
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid, selective and stable HaloTag-LbADH immobilization directly from crude cell extract for the continuous biocatalytic production of chiral alcohols and epoxides†
J.
Döbber
ab,
M.
Pohl
a,
S. V.
Ley
 b and
B.
Musio
*b
b and
B.
Musio
*b
aIBG-1: Biotechnology, Forschungszentrum Jülich, 52425 Jülich, Germany
bDepartment of Chemistry, University of Cambridge, Cambridge, CB21EW, UK. E-mail: bm450@cam.ac.uk
First published on 17th January 2018
Abstract
A strategy for biocatalyst immobilization in flow directly from the crude cell extract is described. The efficiency and the stability of the immobilized enzyme were demonstrated during the asymmetric reduction of a range of ketones. The cascade two-step chemo-enzymatic preparation of chiral epoxides was possible through the initial ketone bioreduction to an intermediate halohydrin followed by its intramolecular cyclization.
The use of biocatalytic methods in continuous processes for more sustainable chemistry applications has been growing over the last few years.1 The benefits arising from the continuous biotransformations are clear in terms of re-usability of non-toxic catalysts, high selectivity, mild and accurately controlled reaction conditions, reduction of protection–deprotection steps and easy scale-up through improved downstream processing.2 However, the implementation of continuous biocatalytic processes demands a high operational stability of enzymes, which is still an existing concern especially in the presence of conventional media such as organic solvents.3 Furthermore, the availability and stability of enzymes can be limiting.
Enzyme immobilization technologies, which lead to the formation of an insoluble biocatalyst, in some cases can aid efficient recycling and an enhanced operational stability. Ideally, enzyme immobilization should proceed rapidly to maintain enzyme activity and result in a biocatalyst formulation that does not leach. Additionally, the ideal arrangement would be cheap to produce and would avoid excessive waste.4
Particularly interesting for flow applications is the prospect of preparing plug-flow reactors containing the desired immobilized biocatalyst using simple procedures.
Recently, we reported on an innovative immobilization strategy based on the commercially available HaloTag™ technology (Promega).5 The HaloTag™ is a modified dehalogenase enzyme from Rhodococcus species6 that can be genetically fused to an enzyme to effect rapid binding from a crude cell extract. This fusion tag recognizes terminal chloroalkane substituents exposed on the surface of the HaloLink™ resin and affords a stable covalent ester bond via a self-terminating mechanism thereby immobilizing the target enzyme (Fig. 1). We applied this concept successfully for the one-step purification and immobilization of the benzaldehyde lyase from Pseudomonas fluorescence (PfBAL) and could prove its usefulness for the efficient recycling of PfBAL in repetitive batch reactions.5
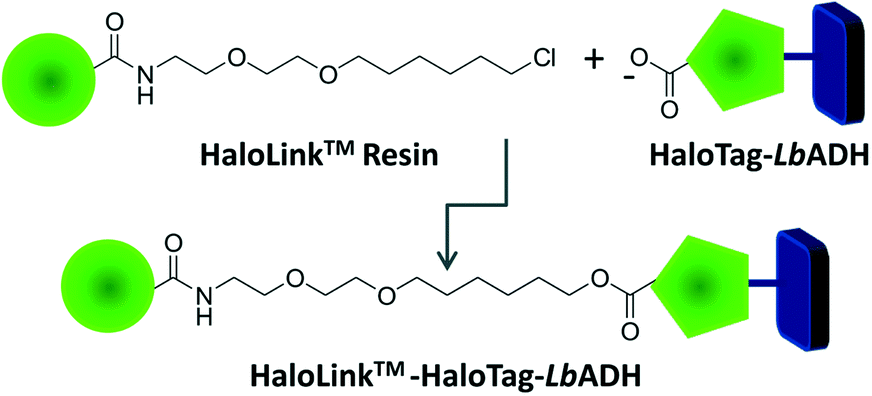 | ||
| Fig. 1 Schematic overview of the HaloTag-LbADH enzyme immobilization on the HaloLink™ resin. The HaloLink™ resin consists of Sepharose beads and is visualized as green spheres. The HaloTag™ was genetically fused to the LbADH and recognizes the terminal chloroalkane ligands to establish a covalent ester bond thereby immobilizing the biocatalyst. | ||
Here we extend the HaloTag™ method to a continuous biocatalytic production process involving the alcohol dehydrogenase from Lactobacillus brevis (LbADH).7LbADH is a complex biocatalyst system consisting of four subunits forming a tetramer to achieve the active conformation. The non-covalently bound cofactor NADPH is also essential for catalysis and must be recycled efficiently to make the corresponding biotransformations economically viable. For the LbADH, different strategies for cofactor regeneration have been developed, including enzyme engineering or substrate-coupled approaches.8
In this study we demonstrate an easy and rapid preparation of a packed-bed flow reactor by immobilizing the fusion enzyme directly from the crude cell extracts from the respective recombinant E. coli production host immediately prior to conducting the biotransformation. Furthermore, we evaluate the performance of the immobilized biocatalyst for the continuous asymmetric reduction of a range of ketones to the corresponding chiral alcohols. Finally, we demonstrate the high operational stability of the new immobilized HaloTag-LbADH and its use in organic solvents for possible multi-step transformations.
In the first stage, the HaloTag™ was genetically fused to the N-terminus of the LbADH resulting in the fusion enzyme HaloTag-LbADH. After successful production of this fusion enzyme in E. coli BL21 (DE3) cells, we verified its activity as a biocatalyst in comparison to the non-immobilized, untagged LbADH. Thus, HaloTag-LbADH was immobilized in batch to the HaloLink™ resin directly from the crude cell extract by incubation for 1 h at 25 °C (see ESI† for more details). The activity was analyzed following the reduction of benzaldehyde to benzyl alcohol in an aqueous buffer (Fig. 2, conditions A). The immobilized HaloTag-LbADH exhibited a residual activity of 35% by comparison to the reference. A similar reduced activity of an immobilized biocatalyst with respect to the free reference system is a common outcome and can be explained by different reasons. First, the physical confinement of the enzyme on the support can lead to a reduced flexibility, thus affecting the activity. Additionally, the immobilization and the high local concentration of an enzyme on a support may cause mass-transfer limitations in comparison to soluble systems. Importantly, in the case of complex tetrameric enzymes carrying four individual HaloTags, such as HaloTag-LbADH, steric hindrance of the active site, may be an issue. In this case the size per subunit of the enzyme was more than doubled by the HaloTag-fusion from 252 amino acids (26.76 KDa) for the native enzyme to 581 amino acids (63.58 KDa) for the HaloTag-LbADH fusion enzyme. By contrast, the previously immobilized tetrameric HaloTag-PfBAL fusion enzyme5 revealed a residual activity of 65% indicating that the influence of the tag cannot be predicted and seems to depend on the relative sizes and overall conformation of the particular fusion partners. Then, we investigated the influence of the sacrificial co-substrate 2-propanol on the activity of the free LbADH and the immobilized HaloTag-LbADH (Fig. 2, conditions B). 2-Propanol is generally used for the substrate-coupled cofactor regeneration, being oxidized to acetone by LbADH thereby reducing NADP+ to NADPH.8a Both enzyme variants were less active in the presence of 2-propanol (10 vol%). However, the activity of the free, untagged LbADH was reduced by 75%, while the immobilized HaloTag-LbADH variant lost only half of its activity. This observation indicates that the immobilized HaloTag-LbADH was relatively more stable against the detrimental effects of 2-propanol and that immobilization of the enzyme led to the formation of a more robust catalyst. We also compared the activity of the free, untagged LbADH with the activity of the immobilized HaloTag-LbADH in a buffer/THF/2-propanol system (Fig. 2, conditions C).
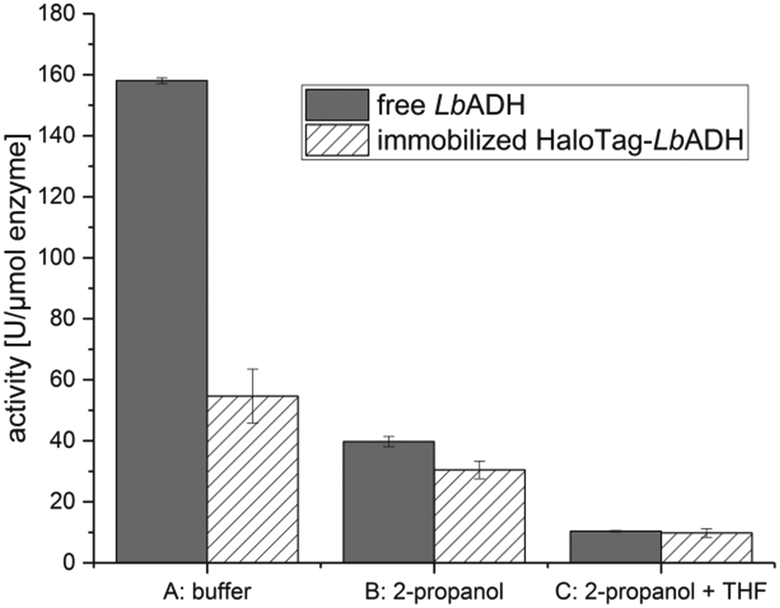 | ||
| Fig. 2 Activity of immobilized HaloTag-LbADH in different solvent systems compared to the free, untagged LbADH. Initial rate activities were analyzed by HPLC following the reduction of benzaldehyde (20 mM) to benzyl alcohol in a total volume of 1 ml at 25 °C. Conditions A: NADPH (20 mM), KPi 50 mM (pH 7.0), MgCl2·H2O (1 mM); conditions B: NADPH (0.5 mM), 2-propanol (10 vol%), KPi 50 mM (pH 7.0), MgCl2·H2O (1 mM); conditions C: NADPH (0.5 mM), 2-propanol (10 vol%), THF (10 vol%), KPi 50 mM (pH 7.0), MgCl2·H2O (1 mM). Error bars represent the variance of three activity measurements from the same sample. | ||
The activity of the free LbADH as well as of the immobilized HaloTag-LbADH decreased further in the presence of THF. However, no difference in activity between both the free and the immobilized enzyme was observed although the difference in the buffered system was shown to be about 65% (Fig. 2, conditions A).
As a next step, the preparation of a packed-bed reactor was investigated. Thus, an Omnifit® column (30 MM × 50 MM) was filled with HaloLink™ resin (360 mg wet weight) and connected to an Asia Syringe Pump (Syrris, Royston, United Kingdom) to drive the flow streams. The column was then loaded with a solution of crude cell extract containing the HaloTag-LbADH fusion enzyme. The efflux was re-circulated and its UV absorption (282 nm) was continuously monitored in-line by a UV/Vis detector (Flow-UVTM, Uniqsis Ltd) provided with a high pressure flow cell and a 220–1050 nm Xenon pulsing lamp (Fig. 3a). The solid packed bed appeared to be saturated with HaloTag-LbADH after 216 min of continuous re-circulation of the crude cell extract solution as was indicated by a constant UV absorbance (Fig. 3b, solid line). Then, the reactor was washed to elute all the non-specifically bound proteins until the absorbance of the efflux reached the baseline (Fig. 3b, dashed line). Finally, the resin was removed from the column to evaluate the immobilization and to determine the purity of the bound enzyme. Thus, the ester bond connecting the HaloTag™ with the HaloLink™ resin was saponified by treatment with sodium dodecyl sulfate (SDS) and NaOH and a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis was performed.
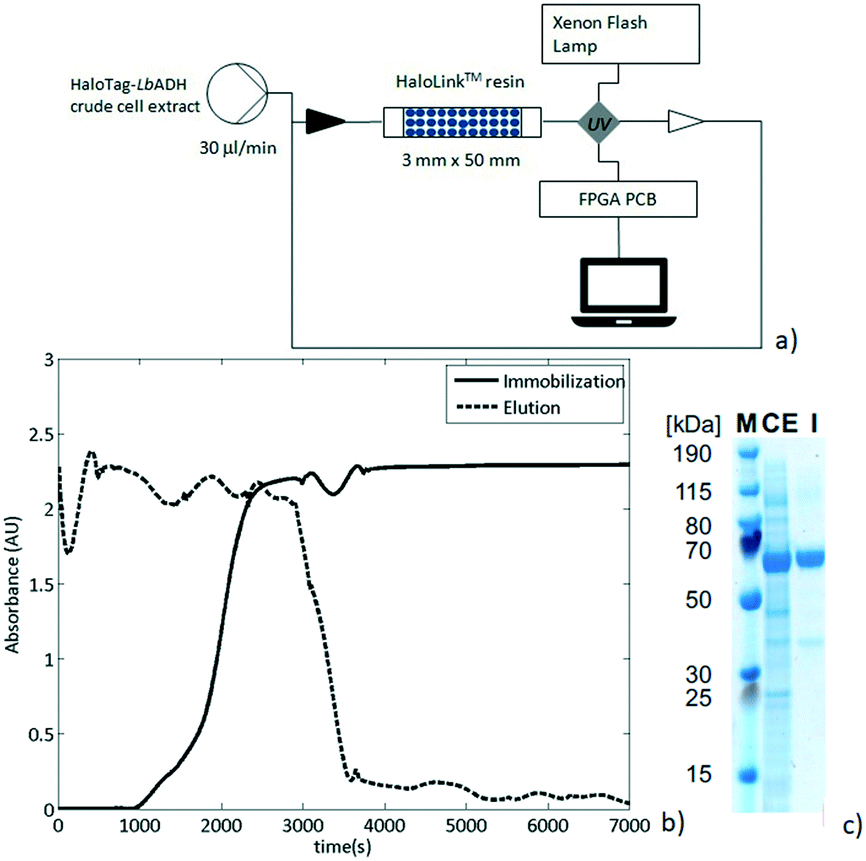 | ||
| Fig. 3 a) Set-up for the immobilization of the HaloTag-LbADH directly from the crude cell extracts with in-line UV analysis at 282 nm (Field Progammable Gate Array, FPGA; Printed Circuit Board, PCB). b) In-line monitoring during the immobilization step (solid line) during the unbound proteins elution step (dashed line). c) SDS-PAGE analysis of HaloTag-LbADH before (CE) and after immobilization (I). M: PageRuler™ Plus Prestained Ladder (ThermoFisher Scientific, Germany). | ||
The HaloTag-LbADH was efficiently immobilized with a very high purity since almost no contamination was observed on the HaloLink™ resin and the protein band corresponding to one monomer of the HaloTag-LbADH (63 kDa per monomer) predominated markedly (Fig. 3c).
The total amount of protein (4 mg) present on 360 mg wet resin was determined by using the bicinchoninic acid (BCA) assay.
Once the feasibility of preparing a biocatalytic packed-bed bioreactor directly from the crude cell extracts was demonstrated, its efficiency in catalyzing the asymmetric reduction of a range of ketones was explored particularly in terms of reaction conditions and stability of the bioreactor. First, we aimed to find the appropriate 2-propanol concentration necessary for optimal cofactor regeneration, thereby enabling highest conversion and maximal enzyme activity and stability. Thus, the reduction of acetophenone 1a to (R)-phenylethanol 2a was first studied in batch mode. As the 2-propanol concentration influences the thermodynamic equilibrium of the reaction, usually an excess is necessary to reach a high conversion. However, a high 2-propanol concentration may affect the enzyme activity as well as stability. We observed a decreased activity of the immobilized HaloTag-LbADH with increasing concentrations of 2-propanol, though the biocatalyst remained remarkably active even in presence of 2-isopropanol 90 vol% (Fig. S1 in ESI†). Aiming at understanding the effect of 2-propanol on the stability of the immobilized HaloTag-LbADH, repetitive reactions were performed by examining the same batch of immobilized biocatalyst in the presence of different concentrations of 2-propanol ranging between 10 vol% and 90 vol%. In general, the enzyme was highly stable under all the test conditions. No loss of activity between the repetitive reactions was observed for reactions with 2-propanol 10–25 vol%, and only a slight decrease after the fourth cycle was observed for reactions performed with higher concentrations up to 90 vol% (Fig. S2–S6 in ESI†). With this evidence in hand, we moved on to study the reduction of acetophenone 1a in flow using the prepared HaloTag-LbADH packed-bed reactor. The corresponding (R)-phenylethanol 2a could be obtained with 95% conversion when a solution of acetophenone (50 mM) and NADPH (0.5 mM) in a mixture of Kpi 50 mM (90 vol%) and 2-propanol (10 vol%) was passed continuously through the packed-bed reactor (flow rate 30 μl min−1). Higher substrate concentrations (50–100 mM) and faster flow rates (up to 100 μl min−1) were also tested, but the reaction occurred in all cases with lower conversion. More importantly, this reaction was continued over several days and stopped after 138 h, with no loss in activity (see ESI† for experimental details).
To enable the reduction of other ketones, which were less soluble in the buffer/2-propanol solvent system, the addition of conventional co-solvents was considered. Methyl tert-butyl ether (MTBE), dimethyl sulfoxide (DMSO), acetonitrile (ACN), dimethylformamide (DMF) and tetrahydrofuran (THF) were evaluated for the reduction of 4′-bromoacetophenone 1b as a test case. It was found that the formation of a biphasic or emulsion solvent system was not favorable for the reaction. Among the solvents screened, only THF (30 vol%) gave encouraging results giving access to the corresponding (R)-4′-bromophenylethanol 2b with high yield (94%) and excellent ee (>99%). When a lower THF concentration was added to the reaction mixture, the substrate and the product were partially trapped within the packed bed, as demonstrated by UV absorption of the efflux and by the low mass recovery after the reaction work-up. Next, starting from a range of prochiral ketones, we demonstrated the generality of this synthetic strategy for the preparation of a number of chiral alcohols (Fig. 4). The immobilized HaloTag-LbADH system exhibited a good catalytic activity towards a range of substrates, including substituted aromatic ketones (1a–1l), an aliphatic ketone (1m, the ee of 2m was determined by HPLC analysis of the corresponding benzoate derivative), a β-ketoester (1n) and an alkynyl ketone (1o).
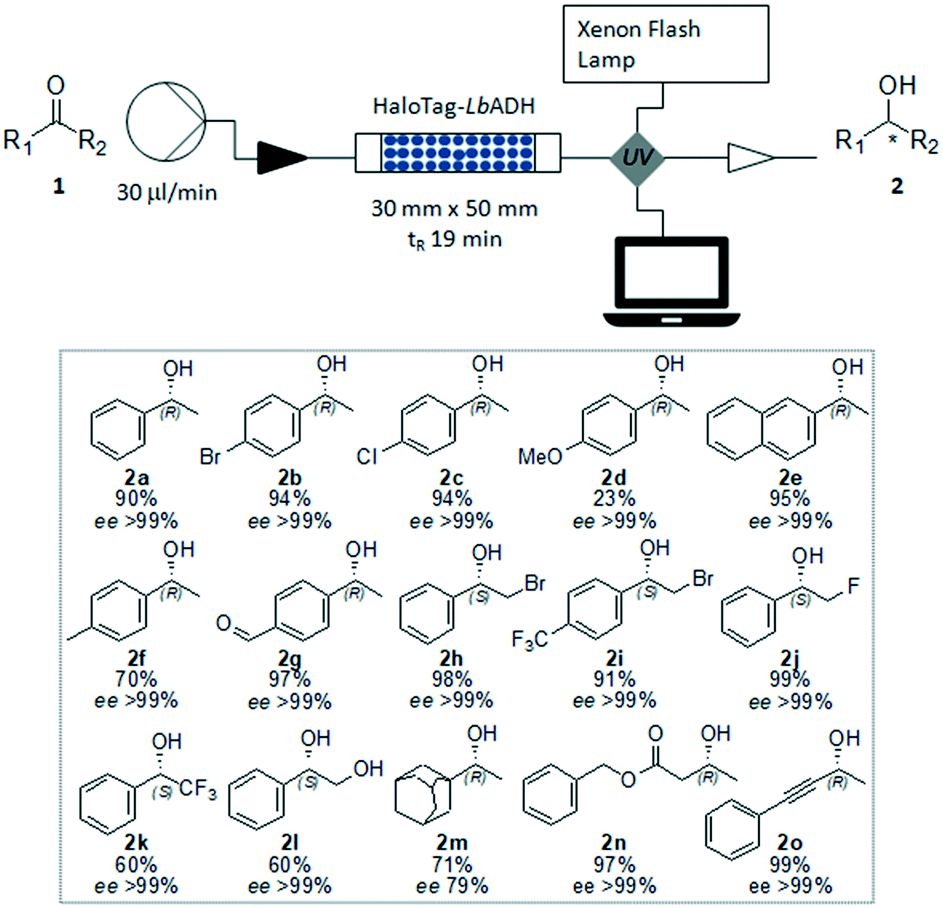 | ||
| Fig. 4 Asymmetric reduction of ketones by using the immobilized HaloTag-LbADH in flow. Reaction conditions: ketones 1a–1o (50 mM), NADPH (0.5 mM), 2-propanol (10 vol%), THF (30 vol%), KPi 50 mM (pH 7.0), MgCl2·H2O (1 mM), flow rate (30 μl min−1). Isolated yields after purification. The ee were determined by GC or HPLC as described in the experimental procedure. | ||
The obtained results are in excellent agreement with the reported data describing the dependence of the relative activity of LbADH on the steric and electronic effects due to the substrate nature and on the thermodynamic stability of the products.9 The activity of the bioreactor remained constant under these reaction conditions, as demonstrated by the continuous production of (R)-(−)-2n starting from benzylacetoacetate 1n, with a total amount of pure compound equated to 420 mg (2.16 mmol) produced over 24 h. After a washing step with buffer, the biocatalytic solid bed demonstrated to be still active and efficient for another reaction.
Finally, we explored the feasibility of an enantioselective two-step chemo-enzymatic synthesis of the chiral epoxide (S)-5, using the bio-reduction to form the intermediate halohydrin 2h, which was immediately cyclized in a glass microreactor under basic conditions (pH 13, Fig. 5). The corresponding (S)-2-phenyloxirane 5 could be obtained in a short time (35 minutes) with high conversion (98%) and excellent ee (98%). This experiment demonstrated the advantages of the flow-plug technology in multi-step processes, allowing a quick change of the reaction conditions and an efficient compartmentalization of the synthetic steps.
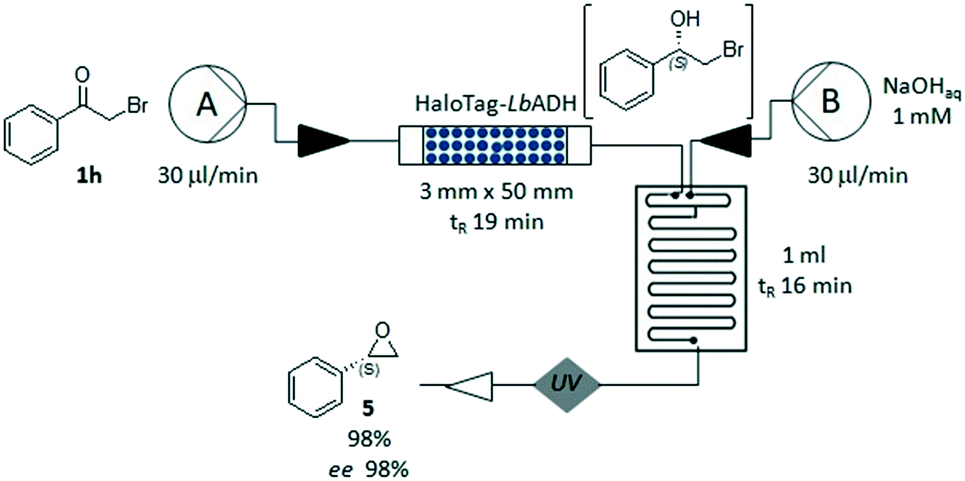 | ||
| Fig. 5 Two-step chemo-enzymatic synthesis of (S)-2-phenyloxirane 5. Pump A loading the packed-bed reactor containing immobilized HaloTag-LbADH: flow rate 30 μl min−1, ketone 1h (50 mM), NADPH (0.5 mM), 2-propanol (10 vol%), THF (30 vol%), KPi 50 mM (pH 7.0), MgCl2·H2O (1 mM). Pump B loading the glass microreactor (2 inputs, 1 ml): flow rate 30 μl min−1, NaOH (1 mM, pH 13). Conversion determined by 1H-NMR. | ||
In conclusion, we have designed a new methodology for the fast preparation of cartridges containing a stable, covalently immobilized enzyme, starting directly from the crude cell extracts without any need of previous enzyme chromatographic purification step. The resulting packed bed bioreactor was then used for the continuous enantioselective production of chiral alcohols with the re-use of the catalyst. The application of this bioreactor to multi-step processes was demonstrated during the two-step chemo-enzymatic transformation of 2-bromoacetophenone into (S)-2-phenyloxirane, with benefits resulting from improved downstream processing. The relative ease of construction of the bioreactor and its robustness, even in the presence of THF as co-solvent, opens up wider opportunities for synthesis.
This work was supported by the EPSRC (Award No. EP/K009494/1 and EP/K039520/1) and by the German Federal Ministry of Education and Research (BMBF) within the project “Molecular Interaction Engineering” (funding code 031A095).
Conflicts of interest
The authors declare no conflicts of interest.Notes and references
- (a) L. Hajba and A. Guttman, J. Flow Chem., 2016, 6, 8–12 CrossRef CAS; (b) A. Brahma, B. Musio, U. Ismayilova, N. Nikbin, S. Kamptmann, P. Siegert, G. Jeromin, S. V. Ley and M. Pohl, Synlett, 2015, 262–266 Search PubMed; (c) J. C. Thomas, M. D. Burich, P. T. Bandeira, A. R. Marques de Oliveira and L. Piovan, Biocatalysis, 2017, 3, 27–36 CrossRef; (d) G. Gasparini, I. Archer, E. Jones and R. Ashe, Org. Process Res. Dev., 2012, 16, 1013–1016 CrossRef CAS; (e) K. Schroer, U. Mackfeld, I. A. W. Tan, C. Wandrey, F. Heuser, S. Bringer-Meyer, A. Weckbecker, W. Hummel, T. Daußmann, R. Pfaller, A. Liese and S. Lütz, J. Biotechnol., 2007, 132, 438–444 CrossRef CAS PubMed.
- (a) P. D. de María and F. Hollmann, Front. Microbiol., 2015, 6, 6–10 Search PubMed; (b) S. Wenda, S. Illner, A. Mell and U. Kragl, Green Chem., 2011, 13, 3007 RSC; (c) M. D. Truppo, ACS Med. Chem. Lett., 2017, 8, 476–480 CrossRef CAS PubMed; (d) P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS; (e) A. Madhavan, R. Sindhu, P. Binod, R. K. Sukumaran and A. Pandey, Bioresour. Technol., 2017, 245(Part B), 1304–1313 CrossRef CAS PubMed; (f) A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed; (g) R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem. – Eur. J., 2017, 1–25 CAS; (h) A. P. Green and N. J. Turner, Perspect. Sci., 2016, 9, 42–48 CrossRef.
- (a) J. M. Guisan, Immobilization of Enzymes and Cells IN Series Editor, 2013, vol. 1051 Search PubMed; (b) V. Bansal, Y. Delgado, M. D. Legault and G. Barletta, Molecules, 2012, 17, 1870–1882 CrossRef CAS PubMed; (c) U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC; (d) R. A. Sheldon and S. Van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- (a) I. Eş, J. D. G. Vieira and A. C. Amaral, Appl. Microbiol. Biotechnol., 2015, 99, 2065–2082 CrossRef PubMed; (b) C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS; (c) R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC; (d) C. Mateo, V. Grazú, B. C. C. Pessela, T. Montes, J. M. Palomo, R. Torres, F. López-Gallego, R. Fernández-Lafuente and J. M. Guisán, Biochem. Soc. Trans., 2007, 35, 1593–1601 CrossRef CAS PubMed; (e) A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- J. Döbber and M. Pohl, J. Biotechnol., 2017, 241, 170–174 CrossRef PubMed.
- L. P. Encell, R. Friedman Ohana, K. Zimmerman, P. Otto, G. Vidugiris, M. G. Wood, G. V. Los, M. G. McDougall, C. Zimprich, N. Karassina, R. D. Learish, R. Hurst, J. Hartnett, S. Wheeler, P. Stecha, J. English, K. Zhao, J. Mendez, H. A. Benink, N. Murphy, D. L. Daniels, M. R. Slater, M. Urh, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, Curr. Chem. Genomics, 2012, 6, 55–71 CrossRef PubMed.
- B. Riebel, PhD dissertation, 1996, Heinrich-Heine University Düsseldorf Search PubMed.
- (a) S. Leuchs and L. Greiner, Chem. Biochem. Eng. Q., 2011, 25, 267–281 CAS; (b) W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- (a) C. Rodríguez, W. Borzęcka, J. H. Sattler, W. Kroutil, I. Lavandera and V. Gotor, Org. Biomol. Chem., 2014, 12, 673–681 RSC; (b) F. R. Bisogno, E. García-Urdiales, H. Valdés, I. Lavandera, W. Kroutil, D. Suárez and V. Gotor, Chem. – A Eur. J., 2010, 16, 11012–11019 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7re00173h |
| This journal is © The Royal Society of Chemistry 2018 |