
Improving the photoluminescence quantum yields of quantum dot films through a donor/acceptor system for near-IR LEDs†
Nathaniel. J. L. K.
Davis
 a,
Jesse R.
Allardice
b,
James
Xiao
b,
Arfa
Karani
b,
Tom C.
Jellicoe
b,
Akshay
Rao
b and
Neil C.
Greenham
*b
a,
Jesse R.
Allardice
b,
James
Xiao
b,
Arfa
Karani
b,
Tom C.
Jellicoe
b,
Akshay
Rao
b and
Neil C.
Greenham
*b
aSchool of Chemical and Physical Sciences, Victoria University of Wellington, Wellington 6140, New Zealand
bCavendish Laboratory, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK. E-mail: ncg11@cam.ac.uk
First published on 9th October 2018
Abstract
Near-infrared light-emitting diodes (LEDs) show potential for telecommunication and medical applications. Quantum dot nanocrystals (QDs), specifically lead chalcogenides, are candidate LED materials since they exhibit tuneable luminescence across the whole near-infrared region, but their surface structure must be carefully controlled to achieve efficient emission. We demonstrate an efficient donor–acceptor QD system by embedding low-energy QDs with high photoluminescence quantum efficiency (PLQE) into a matrix of higher-energy QDs with lower PLQE. We find that the overall PLQE of densely packed cross-linked QD films can be improved by the incorporation of a relatively small fraction of well-passivated acceptor QDs, also leading to improved LED performance. Excitations are transferred into the isolated low-energy acceptor QDs, where they recombine with high radiative efficiency.
Conceptual insightsIn quantum dot (QD) LEDs, surface states can lead to the quenching of excitons. Hence extensive passivation with a higher-bandgap inorganic shell is typically used to improve the luminescence efficiency, adding to the synthetic complexity and cost of the overall product. Here, we report a simple, versatile and highly efficient donor–acceptor QD energy cascade system formed by embedding low-energy core–shell PbS/CdS QDs with high PLQEs into a matrix of higher-energy PbS QDs with lower PLQEs.These composites increase the overall PLQE of densely packed cross-linked QD films by the incorporation of a relatively small fraction of well-passivated acceptor QDs, also leading to improved LED performance. These LEDs out-perform other leading PbS near-infrared QD LEDs based on a similar device structure and compared with devices that purely use high-quality core–shell PbS/CdS QDs, we achieve optimum efficiencies with only 1/20th the concentration of high-quality QDs. This low concentration of high quality passivated dots and the improved device performance makes this system both comparably cheaper and more versatile than systems with passivation of all the QDs. We believe that this demonstration will open new avenues for the community and allow them to integrate such systems into a range of device applications. |
Introduction
Light-emitting diodes (LEDs) based on chemically synthesised semiconductor quantum dots offer the benefits of sharp emission spectra, colour tunability through the quantum size effect, and solution processability.1–3 Their efficiency has improved greatly over the last 30 years, and they can provide emission throughout the visible range for display and lighting applications.2–4 Quantum dot LEDs provide particular advantages over their organic LED competitors for emission in the infrared, since the strong vibronic coupling present in organic molecules typically leads to broad spectra and rapid non-radiative decay.5,6IR-emitting QD LEDs based on InAs and PbS have already been demonstrated,7–10 but, as with other QD LEDs,11–17 careful control of the nanoparticle surfaces and inter-nanoparticle interactions is needed to achieve efficient performance. Surface states can lead to quenching of excitons, hence passivation with a higher-gap inorganic shell and/or an organic ligand shell is typically used to improve the luminescence efficiency.18–22 Increasingly, the focus of QD LED improvement is moving towards densely packed films of QDs as the emissive layer, rather than embedding the QDs in a charge-transporting matrix.23 In this case, it is necessary to obtain good inter-particle electronic contact in order to achieve facile charge transport.24–28 A trade-off therefore exists between achieving good surface passivation and minimising tunnelling barriers between particles.29
It is well known that excitons may move between closely spaced dots by the process of Förster transfer, and may thus explore many dots within a film before decaying. Since Förster transfer is preferentially a down-hill process in energy, it can be used to “funnel” excitons between QDs of different energies.30–32 Although energy transfer has been studied in many QD systems,23–26,33–38 relatively little attention has been paid to its effect on the overall efficiency of emission. Indeed, it is often found that densely packed films have lower luminescence efficiencies than those measured in solution.1,39 One explanation for this effect is that in the film, excitons diffuse between dots until they find a defect site (a “bad dot”) where rapid non-radiative decay occurs.
Here, we show that by introducing a small number of well-passivated low-energy QDs, we are able to achieve efficient emission from a film of closely-spaced QDs that would otherwise have a low PLQE. These films give good electrical performance in LEDs, whilst retaining the benefits of efficient emission. Excitons are able to find a well-passivated “good dot” before non-radiative decay occurs in the surrounding dots.
Results
The concept of an efficient donor–acceptor energy transfer system was explored thought the synthesis of two different types of QDs (Fig. 1(a)). For the donor dots we use PbS QDs of diameter ∼2.1 nm (ESI,† Fig. S1), showing emission at 1.4 eV. These dots are synthesised with native oleic acid ligands. In solution they have a PLQE of 29%; in a film this drops to 7% and 1% when cross-linked with 1,3-benzenedithiol (BDT). For the acceptor dots we used PbS/CdS QDs of diameter ∼3.7 nm (ESI,† Fig. S2), showing emission at 1.1 eV. These dots have a surface cation-exchange treatment with Cd to produce a core–shell structure, and have a PLQE in solution of 71%, 27% in a film, and 9% in a film cross-linked with (BDT). The optical properties of the dots are summarised in Table 1, and their emission and absorption spectra in solution are shown in Fig. 1(b).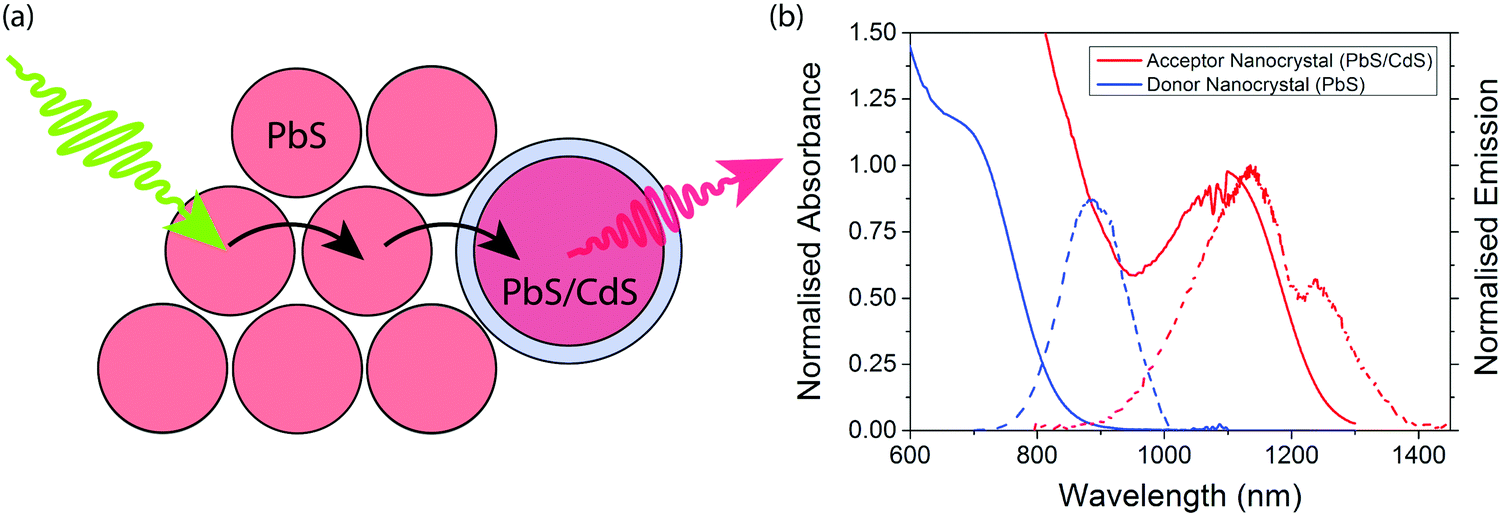 | ||
| Fig. 1 (a) Schematic representation of exciton migration from small-diameter large-bandgap PbS QDs to large-diameter low-bandgap PbS/CdS core–shell QDs. (b) Absorption and emission spectra of the donor and acceptor QDs in toluene. The dip at 1220 nm is an artefact due to toluene absorption. | ||
| Material | Donor nanocrystals | Acceptor nanocrystals |
|---|---|---|
| PbS | PbS/CdS | |
| Bandgap (eV) | 1.75 | 1.23 |
| PLQE (%) | 29 | 71 |
| Size (nm) | 2.1 ± 0.2 | 3.7 ± 0.3 |
Films are formed after mixing donor and acceptor QDs at various ratios (by weight) in solution. The donor and acceptor dots can be clearly distinguished by size and contrast for films spin-coated onto TEM grids (Fig. 2(b–f) and ESI,† Fig. S1). The acceptor dots are randomly distributed, except for the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 donor:acceptor ratio where there is evidence of super-lattice formation.
:1 donor:acceptor ratio where there is evidence of super-lattice formation.
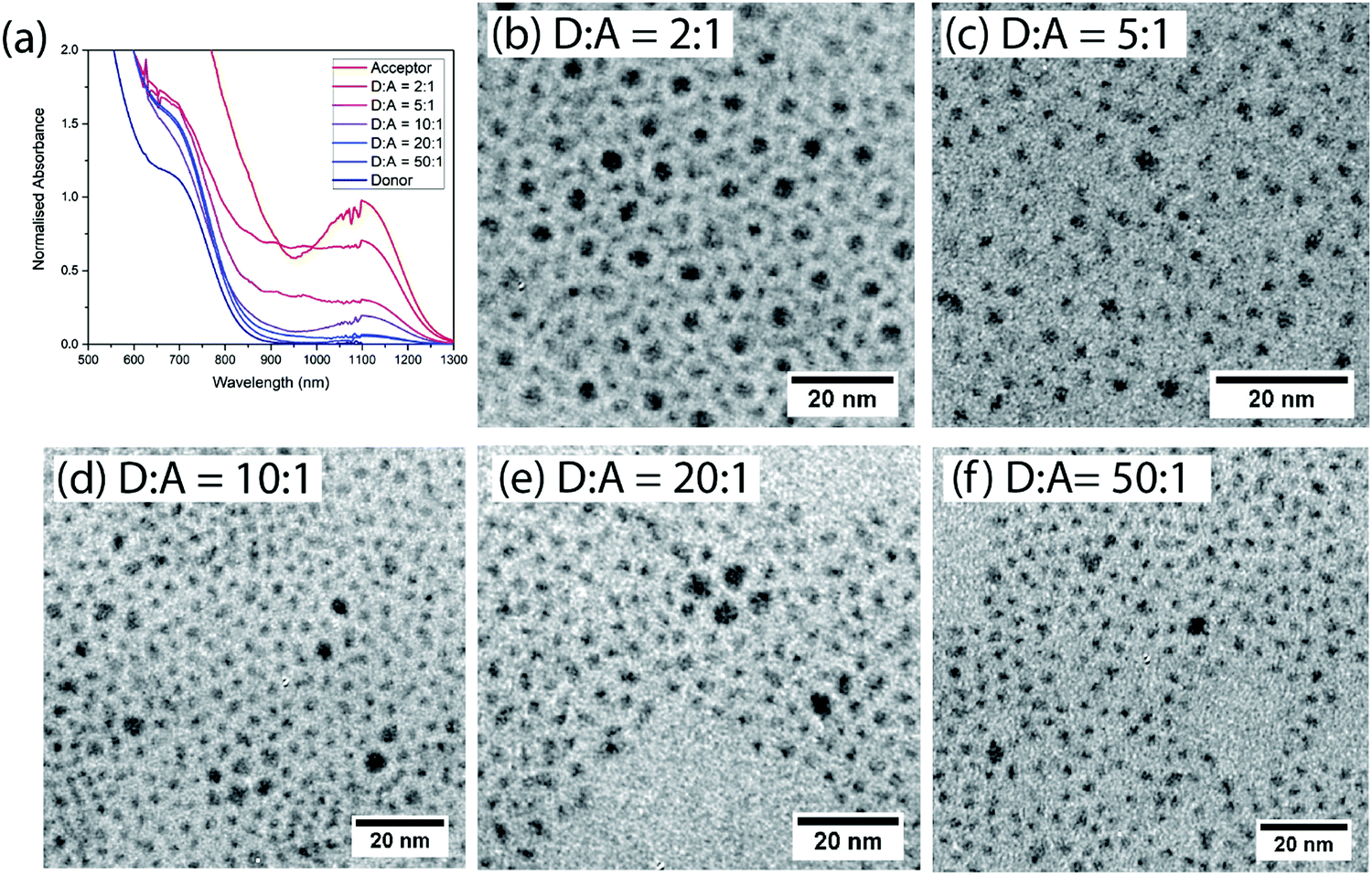 | ||
| Fig. 2 (a) Normalized absorption of films of different donor:acceptor ratios. (b–f) TEM images of the different non-cross-linked donor:acceptor blend films. | ||
To make efficient LED devices with low turn-on voltages the oleic acid ligands must be replaced with shorter ligands.9,27,28 We therefore study films which have been cross-linked using BDT, displacing the original ligands. The absorption spectra of mixed BDT cross-linked films comprise a superposition of the absorption spectra of the constituent dots, consistent with the dots remaining physically distinct on mixing (Fig. 2(a)).
Fig. 3 shows the emission spectra of the mixed films, and the respective PLQEs are shown in Table 2. For films with the native oleic acid ligand, emission from both donor and acceptor dots is seen. Acceptor emission is always more prominent than would be expected based on the amount of absorption in the acceptor QDs, so energy transfer is clearly taking place, but significant donor emission remains even when the acceptor concentration is as high as 17% by weight (5:1 donor:acceptor). More efficient energy transfer is observed in the BDT cross-linked films, consistent with a reduced inter-QD spacing. Fig. 3(b) shows that donor emission is almost completely quenched at acceptor concentrations above 5% (20:1 donor:acceptor) in the cross-linked films.
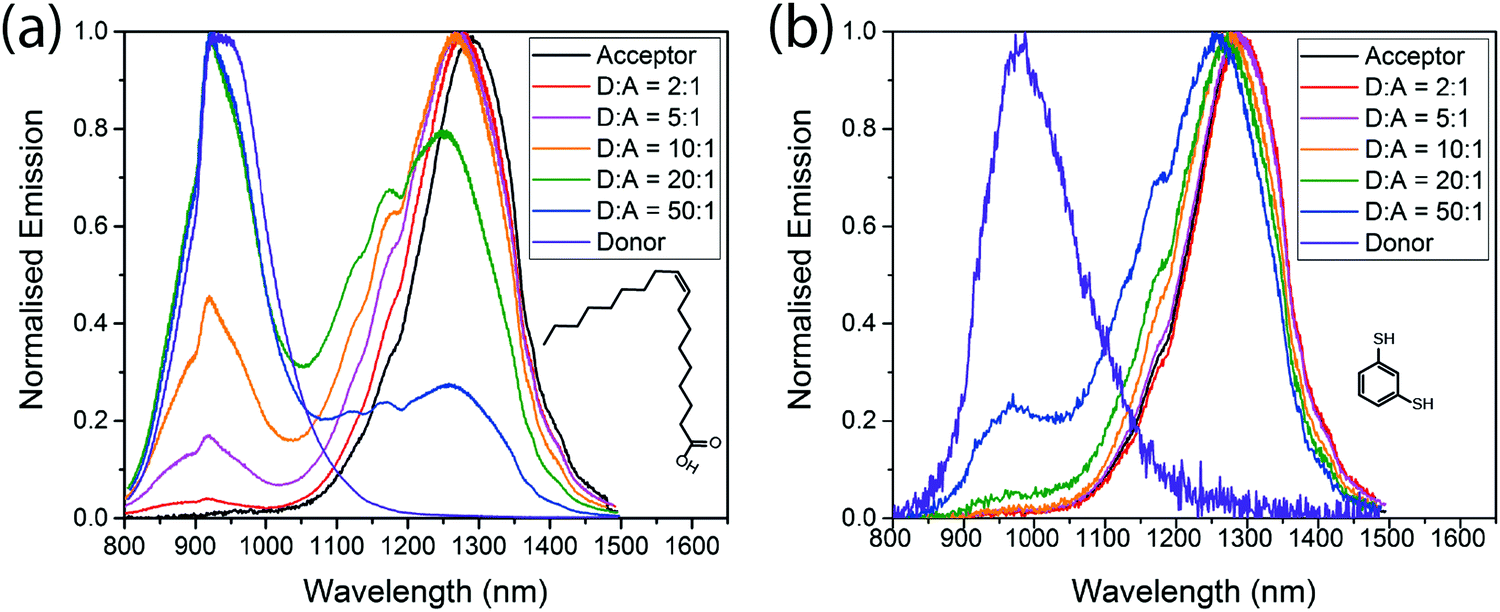 | ||
| Fig. 3 Emission spectra of different donor:acceptor blends, with oleic acid ligands (a) and benzenedithiol ligands (b) excited at 405 nm. | ||
| PLQE in drop cast film with oleic acid ligands (%) | PLQE in spin coted film with BDT ligands (%) | |
|---|---|---|
| Acceptor | 27 | 9 |
| D:A = 2:1 |
14 | 6 |
| D:A = 5:1 |
10 | 7 |
| D:A = 10:1 |
9 | 10 |
| D:A = 20:1 |
8 | 10 |
| D:A = 50:1 |
7 | 6 |
| Donor | 7 | 1 |
For cross-linked and non-cross-linked films, addition of acceptor QDs gives higher PLQE than in pure donor films, consistent with excitons reaching bright acceptor QDs before decaying (Table 2). The effect is particularly marked in cross-linked films, where the pure donor films have PLQEs of only 1%, but addition of only 2% acceptor QDs (50:1 donor:acceptor) is sufficient to increase the PLQE to 6%. At donor:acceptor ratios of 20:1 and 10:1, the PLQE is slightly higher than for pure acceptor films, suggesting that once an exciton reaches an isolated acceptor QD it is unable to explore further, thus reducing the probability of finding a poorly-passivated acceptor QD with rapid non-radiative decay.
We use transient absorption (TA) to further study the transfer of energy from donor to acceptor QDs in cross-linked films with different blend ratios. As TA is a technique that looks at the populations of excited systems, compared to transient photoluminescence which requires emission, we can monitor this transfer even when there is little to no emission from the donor dots. For measurements up to 2 ns (ESI,† Fig. S2 and S3) we pump at 532 nm, and for measurements on nanosecond timescales (ESI,† Fig. S4 and S5) we pump at 530 nm; hence the samples are pumped in a region where both donor and acceptor QDs are absorbing.
Between 1075 and 1350 nm, on picosecond timescales the acceptor QD TA signal is a mixture of a short-lived photoinduced absorption (PIA) feature and the long-lived ground state bleach (GSB) (ESI,† Fig. S2(a)). The donor TA signal shows a broad PIA feature in this region (ESI,† Fig. S2(b)). Photoexcitation of the different blend ratios results in an initial transient absorption signal which is a linear combination of these three spectral features, dependent on the optical density and resulting initial excited state populations of the two QD species (Fig. 4(a) and ESI,† Fig. S2 and S3). Looking specifically at the transient absorption around 1130 nm, excited donors give a negative (PIA) signal and excited acceptors give a positive (GSB) signal. Probing at this wavelength thus gives a convenient way to study excitation transfer from donor to acceptor, as shown in Fig. 4(a). The rising positive signal gives clear evidence of energy transfer from donor to acceptor. Extending these measurements to nanosecond timescales (Fig. 4(b) and ESI,† Fig. S4 and S5) allows the full dynamics of energy transfer to be tracked, followed by decay of the acceptor excitation.
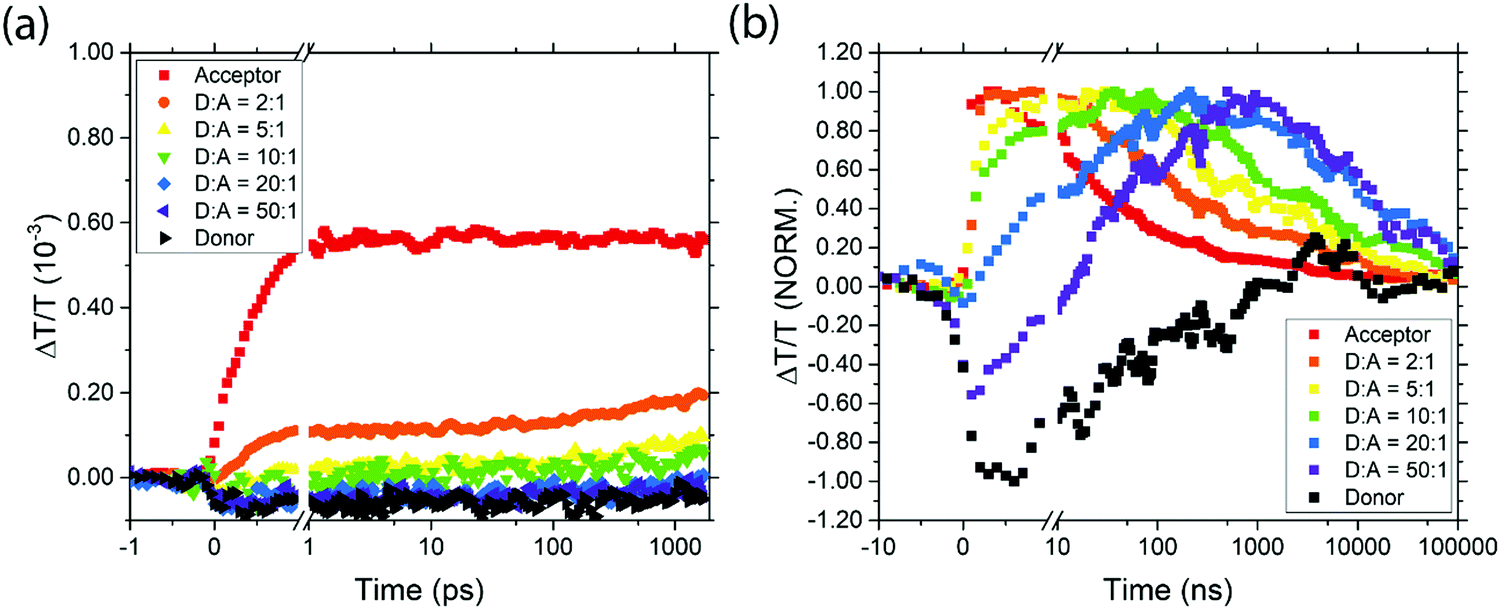 | ||
| Fig. 4 The transient transmission signal strength at 1120–1140 nm for the various samples on picosecond (a) and nanosecond (b) timescales. | ||
It is clear from the data in Fig. 4 that exciton transfer and exciton decay take place over a range of timescales that cannot be described by a single rate constant. This type of dispersive behaviour is typical for QD films which exhibit energetic and spatial disorder together with trapping phenomena. Energy transfer to the acceptor is typically preceded by diffusive exciton hopping between donor QDs, until an acceptor QD is found, and hence in any case it is not expected that the population of excited acceptors will grow with exponential dynamics. Nevertheless, we can draw valuable qualitative conclusions from Fig. 4. First, we note that for pure donor films, the population falls to 50% in about 50 ns, and is negligible by 1 μs. For pure acceptor films, the dynamics are similar to pure donor films, but as the acceptor QDs are diluted in a donor film, the decay dynamics become slower, extending beyond 10 μs. In principle, some of the apparent lifetime increase could be due to energy transfer from donor dots feeding the acceptor population, but since donor excitons will have decayed by 1 μs this effect cannot explain the much slower decay observed in dilute acceptor QDs. Instead, we conclude that the dilution of acceptor QDs prevents excitons from diffusing to other acceptor QDs and thus encountering sites where non-radiative decay may occur.
The timescales for the rise in positive signal clearly show that energy transfer occurs more quickly in films with higher acceptor concentrations, as expected. In films with high acceptor concentrations the energy transfer occurs on sub-nanosecond timescales, consistent with other reports of nonradiative energy transfer in PbS quantum dots with short ligands,25 and significantly faster than the transfer times of 30–400 ns reported in samples with oleic acid ligands.26,40 For films with more dilute acceptor QDs, the timescale for energy transfer to occur is extended due to the hopping required between donor QDs before an acceptor is found. Characteristic timescales of ∼40 ns are seen for the lowest acceptor concentrations. Importantly, even in films with low acceptor concentrations, the energy transfer dynamics are sufficiently rapid to compete with exciton decay in the donor, consistent with the steady-state PL measurements reported above.
To further demonstrate the versatility of our donor–acceptor QD system we create near-infrared QD LEDs. The devices consist of an ITO anode, PEDOT:PSS as the hole-injection layer, poly(9,9-dioctylfluorene-alt-N-(4-sec-butylphenyl)-diphenylamine) (TFB) as the hole-transporting layer, cross-linked PbS and PbS/CdS as the light-emitting active layer and aluminium as the cathode (Fig. 5(a)). Full fabrication and measurement details can be found in the methods. All devices operate with similar current–voltage curves, implying similar charge injection and transport (Fig. 5(b)). Devices made from donor QDs exhibit the lowest efficiencies. Devices made purely from acceptor QDs show slightly higher efficiencies, but are outperformed by the blend devices. Both the EQE and radiance at fixed voltage increase as the donor:acceptor ratios is varied from 2:1 to 20:1, only starting to decrease at ratios of 50:1 (Fig. 5(c and d)). Consistent with the luminescence and transient absorption measurements above, we assign this to the acceptor QDs acting as lower-energy, high-efficiency emissive states. The fact that the current–voltage characteristics are similar for the different blend ratios suggests that charge transport and recombination take place predominantly on the donor, followed by exciton transfer, rather than the acceptor QDs acting directly as centers for charge recombination. The donor:acceptor blend devices shown here out-perform other leading PbS near-infrared QD LEDs9 based on a similar device structure. Compared with devices that purely use high-quality core–shell PbS/CdS QDs,10 we achieve optimum efficiencies with only 1/20th the concentration of high-quality QDs.
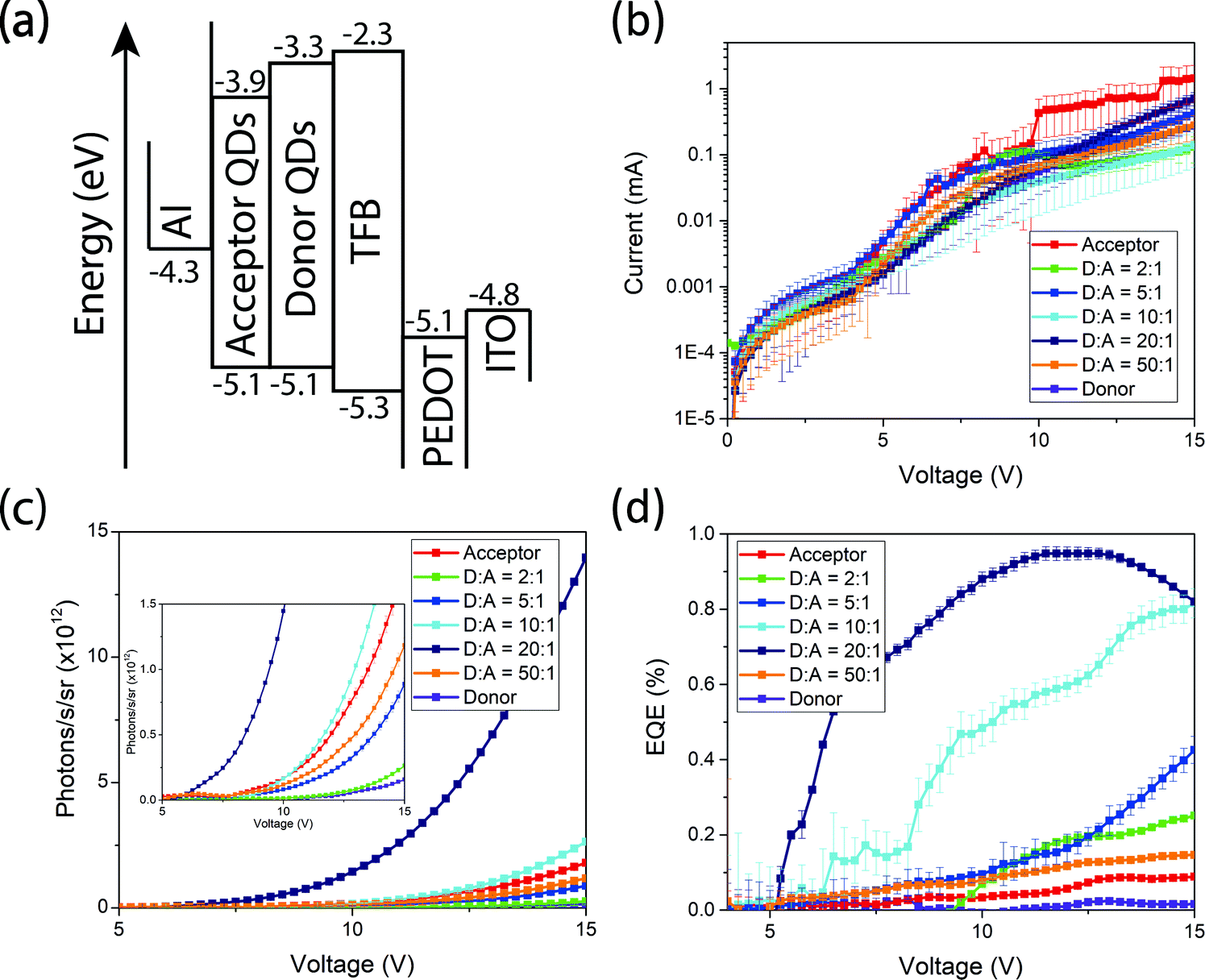 | ||
| Fig. 5 Band structure (a), current–voltage characteristics (b), radiance versus voltage (c) and EQE versus voltage (d) plots of the fabricated near-infrared QD LEDs. | ||
Conclusion
We have demonstrated that incorporation of a relatively low concentration of high-quality PbS/CdS QDs into a densely packed cross-linked film of PbS QDs can lead to significant improvement in luminescence efficiency. Spectroscopic measurements show efficient energy transfer into the acceptor QDs, with reduced non-radiative decay since the excitations are localized on isolated acceptor QDs. The use of these mixed films in IR-emitting LEDs allows efficiencies to be optimized.Methods
Materials
All chemicals were purchased from Sigma-Aldrich and used as delivered.PbS nanocrystal synthesis
The synthesis of PbS NCs was carried out following modified versions of previously reported methods.41 Briefly, PbO (0.62 g, 2.8 mmol), oleic acid (2.1 mL, 6.6 mmol), 1.81 g (acceptor) or 1.5 mL, 4.7 mmol, 1.3 g (donor) and octadecene (25.0 mL, 76.3 mmol, 19.5 g) were combined in a three-neck flask and degassed at 110 °C under vacuum (10−2 mbar or better) for 2h. Subsequently, the reaction flask was flushed with nitrogen and heated to 115°C. A solution of bis(trimethylsilyl)sulphide (286 μL, 1.4 mmol), diphenylphosphine (144 μL, 0.83 mmol) in octadecene (13.9 mL, 42.4 mmol, 10.8 g) was rapidly injected into the lead precursor solution. The bandgap of the PbS NCs was tuned by adjusting the oleic acid concentration as above. The reaction was quenched by placing the reaction flask in an ice-water bath. The PbS nanocrystals were isolated from the reaction mixture by flocculating to turbidity using a 1-butanol/ethanol/hexane solvent system. The purified QDs were then redispersed in octane at a concentration of ∼100 mg mL−l and stored under argon.
Pbs/CdS core shell nanocrystals
The synthesis of the PbS/CdS nanocrystals was performed by cation exchange with a Cd–oleate complex.42 Briefly, cadmium oxide (1.03 g, 99.999%), oleic acid (6.35 mL, 90%) and 1-octadecene (25 mL, 90%) was placed in a three-necked round bottomed flask and degassed under vacuum for 110 °C. The vessel was switched to N2 and heated to 230 °C for 2 hours, resulting in the formation of a clear Cd–oleate solution. The solution was cooled and transferred to a glovebox for storage. Cation exchange was performed with the addition of Cd–oleate solution to PbS nanocrystals. Typical reaction is as follows: PbS nanocrystals in toluene (50 mg, 50 mg mL−1) were heated to 100 °C. Cadmium oleate in ODE (0.33 mL, 0.26 M) was added to the nanocrystal solution. The reaction was quenched after the desired time with the addition of anhydrous acetone and the core–shell dots were precipitated and resuspended twice with acetone and toluene.Continuous wave measurements
Absorption spectra of solutions were measured on nanocrystals samples dispersed in toluene at a concentration of ca. 1 mg mL−1 in a 1 cm × 1 cm cuvette using a Shimadzu UV-3600 Plus spectrometer. Film absorption spectra were measured on Shimadzu UV-3600 Plus spectrometer, the samples were prepared on quartz glass by spin coating from a 10 mg mL−1 solutions at 2000 rpm for 15 s and encapsulated via affixing a glass microscope slide with epoxy. Photoluminescence was measured on an Edinburgh Instruments FLS90 fluorimeter. Film samples were excited by front face illumination at 45° to the surface, detection was at 90° to excitation also at 45° to the surface.PLQE measurements
Nanocrystal films and solutions were placed in an integrating sphere and were photo-excited using a 532 nm continuous-wave laser. The laser and the emission signals were measured and quantified using a calibrated Andor iDus DU490A InGaAs detector and Shamrock SR-303i spectrometer for the determination of PL quantum efficiency. PLQE was calculated as per de Mello et al.43TEM
TEM samples were prepared by drop casting a ca. 5 mg mL−1 nanocrystal solution in toluene on a TEM Grid (200 mesh Cu, Agar Scientific) in an argon-filled glove box and imaged employing a FEI Tecnai F20 microscope operated at 200 kV.Transient absorption
In this technique a pump pulse generates photoexcitations within the films, which are then probed at later times using a broadband probe pulse. Transient absorption spectra were recorded over short (ps) (1 ps–1 ns) and long (ns) (1 ns–100 μs) time delays with a probe pulse covering from 1075–1350 nm. For short-time (ps-TA) measurements a portion of the output of a Ti:Sapphire amplifier system (Spectra-Physics Solstice) operating at 1 kHz, was used to pump a home built non-collinear optical parametric amplifier (NOPA) to generate the pump pulse at 532 nm (FWHM 50 nm, <100 fs). In both short and long-time measurements another portion of the amplifier output was used to pump a home-built non-collinear optical parametric amplifier, to generate the probe pulse. The probe beam was then split to generate a reference beam so that laser fluctuations could be normalized. For short-time measurements the probe is delayed using a mechanical delay-stage (Newport). For long-time (ns-TA) measurements a separate frequency-doubled Q-switched Nd:YVO4 laser (AOT-YVO-25QSPX, Advanced Optical Technologies) is used to generate the pump. This laser produces pulses with a temporal breadth below 1 ns at 530 nm and has an electronically controlled delay. The pump and probe beams are overlapped on the sample adjacent to a reference probe beam. This reference is used to account for any shot-to-shot variation in transmission. The samples were prepared on quartz glass by spin coating from a 10 mg mL−1 solutions at 2000 rpm for 15 s, and encapsulated via affixing a glass microscope slide with epoxy. The beams are focused into an imaging spectrometer (Andor, Shamrock SR 303i) and detected using a pair of linear image sensors (Hamamatsu, G11608) driven and read out at the full laser repetition rate by a custom-built board from Stresing Entwicklungsburo. Initial measurements were recorded at a range of laser fluences (20–80 μJ cm−2) on both the ps transient absorption set-ups. The TA spectra presented in this work were recorded at 20 μJ cm−2 fluence, at which there was no short time (ps regime) Auger recombination observed from multiple excited states occupying the one quantum dot. In all measurements every second pump shot is omitted, either electronically for long-time measurements or using a mechanical chopper for short-time measurements. The fractional differential transmission (ΔT/T) of the probe is calculated for each data point once 1000 shots have been collected.LED device fabrication
Poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) was spin-coated onto an ITO-coated glass substrate at 6000 rpm for 45 s, followed by annealing at 140 °C for 30 min in a nitrogen-filled glovebox. A 10 mg mL−1 poly(2,7-(9,9-di-n-octylfluorene)-alt-(1,4-phenylene-((4-sec-butylphenyl)imino)-1,4-phenylene)) (TFB) in toluene, was spin coated on top the Pedot:PSS layer at 2000 rpm for 60 s followed by annealing at 150 °C for 10 m. Next the PbS nanocrystal layer was spin-coated on the substrate at a concentration of 25 mg mL−1 in octane (1500 rpm for 15 s) after a wait of 5 s. Subsequently, the native oleic acid ligand was exchanged with benzene dithiol (20mmol in acetonitrile) in a second spin-coating step using the same spinning conditions. To remove residual ligand and un-exchanged nanocrystals consecutive spin-rinsing steps using pure acetonitrile and octane were performed. This cycle was repeated three times. The samples were then transferred into a thermal evaporator and aluminium (Al; 80 nm) and was deposited through a shadow mask at 3 × 10−6 mbar or better. The LEDs were encapsulated by affixing a glass slide on top of the contacts using transparent UV epoxy glue.
LED characterization
Current versus voltage characteristics were measured using a Keithley 2400 source measure unit. Photon flux was measured simultaneously using a calibrated germanium photodiode centered over the light-emitting pixel. External quantum efficiency was calculated assuming a Lambertian emission profile.Author contribution
N. J. L. K. D., J. X. and T. C. J. synthesized the nanocrystals. N. J. L. K. D. characterized the nanocrystals and carried out all experiments unless mentioned otherwise. J. R. A. performed the transient absorption measurements. N. J. L. K. D., J. R. A., N. C. G. and A. R. contributed to writing the manuscript, and all authors contributed to discussion and analysis of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. J. L. K. D. acknowledges funding from the Ernest Oppenheimer fund. J. R. A. thanks the Cambridge Commonwealth European and International Trust, and Winton Programme for the Physics of Sustainability for financial support. J. X. thanks the EPSRC CDT in Nanoscience and Nanotechnology (EP/L015978/1). This work was supported by the EPSRC (EP/M005143/1 and EP/M024873/1). The data underlying this publication are available at https://doi.org/10.17863/CAM.30558.References
- C. Murray, Annu. Rev. Mater. Sci., 2000, 545–610 CrossRef CAS.
- E. H. Sargent, Adv. Mater., 2005, 17, 515–522 CrossRef CAS.
- D. V. Talapin, J. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- L. H. Slooff, A. Polman, F. Cacialli, R. H. Friend, G. A. Hebbink, F. C. J. M. Van Veggel and D. N. Reinhoudt, Appl. Phys. Lett., 2001, 78, 2122–2124 CrossRef CAS.
- G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111–116 CrossRef CAS.
- N. Tessler, V. Medvedev, M. Kazes, S. Kan and U. Banin, Science, 2002, 295, 2001–2003 CrossRef PubMed.
- G. Konstantatos, C. Huang, L. Levina, Z. Lu and E. H. Sargent, Adv. Funct. Mater., 2005, 15, 1865–1869 CrossRef CAS.
- X. Ma, F. Xu, J. Benavides and S. G. Cloutier, Org. Electron., 2012, 13, 525–531 CrossRef CAS.
- G. J. Supran, K. W. Song, G. W. Hwang, R. E. Correa, J. Scherer, E. A. Dauler, Y. Shirasaki, M. G. Bawendi and V. Bulovic, Adv. Mater., 2015, 27, 1437–1442 CrossRef CAS PubMed.
- P. O. Anikeena, J. E. Halpert, M. G. Bawendi and V. Bulovic, Nano Lett., 2009, 9, 2532–2536 CrossRef PubMed.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 CrossRef CAS.
- B. O. Dabbousi, M. G. Bawendi, O. Onitsuka and M. F. Rubner, Appl. Phys. Lett., 1995, 66, 1316–1318 CrossRef CAS.
- S. Coe, W.-K. Woo, M. Bawendi, V. Bulović and V. Bulovic, Nat. Lett., 2002, 420, 800–803 CrossRef CAS PubMed.
- J. S. Steckel, S. Coe-Sullivan, V. Bulović and M. G. Bawendi, Adv. Mater., 2003, 15, 1862–1866 CrossRef CAS.
- J. Zhao, J. A. Bardecker, A. M. Munro, M. S. Liu, Y. Niu, I.-K. Ding, J. Luo, B. Chen, A. K.-Y. Jen and D. S. Ginger, Nano Lett., 2006, 6, 463–467 CrossRef CAS PubMed.
- K. N. Bourdakos, D. M. N. M. Dissanayake, T. Lutz, S. R. P. Silva and R. J. Curry, Appl. Phys. Lett., 2008, 92, 1–4 Search PubMed.
- A. H. Ip, S. M. Thon, S. Hoogland, O. Voznyy, D. Zhitomirsky, R. Debnath, L. Levina, L. R. Rollny, G. H. Carey, A. Fischer, K. W. Kemp, I. J. Kramer, Z. Ning, A. J. Labelle, K. W. Chou, A. Amassian and E. H. Sargent, Nat. Nanotechnol., 2012, 7, 577–582 CrossRef CAS PubMed.
- P. Reiss, M. Protière and L. Li, Small, 2009, 5, 154–168 CrossRef CAS PubMed.
- W. K. Bae, J. Joo, L. A. Padilha, J. Won, D. C. Lee, Q. Lin, W. Koh, H. Luo, V. I. Klimov and J. M. Pietryga, J. Am. Chem. Soc., 2012, 134, 20160–20168 CrossRef CAS PubMed.
- J. M. Pietryga, D. J. Werder, D. J. Williams, J. L. Casson, R. D. Schaller, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 4879–4885 CrossRef CAS PubMed.
- X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019–7029 CrossRef CAS.
- C. Kagan, C. Murray, M. Nirmal and M. Bawendi, Phys. Rev. Lett., 1996, 76, 1517–1520 CrossRef CAS PubMed.
- Z. Lingley, S. Lu and A. Madhukar, Nano Lett., 2011, 11, 2887–2891 CrossRef CAS PubMed.
- Z. Lingley, S. Lu and A. Madhukar, J. Appl. Phys., 2014, 115, 084302 CrossRef.
- S. Kitamura, M. Senshu, H. Tokushige, T. Katsuyama, N. Ozaki, I. Tanaka and Y. Sugimoto, Superlattices Microstruct., 2015, 79, 123–134 CrossRef CAS.
- K.-S. Cho, E. K. Lee, W.-J. Joo, E. Jang, T.-H. Kim, S. J. Lee, S.-J. Kwon, J. Y. Han, B.-K. Kim, B. L. Choi and J. M. Kim, Nat. Photonics, 2009, 3, 341–345 CrossRef CAS.
- J. M. Caruge, J. E. Halpert, V. Wood, V. Bulović and M. G. Bawendi, Nat. Photonics, 2008, 2, 247–250 CrossRef CAS.
- R. Vinayakan, T. Shanmugapriya, P. V. Nair, P. Ramamurthy and K. G. Thomas, J. Phys. Chem. C, 2007, 111, 10146–10149 CrossRef CAS.
- A. L. Rogach, T. A. Klar, J. M. Lupton, A. Meijerink and J. Feldmann, J. Mater. Chem., 2009, 19, 1208 RSC.
- F. Xu, X. Ma, C. R. Haughn, J. Benavides, M. F. Doty and S. G. Cloutier, ACS Macro Lett., 2011, 5, 9950–9957 CAS.
- A. P. Litvin, P. S. Parfenov, E. V. Ushakova, T. A. Vorsina, A. L. Simões Gamboa, A. V. Fedorov and A. V. Baranov, J. Phys. Chem. C, 2015, 119, 17016–17022 CrossRef CAS.
- D. Kim, S. Okahara, M. Nakayama and Y. Shim, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 1–4 Search PubMed.
- S. A. Crooker, J. A. Hollingsworth, S. Tretiak and V. I. Klimov, Phys. Rev. Lett., 2002, 89, 186802 CrossRef CAS PubMed.
- M. Achermann, M. A. Petruska, S. A. Crooker and V. I. Klimov, J. Phys. Chem. B, 2003, 107, 13782–13787 CrossRef CAS.
- D. Kim, K. Okazaki and M. Nakayama, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 1–5 Search PubMed.
- T. Franzl, D. S. Koktysh, T. A. Klar, A. L. Rogach, J. Feldmann and N. Gaponik, Appl. Phys. Lett., 2004, 84, 2904–2906 CrossRef CAS.
- T. Franzl, A. Shavel, A. L. Rogach, N. Gaponik, T. A. Klar, A. Eychmüller and J. Feldmann, Small, 2005, 1, 392–395 CrossRef CAS PubMed.
- J. M. Luther, M. Law, Q. Song, C. L. Perkins, M. C. Beard and A. J. Nozik, ACS Nano, 2008, 2, 271–280 CrossRef CAS PubMed.
- S. W. Clark, J. M. Harbold and F. W. Wise, J. Phys. Chem. C, 2007, 111, 7302–7305 CrossRef CAS.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- M. S. Neo, N. Venkatram, G. S. Li, W. S. Chin and W. Ji, J. Phys. Chem. C, 2010, 114, 18037–18044 CrossRef CAS.
- J. de Mello, H. Wittmann and R. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh01122b |
| This journal is © The Royal Society of Chemistry 2019 |