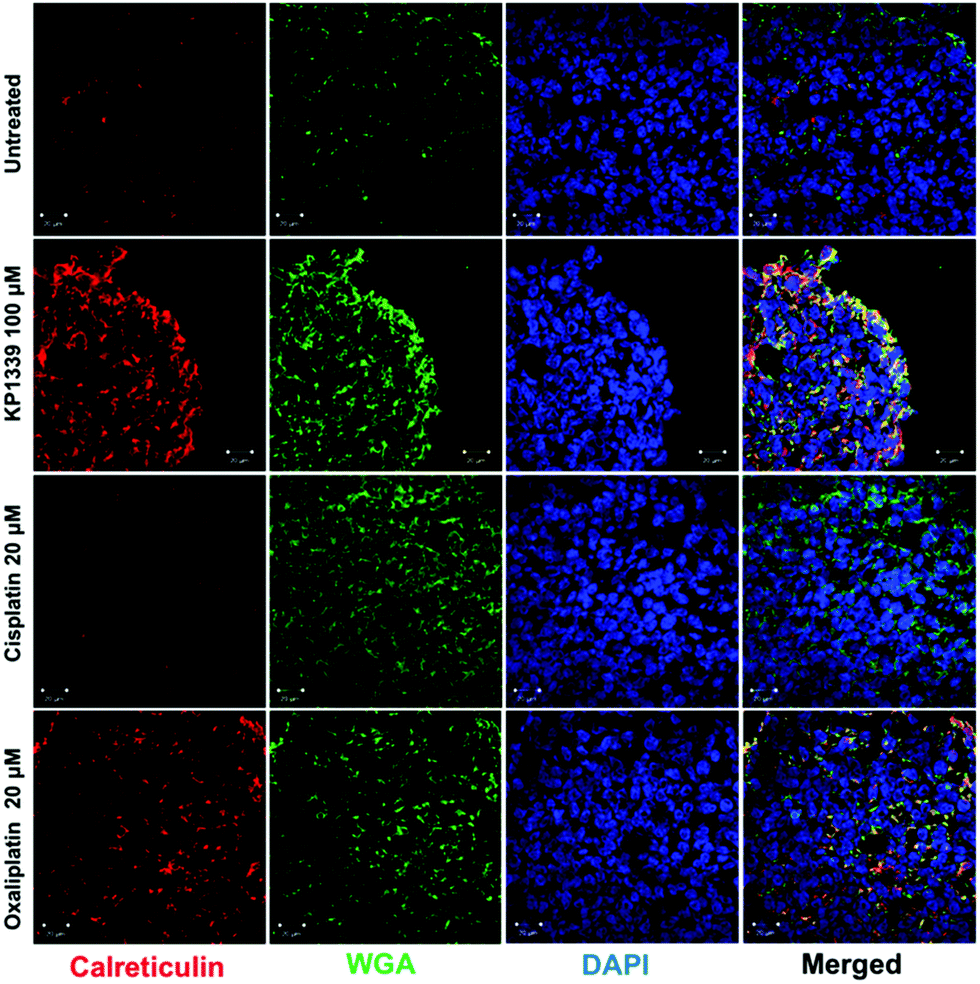 | ||
| Fig. 3 Representative immunofluorescence analysis (confocal microscopy) of paraformaldehyde-fixed (PFA) HCT-116 spheroids. Exposure of calreticulin on the cell membrane is observed after 24 h treatment with KP1339 (100 μM) and oxaliplatin (20 μM), as indicated by co-localization of calreticulin and membrane associated wheat germ agglutinin (WGA). | ||
We next evaluated HMGB-1 release from cells treated with KP1339. This protein is present in the nucleus, then translocates to the cytoplasm and is eventually released to the extracellular space upon treatment with ICD inducers.37 We measured HMGB-1 content in supernatants originated from treated and untreated spheroids by means of ELISA. Indeed, upon treatment with oxaliplatin, cisplatin and KP1339 all three cell lines showed a pronounced release of HMGB-1 from the corresponding tumor spheroids (SF7, ESI†).
We also assessed HMGB-1 localization by using immunofluorescence and super-resolution confocal microscopy. Cryosections of KP1339 treated spheroids showed an enhanced signal of this protein in the cytoplasm, confirming the findings obtained by ELISA (Fig. 4, SF8 and SF9, ESI†).
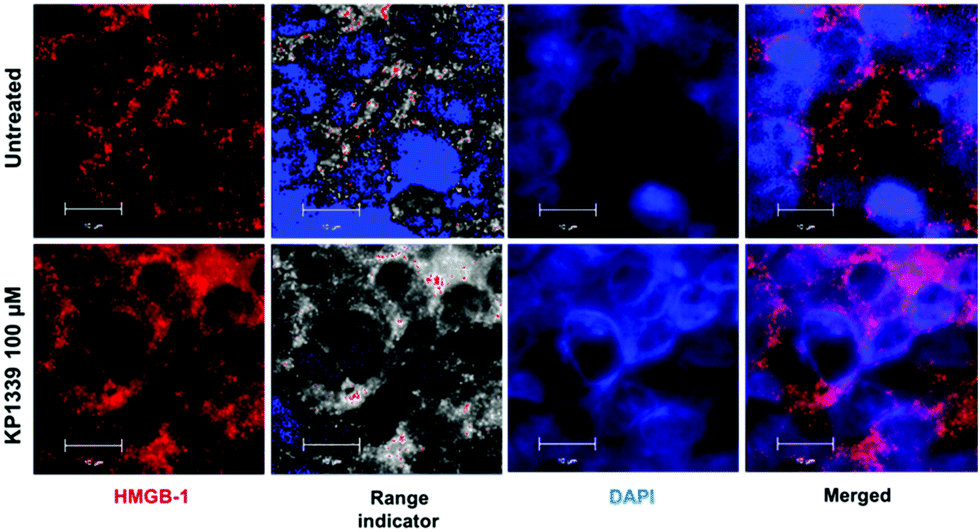 | ||
| Fig. 4 Representative immunofluorescence analysis (structure illumination microscopy) of paraformaldehyde-fixed (PFA) spheroids. Release of HMGB-1 (red and range indicator) into the cytoplasm of HCT-116 spheroids is observed after 24 h treatment with KP1339 (100 μM). | ||
In the context of verifying ATP release, we first investigated whether KP1339 is able to induce autophagy. It has been previously shown that ATP release during chemotherapy-driven ICD relies on activation of the autophagic machinery.32 Therefore, we probed the expression of some key autophagy markers such as Beclin-1 and LC3A/B-II. Upregulation of Beclin-1 and LC3A/B-II in spheroids treated with KP1339 was clearly observed as early as 24 hours after drug treatment. In contrast, cisplatin treated spheroids did not show any pronounced effect, confirming the findings of other studies where cisplatin failed to induce autophagy (SF2, ESI†). The supernatants of treated and untreated spheroids were collected to determine ATP levels by using a luciferase based assay. All drugs induced an increase in ATP release with KP1339-treated cells exhibiting a pronounced release (Fig. 5).
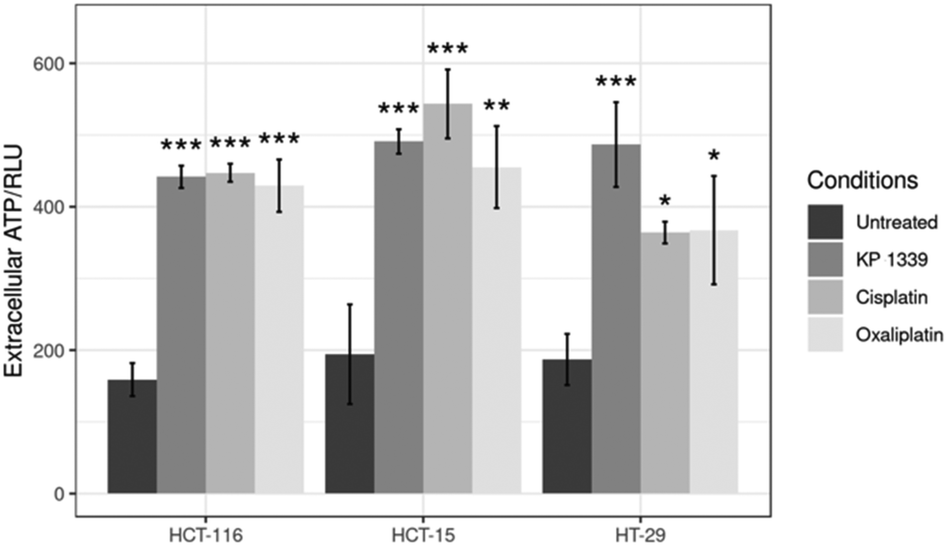 | ||
| Fig. 5 Analysis of ATP levels in supernatants from treated spheroids after drug exposure for 24 h. Mean + STD (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Tukey's range test). | ||
The production of reactive oxygen species (ROS) was subsequently assessed. To demonstrate the oxidative stress response, ROS production was tracked by using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) as an indicator. All drug treatments led to ROS production, with KP1339 exhibiting the most marked effect amongst all treatments (SF10, ESI†).
Our data collectively demonstrate that KP1339, in contrast to cisplatin, induces the full ICD signature in 3D colon cancer spheroids in vitro. ERS is a pre-requisite for ICD; however, not all ERS inducers are able to induce ICD, as demonstrated here in the case of cisplatin.36
In type I ICD, ERS is induced as a collateral effect of the actual mode of action of a compound. Oxaliplatin induces type I ICD due to DNA damage resulting in ERS. Type II ICD inducers, on the other hand, trigger a focused ERS and are known to more efficiently elicit danger signals, e.g., photodynamic therapy with hypericin and oncolytic viruses. There is only a limited number of type II ICD inducers reported in the literature, and for a better understanding of this specific type of ICD, more inducers are required for further studies.38 Based on our findings, KP1339 may qualify as a type II ICD inducer due to its main mode of action that relies on ERS16 (SF2, ESI†) and oxidative stress (SF10 and SF11, ESI†). Most of the chemotherapeutic agents currently used in the clinic, are unable to trigger ICD, often because they do not induce ERS and the consequent CRT exposure.
In accordance with other studies, we confirm here that oxaliplatin induces all the key hallmarks of ICD, while cisplatin failed to stimulate CRT translocation to the cell membrane despite inducing other DAMPs.15,17,20,37 Chemically closely related drugs often exhibit an unexpected degree of heterogeneity in their capacity to trigger ICD, e.g., cisplatin and oxaliplatin.29 Failure to elicit one of the key events in the course of ICD is sufficient to impair the immunogenicity of cell death.21,36 To our knowledge, this is the first study employing a 3D model, where a ruthenium-based complex is shown to induce the ICD signature in vitro.
This work highlights the immunogenic potential of a new class of anticancer chemotherapeutics and warrants the further study of other ruthenium complexes which have reached phase I clinical trials (NAMI-A, KP1090), as well as emerging Ru anticancer agents, such as plecstatin-I and plecstatin-II.26,39
Many chemotherapeutic agents are particularly effective when administered in combination regimens. Immunological effects may explain the success of such combinations, e.g., 5-fluorouracil (with folinic acid) and oxaliplatin (FOLFOX), where the individual agents can target malignant cells through their different modes of action, but can also exert a combined long lasting immunostimulatory effect.
Therefore, further studies would be required to also explore the immunogenic potential of ruthenium-based complexes when used in combination with other chemotherapeutic agents.12,40,41
The spheroid model can be used for the evaluation of drug candidates in drug screening prior to in vivo preclinical animal experiments for fine tuning the drug development process before embarking on clinical investigations. Although further technical optimization is required, the implementation of spheroid models of different complexities in early drug development represents an important technology for improving the overall screening process.42,43
In summary, we have employed for the first time the use of an alternative in vitro model to assess the potential of compounds for ICD in vitro, and have shown that KP1339, a ruthenium-based investigational anticancer drug, induces the ICD signature. The immunogenic potential of KP1339 paves the way for further in vitro and in vivo investigations. However, more complex 3D cultures (e.g., organoids plus immune cells45) would be required to explore in more detail other important aspects of the interactions among cancer cells, tumor microenvironment and immune cells in the course of ICD, in particular the executor cells of ICD, DCs and T cells. Therefore, major efforts should be made to establish organoid cultures more faithfully reflecting the human pathology, which will bring new insights into the molecular events taking place during ICD. Those models with increasing complexity are essential to better translate basic preclinical research into novel treatments for the benefit of cancer patients.42,44
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Andreas Wanninger and his team (Institute of Zoology, University of Vienna) as well as John Soltys (Department of Chemical Catalysis, University of Vienna), Dr Wolfgang Kandioller (Institute of Inorganic Chemistry, University of Vienna), Christina Puri and Oliver Bergner (Boehringer Ingelheim RCV, Vienna) for helpful discussions and for supporting this work.References
- J. B. Swann and M. J. Smyth, J. Clin. Invest., 2007, 117(5), 1137–1146 CrossRef CAS PubMed.
- J. Stagg, R. W. Johnstone and M. J. Smyth, Immunol. Rev., 2007, 220, 82–101 CrossRef CAS PubMed.
- R. D. Schreiber, L. J. Old and M. J. Smyth, Science, 2011, 331, 6024 CrossRef PubMed.
- D. Mittal, M. M. Gubin, R. D. Schreiber and M. J. Smyth, Curr. Opin. Immunol., 2014, 16–25 CrossRef CAS PubMed.
- A. Rudisch, M. R. Dewhurst, L.-G. Horga, N. Kramer, N. Harrer, M. Dong, H. van der Kuip, A. Wernitznig, A. Bernthaler, H. Dolznig and W. Sommergruber, PLoS One, 2015, 10(4), e0124283 CrossRef PubMed.
- E. Gottfried, S. Faust, J. Fritsche, L. A. Kunz-Schughart, R. Andreesen, K. Miyake and M. Kreutz, Immunobiology, 2003, 207(5), 351–359 CrossRef CAS PubMed.
- S. Turley, V. Cremasco and J. L. Astarita, Nat. Rev. Immunol., 2015, 15, 669–682 CrossRef CAS PubMed.
- A. Mantovani and P. Allavena, J. Exp. Med., 2015, 212(4), 435–445 CrossRef CAS PubMed.
- Z. Mei, Y. Liu, C. Liu and L. Cui, Br. J. Cancer, 2014, 111(12), 2372 CrossRef CAS PubMed.
- H. T. Marshall and M. B. A. Djamgoz, Front. Oncol., 2018, 8, 315 CrossRef PubMed.
- J. Berntsson, J. Eberhard, B. Nodin, K. Leandersson, A. H. Larsson and K. Jirström, OncoImmunology, 2018, 7(8), e1465165 CrossRef PubMed.
- D. M. S. Hossain, S. Javaid, M. Cai, C. Zhang, A. Sawant, M. Hinton, M. Sathe, J. Grein, W. Blumenschein, E. M. Pinheiro and A. Chackerian, J. Clin. Invest., 2018, 128(2), 644–654 CrossRef PubMed.
- J. Yao, W. Xi, Y. Zhu, H. Wang, X. Hu and J. Guo, Cancer Manage. Res., 2018, 11(10), 3419–3431 CrossRef PubMed.
- A. Tesniere, T. Panaretakis, O. Kepp, L. Apetoh, F. Ghiringhelli, L. Zitvogel and G. Kroemer, Cell Death Differ., 2008, 15(1), 3–12 CrossRef CAS PubMed.
- A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L. Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J.-P. Pignon, V. Jooste, P. van Endert, M. Ducreux, L. Zitvogel, F. Piard and G. Kroemer, Oncogene, 2010, 29, 482–491 CrossRef CAS PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef CAS PubMed.
- A. M. Dudek, A. D. Garg, D. V. Krysko, D. De Ruysscher and P. Agostinis, Cytokine Growth Factor Rev., 2013, 4, 319–333 CrossRef PubMed.
- A. D. Garg, D. V. Krysko, P. Vandenabeele and P. Agostinis, Cancer Immunol. Immunother., 2012, 61(2), 215–221 CrossRef CAS PubMed.
- D. Y. Q. Wong, W. W. F. Ong and W. H. Ang, Angew. Chem., 2015, 127, 6583–6587 CrossRef.
- O. Kepp, et al. , OncoImmunology, 2014, 3, 9 CrossRef PubMed.
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2017, 2, 97–111 CrossRef PubMed.
- K. Stock, M. F. Estrada, S. Vidic, K. Gjerde, A. Rudisch, V. E. Santo, M. Barbier, S. Blom, S. C. Arundkar, I. Selvam, A. Osswald, Y. Stein, S. Gruenewald, C. Brito, W. van Weerden, V. Rotter, E. Boghaert, W. Sommergruber, Y. Chong, R. de Hoogt and R. Graeser, Sci. Rep., 2016, 1(6), 28951 CrossRef PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schugart, J. Biotechnol., 2010, 1, 3–15 CrossRef PubMed.
- M. J. Hannon, Pure Appl. Chem., 2007, 12, 2243–2261 Search PubMed.
- H. A. Burris, S. Bakewell, J. C. Bendell, J. Infante, S. F. Jones, D. R. Spigel, G. J. Weiss, R. K. Ramanathan, A. Ogden and D. Von Hoff, ESMO Open, 2017, 1 Search PubMed.
- L. S. Flocke, R. Trondl, M. A. Jakupec and B. K. Keppler, Invest. New Drugs, 2016, 34, 261–268 CrossRef CAS PubMed.
- O. Kepp, L. Menger, E. Vacchelli, C. Locher, S. Adjemian, T. Yamazaki, I. Martins, A. Q. Sukkurwala, M. Michaud, L. Senovilla, L. Galluzzi, G. Kroemer and L. Zitvogel, Cytokine Growth Factor Rev., 2013, 4, 311–318 CrossRef PubMed.
- D. J. Todd, A. H. Lee and L. H. Glimcher, Nat. Rev. Immunol., 2008, 8, 663–674 CrossRef CAS PubMed.
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvoegel and G. Kroemer, Cancer Cell, 2015, 28, 690–714 CrossRef CAS PubMed.
- A. R. Van Vliet, S. Martins, A. D. Garg and P. Agostinis, Semin. Cancer Biol., 2015, 33, 74–85 CrossRef CAS PubMed.
- I. Martins, Y. Wang, M. Michaud, Y. Ma, A. Q. Sukkurwala, S. Shen, O. Kepp, D. Métivier, L. Galluzzi, J.-L. Perfettini, L. Zitvogel and G. Kroemer, Cell Death Differ., 2014, 21, 79–81 CrossRef CAS PubMed.
- A. D. Garg, D. V. Krysko, P. Vandenabeele and P. Agostinis, Cell Death Dis., 2016, 18, 7 Search PubMed.
- W. Hou, J. Han, C. Lu, L. A. Goldstein and H. Rabinowich, Autophagy, 2010, 6, 891–900 CrossRef CAS PubMed.
- P. Liu, L. Zhao, F. Loos, K. Iribarren, S. Lachkar, H. Zhou, L. C. Gomes-da-Silva, G. Chen, L. Bezu, G. Boncompain, F. Perez, L. Zitvogel, O. Kepp and G. Kroemer, Sci. Rep., 2017, 7, 14915 CrossRef PubMed.
- L. Bezu, A. Sauvat, J. Humeau, M. Leduc, O. Kepp and G. Kroemer, OncoImmunology, 2018, 7(6), e1431089 CrossRef PubMed.
- L. Bezu, A. Sauvat, J. Humeau, L. C. Gomes-da-Silva, K. Iribarren, S. Forveille, P. Garcia, L. Zhao, P. Liu, L. Zitvoegel, L. Senovilla, O. Kepp and G. Kroemer, Cell Death Differ., 2018, 25(8), 1375–1393 CrossRef CAS PubMed.
- J. Pol, E. Vacchelli, F. Aranda, F. Castoldi, A. Eggermont, I. Cremer, C. Sautès-Fridman, J. Fucikova, J. Galon, R. Spisek, E. Tartour, L. Zitvogel, G. Kroemer and L. Galluzzi, OncoImmunology, 2015, 4, 4 Search PubMed.
- A. D. Garg, A. M. Dudek-Peric, E. Romano and P. Agostinis, Int. J. Dev. Biol., 2015, 59(1–3), 131–140 CrossRef CAS PubMed.
- S. M. Meier, et al. , Angew. Chem., Int. Ed., 2017, 56, 8267–8271 CrossRef CAS PubMed.
- L. Zitvogel, J. M. Pitt, R. Daillère and M. J. Smyth, Nat. Rev. Cancer, 2016, 16(12), 759–773 CrossRef CAS PubMed.
- C. Pozzi, A. Cuomo, I. Spadoni, E. Magni, A. Silvola, A. Conte, S. Sigismund, P. S. Ravenda, T. Bonaldi, M. G. Zampino, C. Cancelliere, P. P. Di Fiore, A. Bardelli, G. Penna and M. Rescigno, Nat. Med., 2016, 6, 624–631 CrossRef PubMed.
- J. Drost and H. Clevers, Nat. Rev. Cancer, 2018, 18, 2 CrossRef PubMed.
- O. I. Hoffmann, C. Ilmberger, S. Magosch, M. Joka, K. W. Jauch and B. Mayer, J. Biotechnol., 2015, 205, 14–23 CrossRef CAS PubMed.
- A. Nyga, J. B. Neves, K. Stamati, M. Loizidou, M. Emberton and U. Cheema, Drug Discovery Today, 2016, 21(9), 1421–1428 CrossRef PubMed.
- S. P. Rebelo, C. Pinto, T. R. Martins, N. Harrer, M. F. Estrada, P. Loza-Alvarez, J. Cabecadas, P. M. Alves, E. J. Gualda, W. Sommergruber and C. Brito, Biomaterials, 2018, 163, 185–197 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mt00051h |
| This journal is © The Royal Society of Chemistry 2019 |