
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces†
Verena
Pfeifer
ab,
Travis E.
Jones
*a,
Juan J.
Velasco Vélez
ac,
Rosa
Arrigo
*d,
Simone
Piccinin
e,
Michael
Hävecker
ac,
Axel
Knop-Gericke
a and
Robert
Schlögl
ac
aDepartment of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195, Berlin, Germany. E-mail: trjones@fhi-berlin.mpg.de
bCatalysis for Energy, Group EM-GKAT, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Elektronenspeicherring BESSY II, Albert-Einstein-Str. 15, 12489, Berlin, Germany
cDepartment of Heterogeneous Reactions, Max-Planck-Institut für Chemische Energiekonversion, Stiftstr. 34-36, 45470, Mülheim a. d. Ruhr, Germany
dDiamond Light Source Ltd., Harwell Science & Innovation Campus, Didcot, Oxfordshire OX 11 0DE, UK. E-mail: rosa.arrigo@diamond.ac.uk
eConsiglio Nazionale delle Ricerche – Istituto Officina dei Materiali, c/o SISSA, Via Bonomea 265, Trieste, 34136, Italy
First published on 1st December 2016
Abstract
Water splitting performed in acidic media relies on the exceptional performance of iridium-based materials to catalyze the oxygen evolution reaction (OER). In the present work, we use in situ X-ray photoemission and absorption spectroscopy to resolve the long-standing debate about surface species present in iridium-based catalysts during the OER. We find that the surface of an initially metallic iridium model electrode converts into a mixed-valent, conductive iridium oxide matrix during the OER, which contains OII− and electrophilic OI− species. We observe a positive correlation between the OI− concentration and the evolved oxygen, suggesting that these electrophilic oxygen sites may be involved in catalyzing the OER. We can understand this observation by analogy with photosystem II; their electrophilicity renders the OI− species active in O–O bond formation, i.e. the likely potential- and rate-determining step of the OER. The ability of amorphous iridium oxyhydroxides to easily host such reactive, electrophilic species can explain their superior performance when compared to plain iridium metal or crystalline rutile-type IrO2.
1 Introduction
In addition to needing renewable energy technologies, an economy based on sustainable resources requires efficient energy storage options. Water electrolysis presents an attractive solution to the latter requirement; it stores excess energy from renewable sources in chemical bonds.1,2 To be effective, electrolyzers must be capable of adapting to the varying power inputs of intermittent renewable sources like wind and solar power. While electrolyzers based on proton exchange membranes (PEM) meet this demand, they require a corrosive acidic working environment. Therefore, current research on water electrolysis is driven by the need for efficient, stable, and cost-effective electrocatalysts for the sluggish oxygen evolution reaction (OER) in acidic media.3While iridium oxide is rare and precious, it remains the state-of-the-art OER catalyst in acidic media owing to its high activity, low overpotential, and good stability.4 To uncover the reason for iridium oxide's increased activity, surface scientists have long examined its electronic structure. By combining electrochemical measurements with ex situ X-ray photoemission spectroscopy (XPS), they provided chemical information about oxidation states and elemental abundance present after emersion at various potentials.5,6 The major drawback of such ex situ experiments is that the active state of the electrode cannot be observed under reaction conditions. Furthermore, since the active iridium surface state is likely a hydrated and hydroxylated amorphous form of iridium oxide,5,7,8 the electrode's surface morphology and composition likely change when emersed from the electrolyte and brought into a UHV environment.5,9 Thus, the active sites may be lost or modified, and since Fierro et al.'s10 isotope-labeling experiments showed that the reactive oxygen species contained in the iridium-oxide matrix are directly involved in the catalytic OER cycle, an in situ investigation elucidating the chemical nature of these reactive species is highly desirable.
Significant effort has been invested in the development of in situ methodology to assess the active state of electrode materials under working conditions. Owing to these efforts, it is now possible to drive electrochemical reactions and simultaneously record XPS11–13 and X-ray absorption spectroscopy (XAS)14,15 to monitor the electronic structure of oxygen-evolving surfaces in situ. Nevertheless, the interpretation of XPS and XAS measurements of iridium oxides, especially in situ, remains challenging. While the literature agrees that hydrated and hydroxylated amorphous forms of iridium oxide with mixed iridium oxidation states have intrinsically higher OER activities than pristine iridium metal and crystalline rutile-type IrO2,7,12,15,16 dissent remains about which types of iridium surface species are present during the catalytic OER cycle.12,15
The difficulties in pinpointing iridium oxidation states partly originate from the lack of well-defined oxidic iridium reference materials other than the tetravalent Ir in rutile-type IrO2.17 Drawing parallels with iridium species with different oxidation states present in non-conductive, non-oxidic reference materials is hampered by the fact that these species often have only small18,19 or reverse20 shifts in excitation or binding energy in NEXAFS and XPS and usually overlap to a large extent. Therefore, while in situ XPS investigations were interpreted to show the presence of iridium species with oxidation states of IV and V during the OER,12in situ XAS measurements were deconvoluted into contributions of IrIII and IrV.15
In contrast to the difficult identification of the iridium species present, oxygen species (formally OI− and OII−) contained in highly OER-active X-ray amorphous iridium oxide structures show clear fingerprints in the near-edge X-ray absorption fine structure (NEXAFS) of the O K-edge.8,16,21 These fingerprint features are highly sensitive to changes in the electronic structure of iridium oxides.22 Hence, the identification of oxygen species in iridium oxide structures presents a key to interpret features in the electronic structure of active iridium oxide surfaces and to shed light on iridium oxide's remarkable activity. For the oxygen species contained in the highly active, amorphous iridium oxide catalysts, for example, the formally OI− species has recently been identified as a highly reactive, electrophilic oxygen.22
Such electrophiles are prone to nucleophilic attack. This susceptibility to attack by water or OH likely makes OI− active in O–O bond formation, which is often described as both the potential-determining23 and rate-limiting24 step of the OER. Since we know the electronic structure fingerprints of electrophilic OI− species,16,21 we can test if they are indeed forming in oxygen-evolving iridium surfaces by in situ NEXAFS and XPS.
On this account, we make use of a PEM-based in situ technique11 for investigating gas-phase water electrolysis. This technique enables us to record XPS and NEXAFS while a model iridium electrode evolves oxygen. An advantage of our approach is that, by keeping the oxygen chemical potential in our system low, we are able to investigate the initial stages of oxide formation on iridium surfaces during the OER. To ensure these low oxygen chemical potentials yield relevant results, we first demonstrate at high overpotentials how iridium is oxidized during the OER and identify the nature of the iridium and oxygen species formed during the reaction. In the second step, we perform a controlled experiment near the onset of the OER activity of iridium to identify a correlation between the amount of evolved oxygen and the concentration of oxygen species present on oxygen-evolving iridium surfaces.
2 Experimental
The discrepancies between electrochemical measurements in electrolyte and surface-sensitive spectroscopic investigations in UHV have been bridged in recent years by setups combining both techniques.11–13 The present study uses an in situ cell for investigating gas-phase water electrolysis described in detail by Arrigo et al.11 In this cell concept (see Fig. S1 in ESI†), a Nafion® proton exchange membrane (PEM) is used to separate a continuous flow of liquid water/electrolyte from the evacuated measurement chamber of the near-ambient-pressure XPS (NAP-XPS) endstation of the ISISS beam line25 at the synchrotron facility BESSY II/HZB, Berlin, Germany. The cell concept is described in detail in the ESI.†In brief, water diffuses through the desiccation cracks of the ≈10 nm thick, sputter-deposited electrodes and the PEM due to the pressure difference between the liquid on one side of the membrane and the evacuated measurement chamber on the other (see Fig. S2–S4 in ESI†). The water both hydrates the PEM, ensuring good ion conductivity, and delivers the reactant molecules to the working electrode with the resultant equilibrium pressure reaching 0.1–10 Pa. By connecting the working (Ir) and the counter (Pt) electrodes to an external potentiostat, OER-relevant potentials can be applied. A quadrupole mass spectrometer (QMS) monitors the gas composition on-line to test if oxygen is evolving from the iridium surface.
With this setup, we are able to record XPS and NEXAFS of the X-ray-exposed model iridium working electrode while the OER proceeds. To work under more controlled conditions, in the second part of our study, we equipped the cell with a Ag/AgCl reference electrode (see Fig. S1 in ESI†).
3 Results and discussion
Oxygen production on the iridium working electrode is a requirement for our PEM-based approach to deliver relevant observations. Our on-line QMS confirms this OER-active condition of the electrode (see Fig. 1a). When switching the potential from Eoc (open circuit potential) to 2 V, we observe an increase in the oxygen QMS trace. When further increasing the potential to 2.5 V, we detect an additional increase in the oxygen QMS trace. As expected, higher potentials lead to a higher OER activity. By contrast, if we introduce a PEM without Ir coating into the cell, the oxygen QMS trace does not increase when we apply OER-relevant potentials (see Fig. S7 in ESI†). Hence, the oxygen detected in the case of the Ir-coated sample must be produced at the Ir working electrode.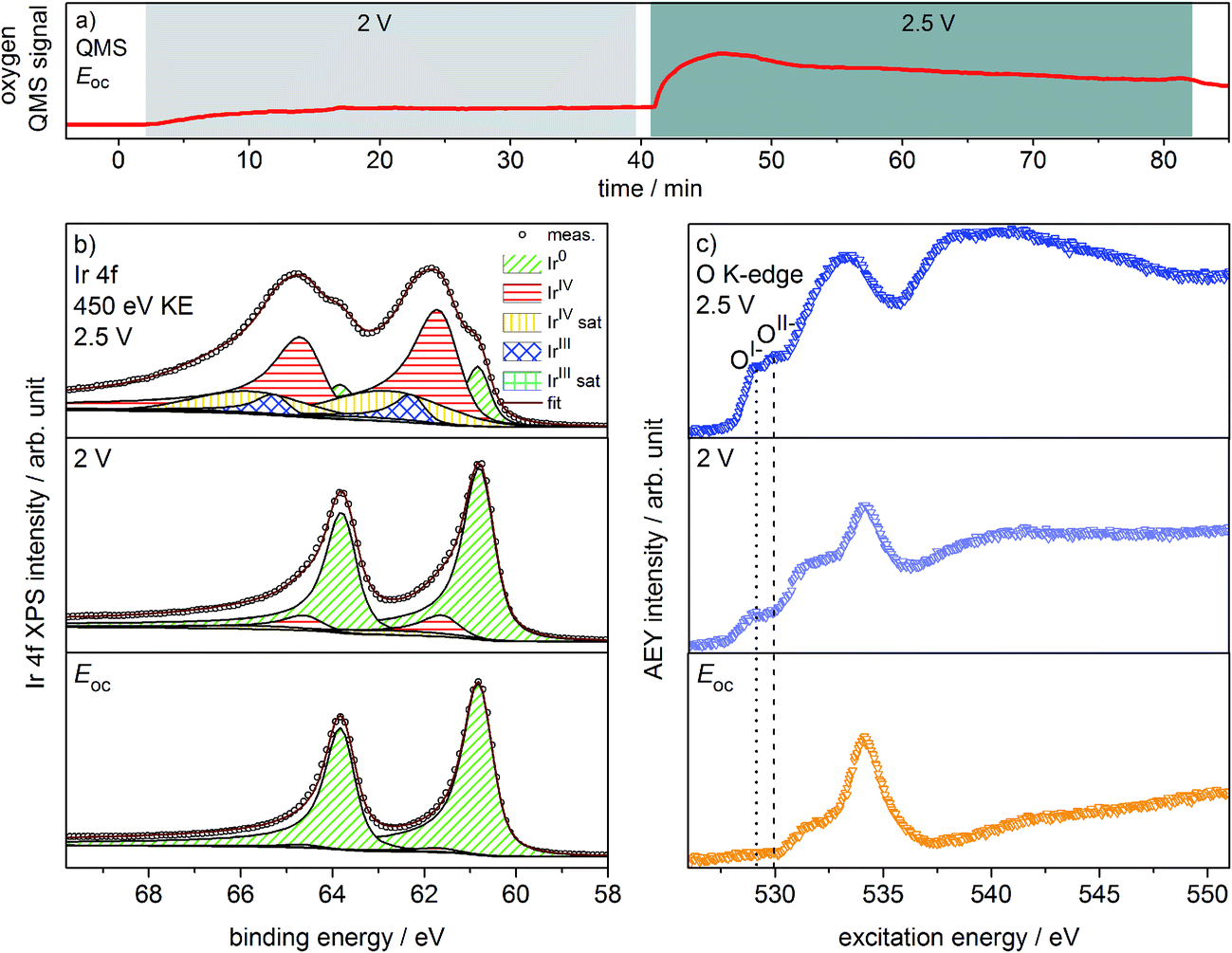 | ||
| Fig. 1 (a) Oxygen QMS trace, (b) Ir 4f spectra and (c) O K-edges of Ir-coated PEM (120 s Ir sputtered) recorded in the two-electrode cell with the indicated potentials applied (p = 5 Pa, H2O). | ||
Ir 4f and O K-edge spectra (probing depths of ≈2 nm and 2–3 nm, respectively) were collected at each of the potentials applied to the Ir working electrode of the two-electrode system to monitor changes in the present iridium and oxygen species (see Fig. 1b and c). A deconvolution of the Ir 4f spectra, i.e. a speciation of the Ir species, can be done employing the fit model for iridium oxides introduced in our previous work.16,21 Good agreement is obtained between the recorded spectra and the fit envelope for all core level spectra (for fit parameters see Table S1 in ESI†).
At Eoc, the Ir 4f spectrum (see Fig. 1b) of a pristine, as-deposited iridium film is dominated by the contribution of metallic Ir at 60.8 eV, which has an asymmetric line shape typical for metallic conductors.26 In addition, a minute oxidic component is present at higher binding energy, likely due to surface oxidation of the sputtered Ir nanoparticles. Increasing the potential to 2 V to start the OER produces only subtle changes in the Ir 4f spectrum. The metallic contribution remains nearly unchanged and the oxidic component grows slightly. Further increasing the potential to 2.5 V leads to pronounced changes, with substantially more intensity emerging at higher binding energies in a broad feature.
The Ir 4f spectrum recorded during the OER at 2.5 V consists of three major contributions. First, there is a residual of the initially metallic Ir at 60.8 eV binding energy. Second, the largest contribution centered at 61.7 eV originates from iridium in the formal oxidation state IV (as in the well-defined reference rutile-type IrO2). This peak has an asymmetric line shape and appears in combination with a satellite at 1 eV higher binding energy.16,21 Finally, there is an additional component at 62.3 eV, i.e. at higher binding energy than IrIV, not found in rutile-type IrO2. The appearance of this higher binding energy iridium oxide feature during OER was also observed by Casalongue et al.12 Based on the shift to higher binding energy, they intuitively assigned this feature to IrV. Nevertheless, in our previous work we combined XPS with theoretical calculations and concluded that IrIII species can exhibit a reverse binding energy shift and that these species are also expected to be found at higher binding energies than IrIV, namely 62.3 eV.16,21 Such IrIII species are present in amorphous, highly OER-active iridium oxyhydroxides. In these materials, the presence of both IrIII and formally OI− has been identified. Hence, an identification of the oxygen species formed during OER may help us shed light on the nature of these additional Ir species present during the OER at 2.5 V.
We can assess the nature of the oxygen species present in the near-surface region of the catalyst by inspecting the O K-edges collected at the different potentials (see Fig. 1c). In the O K-edge spectrum recorded at Eoc, we mainly see contributions of carbonaceous contamination of the surface (532–535 eV) and possibly sulfonic or sulfate groups of the PEM (>537 eV) (see Fig. S8 in ESI†). In accordance with the Ir 4f spectrum collected at Eoc, we observe no clear contribution of iridium oxide species in the corresponding O K-edge. By contrast, at 2 V where we only registered a small contribution of an oxidic component to the Ir 4f spectrum, we see two clearly visible contributions appearing in the low excitation energy region of the O K-edge. The excitation energy values of these two contributions, namely 529 eV and 530 eV, match the main resonances of formally OI− and OII− species identified in the amorphous iridium oxyhydroxide reference material exactly.16,21 When further increasing the potential to 2.5 V, the OI− and OII− contributions to the O K-edge grow in intensity.
While we can imagine O–O groups to be present on the iridium surface as intermediates of the OER, our calculations of the spectroscopic fingerprints of superoxo and peroxo species show that such species cannot account for the observed low excitation energy feature at 529 eV (see Fig. S31 in ESI†). Since the spectral features seen in the iridium surface oxidized during OER coincide with those observed in the amorphous IrIII/IV oxyhydroxide reference, we suggest that these materials are of a similar nature. We tentatively identify the iridium species present on oxygen-evolving iridium surfaces as IrIII and IrIV. Further, we confirm the formation of OI− and OII− species during the OER over iridium. Hence, we have witnessed in situ that, during the OER, electrophilic OI− species form in an amorphous, mixed-valent iridium phase.
The observed formation of OI− species during the OER not only strengthens our previous suggestion that such electrophilic OI− species may be crucial for the OER reactivity16,21,22 but may also explain the long-standing question about the origin of the oxidation signal observed at ≈1.4 V vs. SHE in the CV of iridium (see Fig. 2a). Whereas in the past this signal was often assigned to further oxidation of the metal center, i.e. the transition of IrIV to IrV,5,27,28 we suggest that it reflects the oxidation of oxygen from OII−, contained in the IrOx matrix in form of adsorbed OH groups, to OI−:
| IrOxOII−H ⇌ IrOxOI− + H+ + e− | (1) |
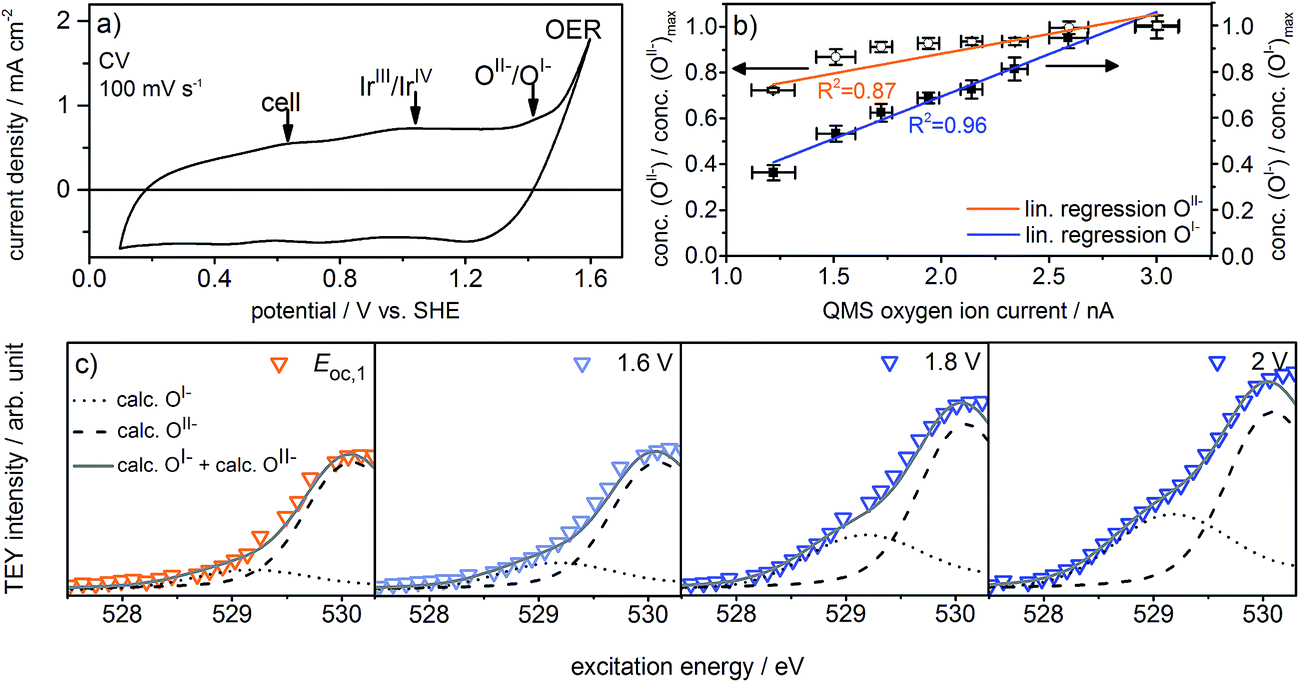 | ||
| Fig. 2 (a) Cyclic voltammogram, (b) normalized OI− and OII− concentrations over QMS oxygen ion current, and (c) zoomed and fitted low excitation energy regions of O K-edges recorded in the three-electrode cell (indicated potentials vs. SHE, ring current = 60 mA, p = 0.3 Pa, 0.1 M H2SO4). | ||
To test this assertion, we used density functional theory (see ESI†) to compute the potential at which OI− forms from surface OH groups, the latter of which give resonances at 532 eV in the O K-edge (see Fig. S28 in ESI†). The computed value of 1.2–1.3 V supports the hypothesis that OI− formation accounts for the ≈1.4 V oxidation signal observed experimentally. While such a redox-active, non-innocent ligand29,30 may at first glance contrast with common intuition, the diffuse nature of the Ir 5d orbitals and the metal's high oxidation state (IV) in IrO2 suggest it is energetically favorable for the oxide to accommodate holes in the O 2p states rather than further oxidize IrIV to IrV.31
With these experiments, we have illustrated the OER-active surface state of an iridium electrode likely consists of an IrIII/IV matrix with oxygen species in the formal oxidation states I− and II−. A comparison of our observations from the recorded Ir 4f spectra and O K-edges highlights that although we only observe subtle changes in the Ir 4f spectrum at 2 V during the OER, the appearance of iridium oxygen compounds is clearly mirrored by the presence of OI− and OII− species in the O K-edge. Hence, the O K-edge is more sensitive to changes in the electronic structure of iridium oxides than the Ir 4f core line. Therefore, we use the O K-edge as basis for the subsequent investigation to determine which species are present near the onset of the OER and how they correlate with activity.
What remains to be clarified is how far the presence of the observed oxygen species is related to iridium's OER activity. For this aim, the reaction needs to be driven under exact potential control at moderate overpotentials. To conduct such experiments near the onset of iridium's OER activity, we upgraded the cell into a three-electrode system by adding a Ag/AgCl reference electrode (Fig. S1 in ESI†), which ensures that the potential applied to the working electrode has exactly the desired value.
Fig. 2a shows the redesigned device works under relevant conditions. The CV of a sputter-deposited Ir electrode displays the characteristic oxidation waves at 1 V vs. SHE (commonly attributed to an oxidation of IrIII to IrIV27,28) and 1.4 V vs. SHE (oxidation of oxygen from OII− to OI−, see the preceding discussion) and the OER onset at 1.5 V vs. SHE. The additional feature visible at 0.6 V vs. SHE originates from the cell itself (see Fig. S6 in ESI†). After a series of 35 CVs to precondition the iridium electrode surface, the potential applied to the working electrode was stepwise increased by 0.05 V starting from 1.6 V vs. SHE. Fig. S18 in ESI† shows the expected concomitant increase of both the current density and the oxygen concentration in the gas phase. A plot of oxygen concentration over current density shows their linear relationship (see Fig. S18 in ESI†) excluding that, within the measured potential region, the ratio between currents due to simple corrosion and oxygen production changes. Furthermore, an estimation of the amount of present Ir atoms (≈1016) and the overall number of electrons passed across the electrode (≈1019) excludes a major contribution to the measured current by simple Ir dissolution/corrosion. By these potential steps, we have created iridium oxide surfaces that evolve different amounts of oxygen. To see whether this increase in oxygen production also shows a correlation with the concentration of the present oxygen species, we recorded the O K-edge at each applied potential (see Fig. S19 in ESI†).
Fig. 2c shows the low excitation energy region of the O K-edge at successively applied potentials. We deconvoluted these regions using the calculated spectra for the OI− and OII− species16,21 to quantify their respective contributions to the overall signal. We used the resulting fits to determine the dependence of the OI− and OII− concentrations on the current density and oxygen QMS ion current (see Fig. 2b, S16 and S21 in ESI†). While we observe a linear correlation between the oxygen evolution activity and the OI− concentration (R2 = 0.96), we see only a weak correlation with the OII− concentration (R2 = 0.87). Hence, the concentration of OI− seems to be tied to the magnitude of the OER reactivity of iridium catalysts whereas the concentration of OII− does not.
If, as we hypothesize, the OI− species in these experiments are stabilized by the applied potential, their concentration should decrease once the potential is reduced below 1.4 V vs. SHE. Aiming to relate the presence of these reactive species to the presence of an applied, OER-relevant potential, we alternatively turned on and off the potential using another fresh sample (see Fig. S22–S24 in ESI†). In the recorded and fitted spectra, we clearly observe that turning on and off the potential also reversibly turns on and off the major contribution to the intensity of the OI− species (see Fig. 3a). The difference of the spectra recorded during and after OER (labeled during-after in Fig. 3b) yields close agreement with the calculated OI− spectrum.
 | ||
| Fig. 3 (a) Low excitation energy region of O K-edges of Ir-coated PEM (60 s Ir sputtered), consecutively recorded (bottom to top) in the three-electrode cell with the indicated potentials vs. SHE applied and (b) difference spectrum of two consecutively recorded O K-edges and comparison with calculated OI− spectrum (ring current = 13 mA, p = 0.45 Pa, 0.1 M H2SO4). | ||
The disappearance of the OI− species once the potential is switched off is likely due to their protonation and shift to higher excitation energies (≈532 eV, see Fig. S28 in ESI†). Unfortunately, owing to the high background signal in this excitation energy region, we are not able to verify such an increase in intensity. Nevertheless, such a protonation and subsequent increase in concentration of hydroxyl groups is in agreement with Reier et al.'s observation that post reaction, highly active OER catalysts have high concentrations of OH-groups at the surface.8 The small residual of OI− still present in our experiments after the potential is turned off is likely due to subsurface sites that are not accessible for protonation. As the thickness of the amorphous oxide layer increases, we would then expect a concomitant increase in the amount of residual OI−, in agreement with its ex situ presence in highly active amorphous iridium oxyhydroxide powders.16,21 However, the fraction of residual OI− in our thin oxide films is minute when compared to the large OI− signal in such bulk amorphous powder catalysts with signal contributions from deeper, possibly inaccessible subsurface OI− species. Hence, while in the present experiment the majority of OI− is likely protonated and forms OH-groups once the potential is switched off, many of the OI− hosted by the bulk oxyhydroxides seem to be unprotonated.
If we assume that the increase in OI− concentration with increasing oxygen evolution activity is not a mere side reaction, we can combine our in situ observations to address the original hypothesis that the electrophilic OI− species participates in O–O bond formation during the OER on iridium. This idea stems from a comparison to photosystem II (PS II). Although in this heavily studied biological system for oxygen generation from water the O–O bond formation process has not yet been finally resolved, theory and experiment strongly agree on the involvement of an electron-deficient oxygen species, either electrophilic oxygen or an oxyl radical, in O–O bond formation.32–37 Also for other water oxidation catalyst systems the presence of such electron-deficient intermediates has been reported based on vibrational measurements.38–41 While, so far, we have not obtained direct experimental evidence for the formation of radicals in the iridium oxide matrix, our previous study22 demonstrated the OI− species, shown in the present study to also form in oxygen-evolving iridium surfaces, to be strong electrophiles. In a mechanism proposed for PS II, which involves such electrophilic oxygen species, an oxygen contained in the Mn water oxidation complex (WOC) transforms into an electrophile (O*) that is subsequently attacked by nucleophilic (bound) water or hydroxides to form the O–O bond.35–37 In a simplified form, this part of the OER process in PS II can be written as:
| WOC–O* + H2O → WOC–OI−–OI−–H + H+ + e− → WOC + O2 + 2H+ + 3e− | (2) |
Under the assumption that a similar mechanism with a ligand-centered oxidation prior to O–O bond formation applies to the OER over iridium oxides, we may write for the reaction between water and the electrophilic OI− species observed on iridium oxides during the OER:
| IrOxOI− + H2O → IrOxOI−–OI−–H + H+ + e− → IrOx + O2 + 2H+ + 3e− | (3) |
The electrophilic nature of the OI− makes this nucleophilic attack of water possible, whereas the additional charge on OII− makes that species less susceptible to such an attack. After the evolution of oxygen, the catalytic cycle can be closed by regenerating IrOxOI− from IrOx and water under the influence of the applied potential. And while we assumed a nucleophilic mechanism in reaction (3) due to suggestions from theory23 and ultra-fast infra-red measurements42 that an OOH intermediate is formed during OER on iridium, we could have formulated the reaction by assuming the OI− observed in this work has significant radical character. The crucial point is that electron-deficient oxygen is required in either mechanism. Thus, the high activity observed for amorphous iridium oxyhydroxide powder catalysts is then apparent from their ability to form large amounts of highly reactive, electron-deficient OI− species.16,21,22 In contrast to these highly active IrOx powders, rutile-type IrO2 does not tend to form a high concentration of OI− and is significantly less active in the OER.16,21 Thus, a catalyst's propensity to accommodate electrophilic OI− species appears to be essential in its ability to catalyze O–O bond formation.
4 Conclusion
In conclusion, using in situ X-ray photoemission and absorption spectroscopies, we have demonstrated for the first time that reactive electrophilic OI− oxygen species form in a mixed-valent iridiumIII/IV matrix during the OER. The observed formation of these OI− species implies that iridium oxide contains redox-active, non-innocent ligands accounting for the redox chemistry of the material. We further found the OI− concentration to increase with the measured oxygen evolution activity and to virtually disappear from our thin oxyhydroxide films in the absence of an applied potential. Both observations agree with our hypotheses that electrophilic OI− species are active in catalyzing the OER on iridium oxides and that enhanced OER performance of iridium oxyhydroxides can be achieved by the accommodation of large amounts of reactive oxygen species serving as precursor sites for the O–O bond formation. In the quest for less expensive materials to catalyze the OER, these findings can guide the way for the design of new high-performance catalysts comprising such reactive oxygen species.Acknowledgements
The authors acknowledge BESSY II/HZB for granting beam time under the proposal #15202526 and Höchst-Leistungs-Rechenzentrum Stuttgart (HLRS) for computational facilities. The authors gratefully acknowledge Eugen Stotz for helping in the construction design of the in situ cells as well as Dr Manfred E. Schuster and Gisela Weinberg for TEM and SEM measurements of the samples. The authors thank Dr Detre Teschner, Dr Cyriac Massué, and Michael Scherzer for fruitful discussions. T. E. J. acknowledges the Alexander-von-Humboldt foundation for financial support. This work was further supported by the Ministry of Education and Science of the Russian Federation (RFMEFI61614X0007) and the Bundesministerium für Bildung und Forschung (05K14EWA) through the joint Russian-German research project “SYnchrotron and NEutron STudies for Energy Storage” (SYNESTESia).References
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS PubMed.
- J. P. Barton and D. G. Infield, IEEE Trans. Energy Convers., 2004, 19, 441–448 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- R. Kötz, H. Neff and S. Stucki, J. Electrochem. Soc., 1984, 131, 72–77 CrossRef.
- R. Kötz and H. Neff, Surf. Sci., 1985, 160, 517–530 CrossRef.
- T. Reier, D. Teschner, T. Lunkenbein, A. Bergmann, S. Selve, R. Kraehnert, R. Schlögl and P. Strasser, J. Electrochem. Soc., 2014, 161, F876–F882 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- Y. H. Hall and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 135–152 RSC.
- S. Fierro, T. Nagel, H. Baltruschat and C. Comninellis, Electrochem. Commun., 2007, 9, 1969–1974 CrossRef CAS.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11660–11664 CrossRef CAS PubMed.
- H. G. Sanchez Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef CAS PubMed.
- J. J. Velasco Vélez, V. Pfeifer, M. Hävecker, R. S. Weatherup, R. Arrigo, C.-H. Chuang, E. Stotz, G. Weinberg, M. Salmeron, R. Schlögl and A. Knop-Gericke, Angew. Chem., Int. Ed., 2015, 54, 14554–14558 CrossRef PubMed.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondinini, Chem. Sci., 2014, 5, 3591–3597 RSC.
- A. Minguzzi, C. Locatelli, O. Lugaresi, E. Achilli, G. Cappelletti, M. Scavini, M. Coduri, P. Masala, B. Sacchi, A. Vertova, P. Ghigna and S. Rondinini, ACS Catal., 2015, 5, 5104–5115 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, M. T. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- A. Holleman and E. Wiberg, Holleman-Wiberg. Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, New York, 1995 Search PubMed.
- J. P. Clancy, N. Chen, C. Y. Kim, W. F. Chen, K. W. Plumb, B. C. Jeon, T. W. Noh and Y.-J. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195131 CrossRef.
- H.-S. Oh, H. N. Nong, D. Teschner, T. Reier, A. Bergmann, M. Gliech, J. Ferreira de Araújo, E. Willinger, R. Schlögl and P. Strasser, J. Am. Chem. Soc., 2016, 138, 12552–12563 CrossRef CAS PubMed.
- B. Folkesson, Acta Chem. Scand., 1973, 27, 287–302 CrossRef CAS.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner, J. Allan, M. Hashagen, G. Weinberg, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. Velasco Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2016, 7, 6791–6795 RSC.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- S. Piccinin, A. Sartorel, G. Aquilanti, A. Goldoni, M. Bonchio and S. Fabris, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4917–4922 CrossRef CAS PubMed.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Academic Press, 2009, vol. 52, pp. 213–272 Search PubMed.
- S. Doniach and M. Šunjić, J. Phys. C: Solid State Phys., 1970, 3, 285 CrossRef CAS.
- M. Hüppauff and B. Lengeler, J. Electrochem. Soc., 1993, 140, 598–602 CrossRef.
- M. A. Petit and V. Plichon, J. Electroanal. Chem., 1998, 444, 247–252 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed.
- J. Zaanen, G. A. Sawatzky and J. W. Allen, Phys. Rev. Lett., 1985, 55, 418–421 CrossRef CAS PubMed.
- N. Cox, M. Retegan, F. Neese, D. A. Pantazis, A. Boussac and W. Lubitz, Science, 2014, 345, 804–808 CrossRef CAS PubMed.
- P. E. Siegbahn, Biochim. Biophys. Acta, 2013, 1827, 1003–1019 CrossRef CAS PubMed.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 CAS.
- J. Messinger, Phys. Chem. Chem. Phys., 2004, 6, 4764–4771 RSC.
- Y. Gao, T. Åkermark, J. Liu, L. Sun and B. Åkermark, J. Am. Chem. Soc., 2009, 131, 8726–8727 CrossRef CAS PubMed.
- J. Limburg, V. A. Szalai and G. W. Brudvig, J. Chem. Soc., Dalton Trans., 1999, 1353–1362 RSC.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362–367 CrossRef CAS PubMed.
- O. Zandi and T. W. Hamann, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed.
- M. Herlihy, M. M. Waegele, X. Chen, C. D. Pemmaraju, D. Prendergast and T. Cuk, Nat. Chem., 2016, 8, 549–555 CrossRef PubMed.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- N. Sivasankar, W. W. Weare and H. Frei, J. Am. Chem. Soc., 2011, 133, 12976–12979 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc04622c |
| This journal is © The Royal Society of Chemistry 2017 |