DOI:
10.1039/C8QM00291F
(Research Article)
Mater. Chem. Front., 2018,
2, 2070-2075
Hydrogen bonding directed co-assembly of polyoxometalates and polymers to core–shell nanoparticles†
Received
12th June 2018
, Accepted 13th September 2018
First published on 13th September 2018
Abstract
A general strategy has been developed here to co-assemble polyoxometalates (POMs) and polymers into core–shell hybrid nanoparticles via hydrogen bonding interaction. Due to the hydrogen bonds between the pyridine groups of poly(4-vinyl pyridine) (P4VP) and the hydrogen bonding donor groups on the POM surface, P4VP is stabilized by the POMs and dispersed as discrete hybrid core–shell nanoparticles in aqueous solution. For these thermodynamically stable nanoparticles, the P4VP cores are covered with hexagonal close-packed POMs. The size of the core–shell particles is controlled by the electrostatic repulsive interaction among the POMs. The introduction of extra salts screens the repulsive force among the POMs, thus increasing the size of the core–shell structures.
Introduction
Due to their rich phase behaviours in both the bulk state and solution, polymers have been widely applied in templating the synthesis of nanoparticles (NPs) with various morphologies and directing the assembly of NPs into hierarchically ordered structures.1–9 Polymers can covalently graft on the surface of NPs with high stability. The obtained hairy NPs show tuneable mechanical properties, better processability, and enhanced compatibility with organic media.10–15 However, the complex synthesis and high cost of purification limit their applications. Non-covalent interactions, including van der Waals forces, hydrophobic interaction, hydrogen bonding, aromatic stacking and electrostatic interaction, have been employed as driving forces to assemble the polymer–NP complex structures.3,16–23 This protocol is feasible from the aspect of synthesis and can build self-healing materials and smart materials that are responsive to external stimuli due to the dynamic nature of the non-covalent bonding.9,20 The search for appropriate polymers and NPs with required surface ligands is the key to this protocol. However, the rational design of non-covalently interacting NP/polymer systems is currently beyond our knowledge. A thorough understanding of the non-covalent interactions is critical to design the building blocks and tune the strength of the polymer template/NP interactions.
Polyoxometalates (POMs) are a group of well-defined, nano-scale molecular clusters composed of early transition metal centers and oxo ligands with tuneable morphologies and surface charge density.24–26 Different from regular NPs, POMs are monodispersed in both solid state and solutions.24–26 Their structures, including surface structures, are well defined and characterized at the atomic level, providing model systems for the study of supramolecular assembly of nano-scale objects.26–28 The surface of POMs is mainly composed of oxo, hydroxyl, and/or water ligands, providing access to the formation of hydrogen bonding.27 The anionic feature of POMs facilitates the study of their electrostatic interaction with cationic molecules.28 As specific examples, POM–organic hybrid materials have been designed to self-assemble into 1D structures via the formation of a hydrogen bonding network.27 Meanwhile, the interaction between anionic POMs and cationic surfactants or polymers leads to the formation of nanostructures with various morphologies.28 Based on the charge interaction between poly(4-vinyl-N-methylpyridinium iodide) and POMs, micelles and vesicles with tuneable sizes can be conveniently prepared.29,30 Therefore, using POMs as models to explore the effect of non-covalent interactions in polymer/NP co-assembly systems is advantageous. Meanwhile, the applications of POMs have been significantly developed and broadened during recent years in the fields of catalysis, medicine, electronic memory devices and single molecular magnets.31–35 The polymer-templated assembly of POMs protocol provides a rational way to extend their practical applications, e.g. in highly efficient heterogeneous catalysis and functional devices.
Herein, we reported the synthesis, structural characterization, and the formation mechanism of core–shell NPs with polymer cores and close-packed POM shells. Poly(4-vinylpyridine) (P4VP) was used as the template material to form complexes with 5 POM clusters with different topologies and charge densities, respectively. The morphologies of the obtained NP complexes were studied by electron microscopy and scattering techniques while the non-covalent interactions between the POMs and polymers were probed by vibration spectroscopy and scattering techniques.
Results and discussion
Synthesis of POM–P4VP complexes
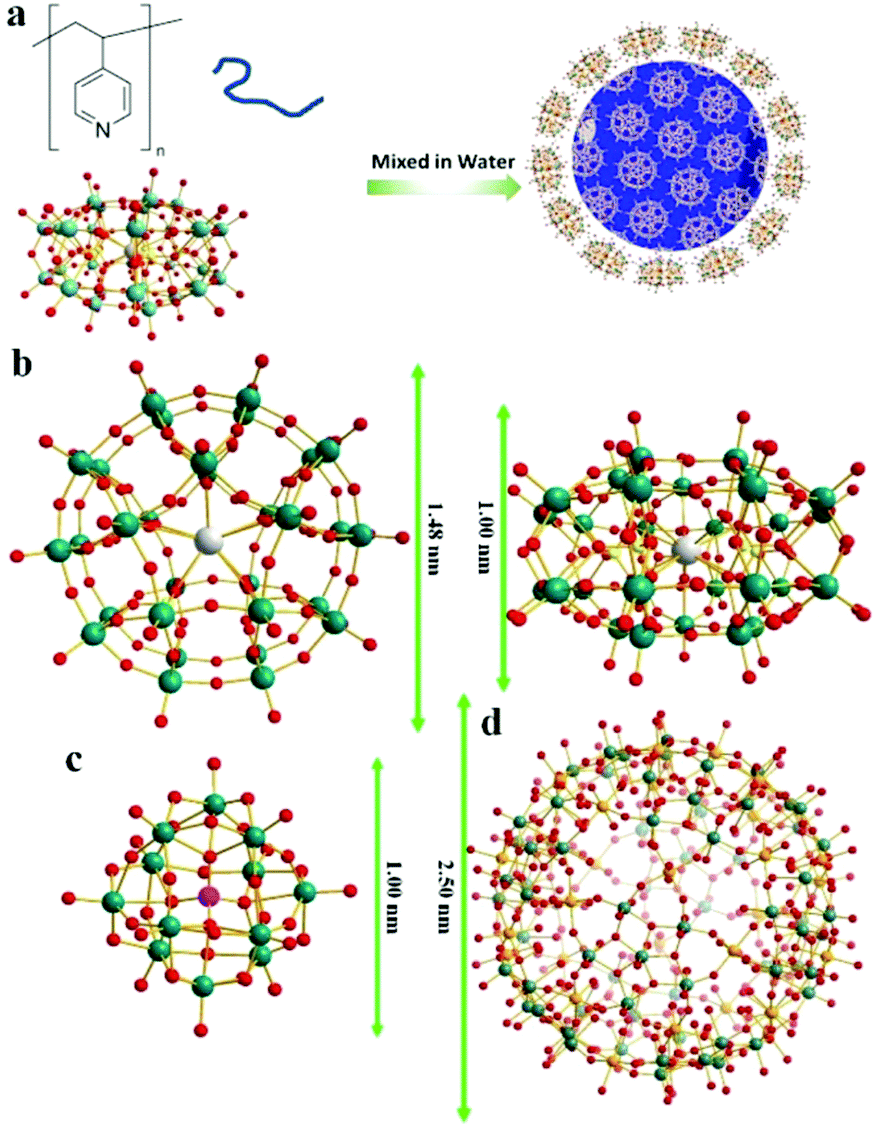
The synthesis of POM–P4VP complexes was convenient and straightforward by mixing ethanol solutions of P4VP with aqueous solutions of POMs (Fig. 1a). In a typical synthetic experiment, 0.5 mL solution of P4VP (Mw = 60 kDa) in ethanol (2 mg mL−1) was added drop-wise to 10 mL aqueous solutions of POMs at different concentrations (Fig. 1). The resulting solutions were kept open to air and stirred for 1 day until ethanol evaporated. The solution turned cloudy and precipitation occurred when the molar ratio of POM/P4VP was below 5. Scanning electron microscopy (SEM) analysis of the precipitates showed they formed irregular aggregates in the mixing process (Fig. 2a). When a larger amount of POMs was introduced into the reaction, clear and stable solutions were obtained (molar ratio of POM/P4VP ≥ 5). P4VP, soluble in polar organic solvents, shows poor solubility in water and is considered as a hydrophobic polymer. Being similar to the formation of Pickering emulsion, water-soluble POMs can be used to stabilize P4VP in aqueous solutions to reduce the surface energy of P4VP in water, which eventually leads to the formation of thermodynamically stable solutions. A certain amount of POMs was required to fully protect the P4VP surface for high solubility and stability. The clear solution obtained after POM/P4VP complexation was investigated by the dynamic light scattering (DLS) technique. The results showed the formation of large nanoparticles with narrow size distribution (see Fig. S1 to S3 in ESI†). More importantly, as a general synthetic protocol, it was successfully applied to prepare stable POM/P4VP complexes with 5 POM clusters of various sizes, including {SW}36 (formula: H50Na10O135S5W30; size: 1.5 nm × 1 nm); {PW}37 (formula: K14NaP5W30O110·31H2O; size: 1.5 nm × 1 nm, plate-shape), {Mo72V30}38 (formula: Na8K14(VO)2[{Mo6O21(H2O)3}10{Mo6O21(H2O)3(SO4)}2{VO(H2O)}20(VO)10({KSO4}5)2]·ca. 150H2O; size: 2.5 nm, hollow sphere), {Mo72Fe30}39 (formula: [Mo72Fe30O252(CH3COO)12{Mo2O7(H2O)}2{H2Mo2O8(H2O)}(H2O)91]·ca. 150H2O; size: 2.5 nm, hollow sphere), and {Keg}40 (formula: H3PW12O40; size: 1 nm, solid sphere) (Fig. 1, 2 and Fig. S2 to S8 in ESI†).
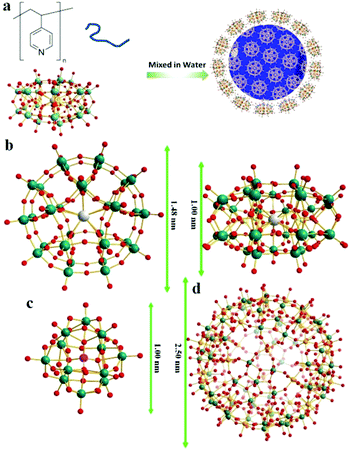 |
| | Fig. 1 (a) Graphical representation of the polymer/POM complexation process; (b) top and side views of the molecular structures of {SW}/{PW}; (c) molecular structure of {Keg}; (d) molecular structure of {Mo72Fe30}/{Mo72V30}. Color codes for spheres: teal, W; red, O; grey, K; yellow, S for {SW} or P for {PW}; orange, Fe for {Mo72Fe30} or V for {Mo72V30}. | |
 |
| | Fig. 2 (a) SEM image of the precipitates formed in {SW}/P4VP complex solution (molar ratio of {SW}/P4VP = 1). Scale bar, 1 μm; (b) TEM image of the NPs formed in {SW}/P4VP complex solution (molar ratio of {SW}/P4VP = 5). Scale bar, 200 nm; (c) zoomed in image of one NP in (b). Scale bar, 20 nm; (d) bright field TEM image (scale bar, 50 nm), (e) Fe mapping (scale bar, 100 nm), and (f) Mo mapping (scale bar, 100 nm) of {Mo72Fe30}/P4VP complex NPs. | |
The structure of the POM-P4VP complexes
The clear solutions of POM/P4VP were further studied by transmission electron microscopy (TEM) to probe the structural details of the formed complex NPs. In a typical TEM study, spherical nanoparticles were observed in the samples prepared from the clear solutions of POM/P4VP mixtures. The average sizes of the nanoparticles measured from the TEM images were consistent with the corresponding size information obtained from DLS studies (Fig. 2, and Fig. S2–S8 in ESI†). A magnified TEM image of a typical nanoparticle with a contrast close to the surface shown in Fig. 2c suggests a core–shell model for the obtained nanoparticles. POM clusters, enriched with heavy metal ions, showed darker contrast than the domain of P4VP in the TEM micrograph of Fig. 2c. Therefore, the inner core in the core–shell model was assigned to P4VP, which was crowded by layers of POMs. To further confirm the model, energy-filtered TEM (EFTEM) was applied to map the distribution of Fe and Mo in the {Mo72Fe30}/P4VP complexes as shown in Fig. 2e and f, respectively, by the three-window method. The complexation of {Mo72Fe30}/P4VP was used herein for EFTEM based on the following consideration: there were two types of heavy metal ions in this POM, providing two different resources for tracking the distribution of POMs. These results not only assured the credibility of the EFTEM results, but also confirmed the stability of POMs during the complexation process. The bright circle means there are a lot more Fe and Mo on the surface, which implies that POM is the major component of the shell structures. (Fig. 2d, e, and f). Because of there being more Mo ions than Fe ions in each POM cluster (Mo/Fe = 72/30) and the higher energy loss of the Fe L edge, the Fe mapping signal is a lot lower than Mo's.
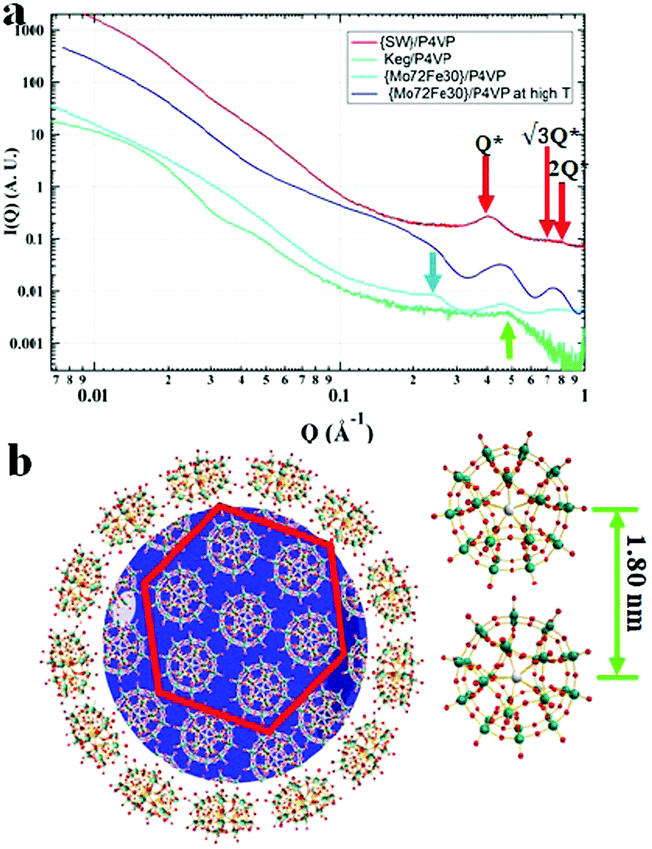
Small angle X-ray scattering (SAXS) techniques, a powerful tool for characterization of nanostructures, were further utilized to explore the packing of POMs on the surface of the P4VP cores.41 The overall SAXS curve of the {SW}/P4VP complexation system showed the feature of poly-dispersed spheres with average sizes of ca. 60 nm (Fig. 3a). Three interaction peaks at 0.40, 0.71 and 0.80 Å−1 were observed at high Q range of the SAXS curve, indicating a hexagonal packing of {SW} on the surface of the P4VP core with a center to center inter-cluster distance of 1.80 nm (Fig. 3a and b). Similar structures have been observed for the other polymer–virus systems.42 The diameter of {SW} was 1.50 nm, suggesting the close contact of {SW} during their aggregation process on the surface of the P4VP core. Such ordered packing on the polymer core surface was a general feature shared by the other POM/P4VP complexation systems. The interaction peaks were also observed in the scattering curves of {PW12}/P4VP and {Mo72Fe30}/P4VP complexes (Fig. 3a). Due to their size differences among {PW12} (1.0 nm), {SW} (1.5 nm), and {Mo72Fe30} (2.5 nm), the positions of their first interaction peaks shifted to low Q range, corresponding to the increasing of inter-POM distance ({Keg}/P4VP, 1.45 nm; {SW}/P4VP, 1.80 nm; {Mo72Fe30}/P4VP, 3.10 nm).
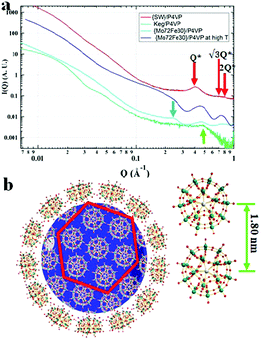 |
| | Fig. 3 (a) SAXS data of concentrated solutions of {SW}/P4VP (red), {Mo72Fe30}/P4VP (blue), and {Keg}/P4VP (green) at room temperature and {Mo72Fe30}/P4VP at 80 °C (light blue); (b) graphical representation of the dense hexagonal packing of {SW} on the surface of the P4VP core. | |
Driving force for POM/P4VP complexation
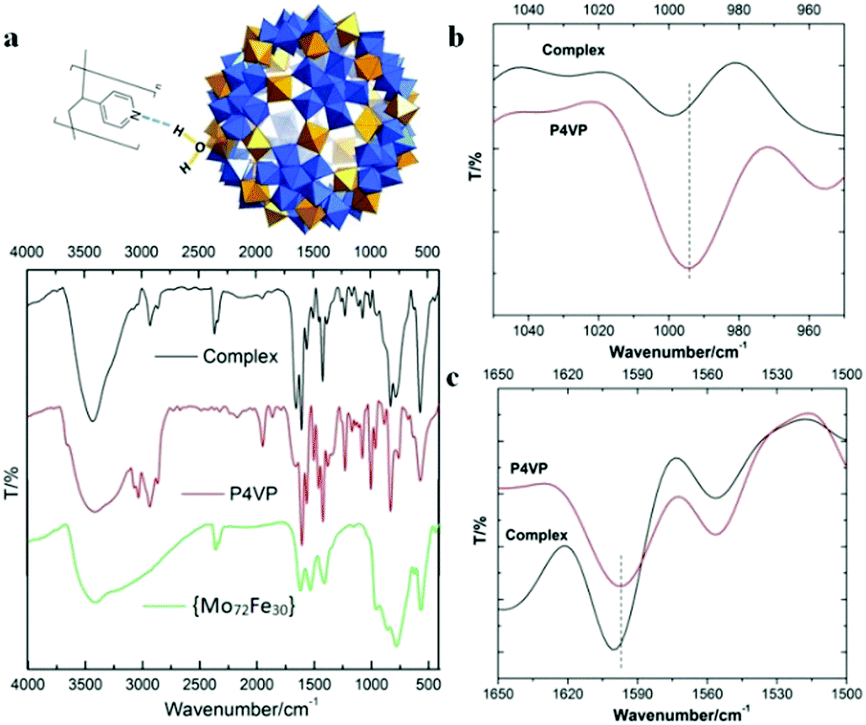
FT-IR techniques, as well as SAXS, were applied to probe the hydrogen bonding interactions between POMs and P4VP during their complexation process. Due to its well-defined hydrogen bond donor sites and corresponding well-studied vibration bands, the {Mo72Fe30} system is selected for FT-IR studies.43 In our previous study, we demonstrated that H-bonding and hydrophobic–hydrophobic interaction are the driving forces for the protein/polymer co-assembly.18,44 According to our previous studies, P4PV is mostly deprotonated at pH > 5. For most of the POM solutions we prepared for complex nanoparticle preparation, their pHs are above 5. Therefore, the contribution from the charge interaction between POMs with protonated P4VP is not the major driving force for the complexation process.44 Because of the hydrophilic property of the POMs, we believe that hydrogen bonding is the main driving force for the assembly of P4VP and POMs.45,46 Hydrogen bonding interactions between the pyridine groups of P4VP and the hydroxyl/water groups on the POM surface were confirmed by FT-IR spectroscopy (Fig. 4). The absorption peak at 3416 cm−1, characteristic of hydroxyl groups in {Mo72Fe30}, shifted to 3427 cm−1 and became sharper and stronger because of the formation of hydrogen bonding (Fig. 4a). The characteristic stretching vibration bands of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond appeared at 1597 cm−1 and 1557 cm−1 for pure P4VP sample, respectively.2 As evidence of the formation of hydrogen bonding, the peak intensity at 1557 cm−1 decreased while the peak at 1597 cm−1 showed a clear shift to 1601 cm−1 for the corresponding spectrum of the complex sample (Fig. 4c).47–49 At the 950–1050 cm−1 region: the peak at 993 cm−1 of the P4VP sample shifted to 1003 cm−1 in the complex samples with higher intensity (Fig. 4b).47–49 Meanwhile, SAXS was used to monitor the solution of {Mo72Fe30}/P4VP complex at high temperature. The sharp peak at 0.23 Å−1 resulting from the interaction among {Mo72Fe30} on the surface of P4VP core disappeared when the solution temperature was elevated to 80 °C (Fig. 3a). The overall particle size was observed to increase by analyzing the low Q range of the SAXS data, suggesting the disassembly of the {Mo72Fe30}/P4VP complex (Fig. 3a). At a high temperature, the hydrogen bonds between {Mo72Fe30} and P4VP were destroyed and the co-assemblies stabilized by this hydrogen bonding were disassembled, leading to a size increase and finally precipitation of P4VP.5,50 This is different from previous studies on the charge interaction between positively charged poly(4-vinyl-N-methylpyridinium) Keggin type POMs. In this report, the hydrogen bonding interaction led to the formation of core–shell nanoparticles while the charge interaction resulted in the formation of micelles and vesicles.29,30
N bond appeared at 1597 cm−1 and 1557 cm−1 for pure P4VP sample, respectively.2 As evidence of the formation of hydrogen bonding, the peak intensity at 1557 cm−1 decreased while the peak at 1597 cm−1 showed a clear shift to 1601 cm−1 for the corresponding spectrum of the complex sample (Fig. 4c).47–49 At the 950–1050 cm−1 region: the peak at 993 cm−1 of the P4VP sample shifted to 1003 cm−1 in the complex samples with higher intensity (Fig. 4b).47–49 Meanwhile, SAXS was used to monitor the solution of {Mo72Fe30}/P4VP complex at high temperature. The sharp peak at 0.23 Å−1 resulting from the interaction among {Mo72Fe30} on the surface of P4VP core disappeared when the solution temperature was elevated to 80 °C (Fig. 3a). The overall particle size was observed to increase by analyzing the low Q range of the SAXS data, suggesting the disassembly of the {Mo72Fe30}/P4VP complex (Fig. 3a). At a high temperature, the hydrogen bonds between {Mo72Fe30} and P4VP were destroyed and the co-assemblies stabilized by this hydrogen bonding were disassembled, leading to a size increase and finally precipitation of P4VP.5,50 This is different from previous studies on the charge interaction between positively charged poly(4-vinyl-N-methylpyridinium) Keggin type POMs. In this report, the hydrogen bonding interaction led to the formation of core–shell nanoparticles while the charge interaction resulted in the formation of micelles and vesicles.29,30
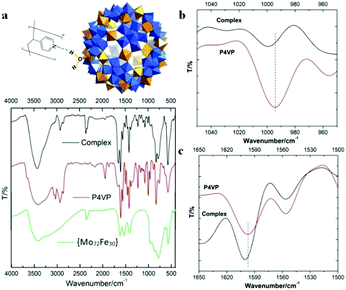 |
| | Fig. 4 (a) The mode of hydrogen bond formation between {Mo72Fe30} and P4VP and the FT-IR spectra of {Mo72Fe30}, P4VP and their complex NPs; (b) and (c) FT-IR spectra of P4VP and complex NPs at selected regions. | |
Further justification of the core–shell character of the POM/P4VP complex NPs
It is directly confirmed from the above structural analysis results that large NPs are formed in the complex aqueous solutions of POMs and P4VP with POMs close packed on the shells of NPs. The existence of P4VP in the final complex NPs is supported by the FT-IR results. The further justification that the cores of the complex NPs are made up of P4VP is mainly based on the hydrophobic nature of P4VP and the existence of hydrogen bonding between POMs and P4VP. In an aqueous environment, the hydrophobicity of P4VP will drive the aggregation of P4VP to irregular precipitates. Therefore, to avoid contacting water, P4VP should be fully covered by hydrophilic POMs. This process is energetically favored because of the multiple hydrogen bonding interactions between P4VP and POMs. It is, thus, natural to conjecture the core–shell picture of the P4VP–POM complex NPs.
Inter-POM interactions on the surface of the polymer core
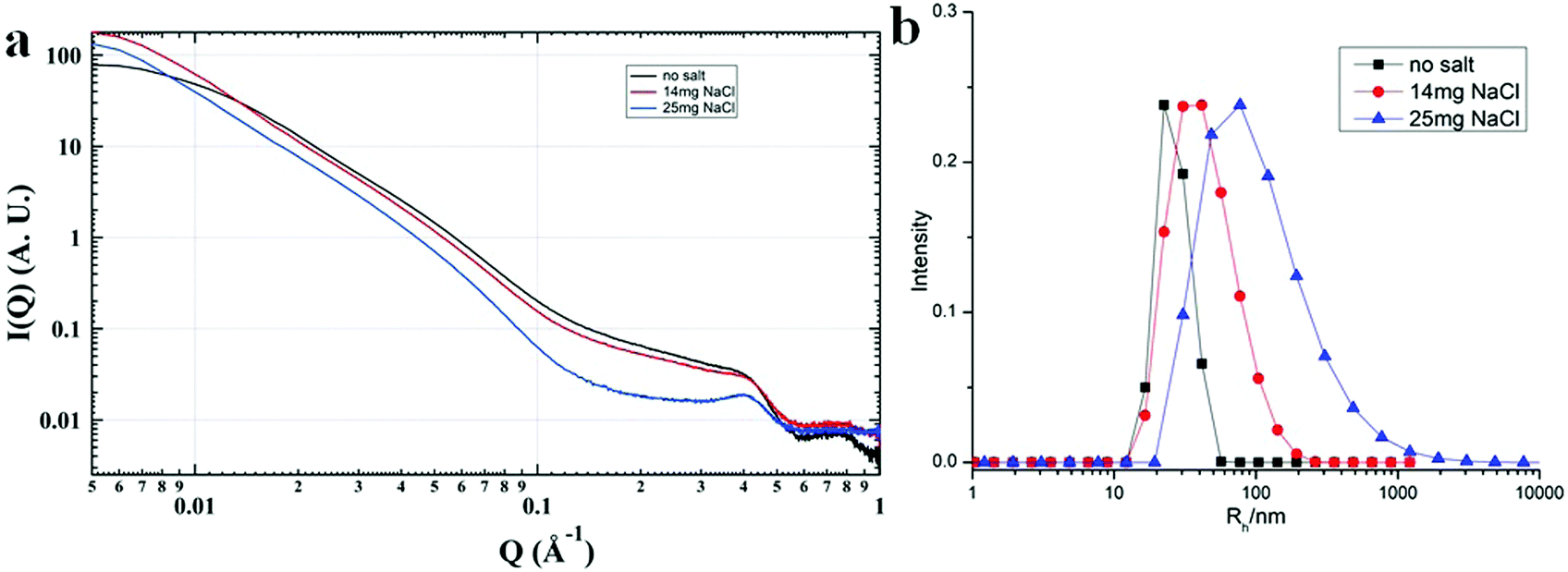
Most of the POMs are highly negatively charged and repulsive with each other in solution. In the aqueous solutions of POM/P4VP complex, POMs strongly interacted with each other with ordered packing on the surface of the polymer core, which is similar to the packing of POMs on the surface of NPs.51 The corresponding structure factors, sharp peaks, were observed in their SAXS data. With the addition of extra salt to the POM/P4VP complex solutions, the interaction peaks became sharper and shifted to high Q direction, suggesting the closer inter-POM distance in their ordered packing structures (Fig. 5a). The introduction of additional salts resulted in the screening of repulsion among these negatively charged POM clusters. Therefore, the inter-POM distance became shorter. Light scattering and TEM studies on these POM/P4VP core–shell structures indicated that the sizes of the complex particles increased with the additional salt (Fig. 5b and Fig. S8 in ESI†). The curvature of the POM/P4VP core–shell complex was defined by the repulsive interaction among POMs in the shell structures. The screening effect weakened the repulsion and decreased the curvature, leading to an increase in particle size.
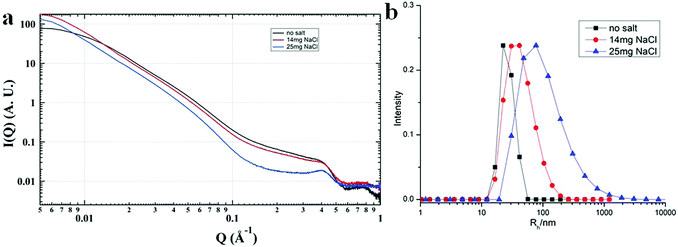 |
| | Fig. 5 (a) SAXS and (b) light scattering studies of the {PW}/P4VP system with the addition of different amounts of extra NaCl. | |
Experimental section
P4VP (Mw = 60 kDa), {Keg}, and ethanol were purchased from Sigma Aldrich and used without further purification. {SW}, {PW}, {Mo72V30}, and {Mo72Fe30} were synthesized and purified according to previous literature.36–39 D. I. water was obtained using a Milli-Q water purification system. Infrared spectra (IR) were recorded as KBr pellets on a Nicolet 510 FT-IR. For TEM measurements, the bright-field images were recorded on a digital CCD camera in a JEOL-1230 microscope with an accelerating voltage of 120 keV. A commercial Brookhaven Instrument LLS spectrometer equipped with a solid-state laser operating at 637 nm was used for measurement of DLS.
The preparation of POM–P4VP complexes
The P4VP stock solution was prepared by dissolving P4VP in ethanol to a concentration of 2 mg mL−1. For the POM solution, the quantity of POM used in the preparation of the aqueous solution was calculated according to the molar ratio of POM ({SW}, 1.4–2.0 mg; {PW}, 1.4–2.0 mg; {Mo72V30}, 1.6–2.0 mg; {Mo72Fe30}, 1.6–2.0 mg; {Keg}, 0.4–2.0 mg) to P4VP. 1 mL of the P4VP stock solution was added drop-wise to 10 mL aqueous solutions of a certain POM sample while the POM solution was kept vigorously stirring. The obtained solution was kept stirring for another 1 h and the solution was left open to air overnight.
SAXS experiments
The complex solution was concentrated to ca. 1 mL using a Macrosep Advance Centrifugal Device (10 K MWCO). The SAXS experiments of these concentrated solutions were performed in a capillary at the 12-ID-B and C beamline at the Advanced Photon Source of the Argonne National Laboratory. The sample to detector distance was about 2 m. A Pilatus detector (Dectris Ltd) was used to acquire images with typical exposure times in the range of 0.1 s.
Conclusions
Nanoscale POM clusters strongly associate and stabilize with P4VP polymers in aqueous solutions and form core–shell structures. The core of P4VP interacted with hydroxyl/water sites on the POM surface via hydrogen bonding, which drives the formation of core–shell NPs. The size of the core–shell was defined by the electrostatic repulsion among POMs in the shell structures. The particle size was tuneable upon the introduction of extra salts because of their screening effect of the repulsive interactions. Our POM/polymer complexation protocol provided a convenient way to prepare POM-based quasi-homogeneous catalysts and bio-compatible materials. The testing of the catalytic efficiency and recyclability of these complex structures is currently underway in our lab.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
P. Y. gratefully acknowledges the support of the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (No. 2016ZT06C322), the Thousand Talents Plan for Young Professionals from Chinese Government, and the startup support from South China University of Technology. T. Liu acknowledges the support from the NSF (CHE1607138) and the University of Akron and the use of the Center for Nanoscale Materials and the synchrotron-based SAXS study in 12-ID-C of the Advanced Photon Source, both are U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This material is based upon work supported by Laboratory Directed Research and Development (LDRD) funding from Argonne National Laboratory, provided by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC02-06CH11357.
Notes and references
- S. A. Jenekhe, Science, 1998, 279, 1903–1907 CrossRef PubMed.
- N. Fujita, T. Yamashita, M. Asai and S. Shinkai, Angew. Chem., Int. Ed. Engl., 2005, 44, 1257–1261 CrossRef PubMed.
- Z. C. Sun, F. Bai, H. M. Wu, S. K. Schmitt, D. M. Boye and H. Y. Fan, J. Am. Chem. Soc., 2009, 131, 13594–13595 CrossRef PubMed.
- S. Sanwaria, A. Horechyy, D. Wolf, C.-Y. Chu, H.-L. Chen, P. Formanek, M. Stamm, R. Srivastava and B. Nandan, Angew. Chem., Int. Ed., 2014, 53, 9090–9093 CrossRef PubMed.
- Y. Zhao, K. Thorkelsson, A. J. Mastroianni, T. Schilling, J. M. Luther, B. J. Rancatore, K. Matsunaga, H. Jinnai, Y. Wu, D. Poulsen, J. M. Frechet, A. P. Alivisatos and T. Xu, Nat. Mater., 2009, 8, 979–985 CrossRef PubMed.
- X. Pang, Y. He, J. Jung and Z. Lin, Science, 2016, 353, 1268–1272 CrossRef PubMed.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef PubMed.
- P. Maury, M. Escalante, D. N. Reinhoudt and J. Huskens, Adv. Mater., 2005, 17, 2718–2723 CrossRef.
- M. Zhang, M. Drechsler and A. H. E. Müller, Chem. Mater., 2004, 16, 537–543 CrossRef.
- A. C. Balazs, T. Emrick and T. P. Russell, Science, 2006, 314, 1107–1110 CrossRef PubMed.
- M. E. Mackay, A. Tuteja, P. M. Duxbury, C. J. Hawker, B. Van Horn, Z. Guan, G. Chen and R. S. Krishnan, Science, 2006, 311, 1740–1743 CrossRef PubMed.
- J. A. Balmer, O. O. Mykhaylyk, S. P. Armes, J. P. A. Fairclough, A. J. Ryan, J. Gummel, M. W. Murray, K. A. Murray and N. S. J. Williams, J. Am. Chem. Soc., 2011, 133, 826–837 CrossRef PubMed.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef.
- D. Sunday, J. Ilavsky and D. L. Green, Macromolecules, 2012, 45, 4007–4011 CrossRef.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973 CrossRef PubMed.
- S. Bhattacharya and S. K. Samanta, Chem. Rev., 2016, 116, 11967–12028 CrossRef PubMed.
- J. B. Carroll, B. L. Frankamp and V. M. Rotello, Chem. Commun., 2002, 1892–1893 RSC.
- T. Li, B. Ye, Z. W. Niu, P. Thompson, S. Seifert, B. Lee and Q. Wang, Chem. Mater., 2009, 21, 1046–1050 CrossRef.
- A. D. Celiz, T.-C. Lee and O. A. Scherman, Adv. Mater., 2009, 21, 3937–3940 CrossRef.
- C. Wang, H. Wu, Z. Chen, M. T. McDowell, Y. Cui and Z. Bao, Nat. Chem., 2013, 5, 1042 CrossRef PubMed.
- J. Kao and T. Xu, J. Am. Chem. Soc., 2015, 137, 6356–6365 CrossRef PubMed.
- M. Kopp, S. Kollenda and M. Epple, Acc. Chem. Res., 2017, 50, 1383–1390 CrossRef PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef PubMed.
- L. Cronin and A. Müller, Chem. Soc. Rev., 2012, 41, 7333–7334 RSC.
- A. Müller and P. Gouzerh, Chem. Soc. Rev., 2012, 41, 7431–7463 RSC.
- P. Yin, D. Li and T. Liu, Chem. Soc. Rev., 2012, 41, 7368–7383 RSC.
- P. Yin, A. Bayaguud, P. Cheng, F. Haso, L. Hu, J. Wang, D. Vezenov, R. E. Winans, J. Hao, T. Li, Y. Wei and T. Liu, Chem. – Eur. J., 2014, 20, 9589–9595 CrossRef PubMed.
- B. Li, W. Li, H. Li and L. Wu, Acc. Chem. Res., 2017, 50, 1391–1399 CrossRef PubMed.
- B. Weifeng, U. Sayaka and M. Noritaka, Angew. Chem., Int. Ed., 2009, 48, 8281–8284 CrossRef PubMed.
- Q. Zhang, Y. Liao and W. Bu, Langmuir, 2013, 29, 10630–10634 CrossRef PubMed.
- S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef PubMed.
- J. M. Clemente-Juan, E. Coronado and A. Gaita-Arino, Chem. Soc. Rev., 2012, 41, 7464–7478 RSC.
- Y.-F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- J. T. Rhule, C. L. Hill, D. A. Judd and R. F. Schinazi, Chem. Rev., 1998, 98, 327–358 CrossRef PubMed.
- Z.-M. Zhang, S. Yao, Y.-G. Li, X.-B. Han, Z.-M. Su, Z.-S. Wang and E.-B. Wang, Chem. – Eur. J., 2012, 18, 9184–9188 CrossRef PubMed.
- M. H. Alizadeh, S. P. Harmalker, Y. Jeannin, J. Martin-Frere and M. T. Pope, J. Am. Chem. Soc., 1985, 107, 2662–2669 CrossRef.
- B. Botar, P. Kogerler and C. L. Hill, Chem. Commun., 2005, 3138–3140 RSC.
- A. Müller, E. Krickemeyer, S. K. Das, P. Kögerler, S. Sarkar, H. Bögge, M. Schmidtmann and S. Sarkar, Angew. Chem., Int. Ed., 2000, 39, 1612–1614 CrossRef.
-
M. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin Heidelberg, Berlin, 1983 Search PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef PubMed.
- T. Li, B. Ye, Z. Niu, P. Thompson, S. Seifert, B. Lee and Q. Wang, Chem. Mater., 2009, 21, 1046–1050 CrossRef.
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann and F. Peters, Angew. Chem., Int. Ed., 1998, 37, 3359–3363 CrossRef.
- N. Suthiwangcharoen, T. Li, L. Wu, H. B. Reno, P. Thompson and Q. Wang, Biomacromolecules, 2014, 15, 948–956 CrossRef PubMed.
- T. Li, L. Wu, N. Suthiwangcharoen, M. A. Bruckman, D. Cash, J. S. Hudson, S. Ghoshroy and Q. Wang, Chem. Commun., 2009, 2869–2871 RSC.
- T. Li, Z. Niu, T. Emrick, T. P. Russell and Q. Wang, Small, 2008, 4, 1624–1629 CrossRef PubMed.
- J. Ruokolainen, J. Tanner, O. Ikkala, G. ten Brinke and E. L. Thomas, Macromolecules, 1998, 31, 3532–3536 CrossRef.
- H. Etxeberria, R. Fernandez, I. Zalakain, I. Mondragon, A. Eceiza and G. Kortaberria, J. Colloid Interface Sci., 2014, 416, 25–29 CrossRef PubMed.
- A. Sidorenko, I. Tokarev, S. Minko and M. Stamm, J. Am. Chem. Soc., 2003, 125, 12211–12216 CrossRef PubMed.
- S. Liu and T. Xu, Macromolecules, 2016, 49, 6075–6083 CrossRef.
- Y. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00291f |
|
| This journal is © the Partner Organisations 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a