
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hollow polymer particles: a review
Ros Azlinawati Ramli
 *
*
Material Technology Program, Faculty of Industrial Sciences & Technology, Universiti Malaysia Pahang (UMP), Lebuhraya Tun Razak, 26300 Gambang, Kuantan, Pahang, Malaysia. E-mail: azlinawati@ump.edu.my; Fax: +06-095492766; Tel: +06-0172949907
First published on 14th November 2017
Abstract
Herein, the basic principles, such as the definitions, classifications, and properties, of hollow polymer particles (HPPs) are critically investigated. HPP preparation methods, such as template, self-assembly, and acid/alkali swelling methods, have been briefly introduced. Moreover, advanced methods to prepare HPPs have been highlighted. In addition, recent studies and applications of HPPs have been briefly described. HPPs are polymeric spheres with size in the range from nanometer to micron and a single pore inside the particles. This review is focused on the classification of HPPs based on the core–shell, single particles, and stimuli-responsiveness. A single pore structure is the most desirable characteristic that allows this class of polymers to encapsulate large guest molecules and thus to be used as a drug delivery system.
 Ros Azlinawati Ramli | Ros Azlinawati Ramli received her BSc. (Hons) from Universiti Kebangsaan Malaysia (UKM) in 2004, and obtained her MSc. (2012) and Ph.D (2014) in Polymer Engineering from Universiti Teknologi Malaysia (UTM). She was awarded her PhD scholarship from the Ministry of Higher Education Malaysia (MOHE) and Best Student Doctor of Philosophy (Polymer Engineering) from UTM. Previously, she was a Senior Chemist for Synthomer R&D Asia. Currently, she is a senior lecturer at Material Technology Program, Universiti Malaysia Pahang (UMP). Her research interests include emulsion polymers, polymer composites, microgels for biomedical applications, and hydrogels for agricultural applications. |
1. Introduction
Hollow polymer particles (HPPs) are polymeric spheres with size in the range from nanometer to micron (1–1000 nm) and a single pore inside the particle; on the other hand, polymers with many pores are called porous polymers.1,2 Interest in HPPs has increased steadily over the past few decades because HPPs can readily be used to upload drugs and active species and can efficiently sequester specific target molecules into the hollow interiors by chemical modification.3 Furthermore, they have a variety of applications, such as they are used as coatings,8–13 drug delivery systems,14–17 catalysts,18–20 and photonic crystals,21–23 in chemistry, biotechnology, and materials science.4–7 Core–shell HPPs are the most widely studied polymer particles in this class of polymers. The combination of inorganic–organic materials holds great promise for novel core–shell hybrid micro- and nanostructured HPPs.24,25HPPs are usually prepared in a spherical form using different methods to obtain the hollow part. HPPs can be synthesized in a liquid or solid form in the range of tens of nanometers to several millimeters.26,27 A variety of monomers, including methyl methacrylate (MMA), butyl acrylate (BA), butyl methacrylate (BMA), and vinyl acetate (VAc), have been used to synthesize HPPs. Moreover, one of the earliest processes for making hollow polymer particles was developed by Kowalski and co-workers in 1984 at Rohm and Haas company using an acid/alkali swelling method.28,29 The common methods that are used to form hollow structures of HPPs are template,1,30–32 self-assembly,33,34 and acid/alkali swelling,35 with different synthesis techniques such as suspension, dispersion, emulsion, and precipitation polymerization. Advanced methods for the preparation of hollow polymers that have been reported recently include urea treatment,36 an in situ reduction method,37,38 inverse mini-emulsion periphery RAFT polymerization (IMEPP),39 and coaxial electrohydrodynamic and atomization (CEHDA).40 Different types of materials, such as monomers,41 block copolymers,42 copolymers,43 and inorganic nanoparticles,44 have been used to synthesize HPPs. Advanced techniques, such as scanning electron microscopy, transmission electron microscopy, atomic force microscopy, and dynamic light scattering have been used to study the morphology system of HPPs.
2. Classifications
Hollow polymer particles (HPPs) can be classified depending on their morphology into core–shell and single-particles; in addition, depending on their state phenomenon, they can be classified into stimuli-responsive, dispersion, and non-dispersion. Moreover, core–shell HPPs can be classified into solid shell and porous shell, as shown in Fig. 1. This section of this study emphasizes on core–shell, single-particle, and stimuli-responsive HPPs. | ||
| Fig. 1 Classification of hollow polymer particles (HPPs) depending on different parameters. | ||
2.1 Core–shell HPPs
Core–shell HPPs can be divided into solid shell and porous shell HPPs. The hollow part can be the core or in the shell of the particles. Hollow spheres with complex core–shell structures have attracted significant interest due to their superior properties such as tunable diameter, cavity size, large surface area, low density, and precise functionality.41,45 Efforts have been made to fabricate core–shell and hollow particles in the past 10–15 years.46 The simplest method to fabricate core–shell HPPs is to coat a sacrificial core particle with a shell, followed by removal of the sacrificial core.21,47,48 The core can be removed by dissolution, thermolysis, or chemical etching to create a hollow interior.1,49Yuan and co-workers prepared HPPs using seeded emulsion polymerization and alkali swelling methods. MMA–MAA–DVB was prepared as core particles, and styrene, acrylonitrile, and divinylbenzene (DVB) were then synthesized onto the cores, followed by alkalization treatment to obtain hollow particles. They prepared hollow particles at different shell thicknesses with different weight ratios of the core monomers to shell monomers. The shell thickness increased with a decrease in the core/shell ratio. When the core/shell ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, the hollow emulsion particles obtained were monodispersed and maintained a more preferable balance between the rigidity of the shell polymers and the hollow percentages.52 Tan et al. developed novel core–double-shell particles with poly(methyl methacrylate-co-butyl acrylate) P(MMA-co-BA) as the core, poly(methyl methacrylate-co-butyl acrylate-co-methacrylic acid) P(MMA-co-BA-co-MAA) as the inner shells, and poly(styrene-co-methyl methacrylate) P(St-co-MMA) as the outer shells via a soap-free emulsion polymerization and acid–alkali osmotic swelling process.55 Mandal et al. prepared HPPs by surface-confined living radical polymerization on silica templates. Using a silica microsphere as a sacrificial core, hollow poly(benzyl methacrylate) (PBzMA) microspheres were produced by core dissolution. Shell thicknesses were measured from the scanning electron microscopy (SEM) images, which are provided in Table 1. The data reveal that the shell thickness increases with the increasing polymerization time.56
:8, the hollow emulsion particles obtained were monodispersed and maintained a more preferable balance between the rigidity of the shell polymers and the hollow percentages.52 Tan et al. developed novel core–double-shell particles with poly(methyl methacrylate-co-butyl acrylate) P(MMA-co-BA) as the core, poly(methyl methacrylate-co-butyl acrylate-co-methacrylic acid) P(MMA-co-BA-co-MAA) as the inner shells, and poly(styrene-co-methyl methacrylate) P(St-co-MMA) as the outer shells via a soap-free emulsion polymerization and acid–alkali osmotic swelling process.55 Mandal et al. prepared HPPs by surface-confined living radical polymerization on silica templates. Using a silica microsphere as a sacrificial core, hollow poly(benzyl methacrylate) (PBzMA) microspheres were produced by core dissolution. Shell thicknesses were measured from the scanning electron microscopy (SEM) images, which are provided in Table 1. The data reveal that the shell thickness increases with the increasing polymerization time.56
| Polymerization time (h) | Shell thickness (nm) | Molecular weight (Mn) | PDI (Mw/Mn) |
|---|---|---|---|
| 3.5 | 175–225 | 9150 | 1.56 |
| 6.5 | 350–400 | 13450 |
1.37 |
| 14.0 | 550–600 | 26500 |
1.26 |
Xu and Asher prepared 150–700 nm HPP particles with 100–500 nm hollow cores via dispersion polymerization and HF etching. Monodisperse HPPs with outside diameters between 200 and 700 nm and shell thicknesses between 20 and 90 nm were synthesized by varying the silica core size, while maintaining the monomer/silica weight ratio at 0.9 and keeping all the other conditions constant.22 Minami et al. prepared HPPs with a single hole in the shell by the self-assembling of phase-separated polymer (SaPSeP) method. The formation of the hole in the shell is due to the emulsifier adsorbed at the interface of the droplet that affects the self-assembly of PDVB molecules at the interface. Fig. 2 shows the SEM images of PS/PDVB particles in the absence (a and c) and presence (b and d) of sodium dodecyl sulfate (SDS) and transmission electron microscopy (TEM) (c and d) images of their ultrathin cross-sections. A single hole was observed in the shell of the hollow polymer particles prepared in the presence of SDS (Fig. 2b).57
 | ||
| Fig. 2 SEM images (a) and (b) of PS/PDVB particles and TEM images (c) and (d) of ultrathin cross-sections of PS/PDVB particles prepared by seeded polymerizations in the absence (a) and (c) and presence (b) and (d) of SDS. Reproduced from ref. 57 with permission from the American Chemical Society. | ||
Guan et al. prepared HPPs through direct polymerization and cross-linking reactions occurring on the surface of carboxyl-capping polystyrene (PS) colloids. The formation mechanism of holes that is based on the concomitant microphase separation and symmetrical volume shrinkage of shell materials provides a reproducible approach for creating holes in polymer shells. To obtain a better understanding of the formation mechanism of the hole, they obtained the reaction products at different polymerization stages. Core–shell microspheres with a single hole covered with a layer of an ultrathin film were observed after 12 h, and core–shell microspheres with open holes were observed after 20 h.3 Fig. 3 shows the schematic for the formation process of holes in the shells of PS involving: (1) the formation of a prepolymer layer, (2) the microphase separation of two polymer shell layers, (3) the development of a small interior void in the shell, (4) formation of a film-covered hole, and (5) formation of an open hole in the polymer shells.
 | ||
| Fig. 3 Schematic for the formation process of holes in the shells of polystyrene (PS). Reproduced from ref. 3 with permission from John Wiley and Sons. | ||
He et al. prepared HPPs with a porous shell by self-assembling of sulfonated polystyrene (PS) emulsion particles at the monomer droplets interface. The swelling of the PS emulsion particles by the oil phase provided a driving force to develop the hollow core. The increase in the polymerization rate led to a thicker shell formation. After polymerization, the self-disengagement of the adsorbed PS particles from the polymeric shell resulted in the porous shell. Fig. 4 shows the SEM (A) and TEM (B) images of PVAc. The TEM images show that the microspheres have hollow core/porous-shell structures, and the pores are distributed compactly and evenly in the shell, which can be seen from the contrast in the bright and dark fields. The porous shell was very thin, with an approximately 35–172 nm thickness for all the system.73
 | ||
| Fig. 4 SEM (A) and TEM (B) images of the hollow core/porous shell of PVAc. Reproduced from ref. 73 with permission from John Wiley and Sons. | ||
Chen et al. prepared poly(acrylic acid) (PAA) nanogels with a hollow core-porous shell structure by the direct polymerization of an acrylic acid monomer in the presence of hydroxypropyl cellulose (HPC) and a cross-linking agent, N,N-methylenebisacrylamide (MBAAm), followed by removal of HPC at a medium pH (pH > 6.0) from the generated HPC-PAA nanoparticles.64 Guan et al. prepared HPPs with a mesoporous shell by Pickering emulsion polymerization as a potential colloidal collector for organic contaminants in water. Isopentyl acetate (PEA) was included to generate a hydrophobic core that could be easily removed during the drying process, resulting in hollow particles. The hollow microspheres were reinforced with silica nanoparticles; this resulted in an amphiphilic surface and nanotunnels in the shell.70 Han et al. developed HPPs with a single Aurum (Au) nanoparticle encapsulated in the porous shell with superior catalytic efficiency and recyclability. At first, a Au-poly(o-methoxyaniline) (POMA) core–shell hybrid was synthesized, followed by swelling treatment in a suitable swelling solvent. Finally, evaporation of the swollen Au–POMA core–shell hybrids resulted in a hollow nanostructure formation.65
2.2 Single-particle HPPs
All the aforementioned HPPs have a closed-shell structure; this means that the encapsulated materials need to diffuse through the shell to reach the interior, which is a slow and ineffective process. Accordingly, single-particle HPPs with one hole on the surface were developed to easily and quickly load a variety of functional materials (including molecular species, proteins, and nanoparticles) into the interiors. The size of the hollow spheres and holes can be controlled by the temperature, amount of solvent, and the initiator to monomer ratio. The special characteristic of single-particle HPPs is that the hole can be closed via a thermal or solvent-induced annealing process after loading the materials.74,75 This class of HPPs exhibits promising applications in drug encapsulation and controlled release systems.Jeong et al. prepared hollow particles with engineered holes on their surfaces (microscale fish bowls) using two different routes. The first route required the use of pre-synthesized latex beads and was mainly limited to a few amorphous polymers such as PS and PMMA. The second route involved the use of an emulsion derived from a polymer solution. This route was more generic and could be directly applied to both amorphous and semicrystalline polymers such as polycaprolactone (PCL) and poly (L-lactide) (PLLA).74 Fig. 5 shows the SEM images of microscale fish bowls prepared from both routes.
 | ||
| Fig. 5 (A) SEM images of microscale fish bowls derived from PS bead swelling by styrene and (B) fish bowls prepared from PMMA emulsions containing different amounts of toluene. Reproduced from ref. 74 with permission from the American Chemical Society. | ||
Im et al. prepared another class of macroporous capsule-polymer shells with controllable holes on their surfaces. The existence of holes (224 nm in radius) on the surfaces of the PS hollow particles allowed for direct loading of the functional materials. After loading, the holes could be closed by annealing the system at a temperature slightly above the glass-transition temperature (Tg) of PS. Fig. 6 shows an SEM image demonstrating the possibility to load the bowl-shaped PS particles with solid PS beads of 260 nm diameter.75
 | ||
| Fig. 6 Demonstration of the possibility to load bowl-shaped PS particles with solid PS beads of 260 nm diameter. Reproduced from ref. 75 with permission from the Nature Publishing Group. | ||
Han et al. prepared HPPs of poly(o-toluidine) (POT) in an aqueous solution by a droplet template without surfactant stabilization. By simply changing the concentration of o-toluidine, the size of the resulting hollow spheres ranged from nanometers to micrometers. Temperature was found to have a significant effect on the size of the hollow spheres. The size of the hole on the surface of each hollow microsphere could be controlled by changing the molar ratio of the initiator to monomer. The droplets formed by the monomer in an aqueous solution were proposed to act as a template during the polymerization process. Fig. 7 shows the TEM images of the POT synthesized at different initiator to monomer ratios (a and b) and at different temperatures (c and d). The results show that the size of the hole on the surface of each hollow microsphere increased with the decreasing [APS]/[o-toluidine] ratio. Temperature is another important factor that influences the sizes of the POT hollow microspheres. When the polymerization reaction temperature was maintained above 15 °C, irregular nanoparticles as well as the hollow microspheres were observed. If the temperature is between 0 and 10 °C, the size of the hollow spheres slightly changes, whereas the size decreases significantly at lower temperatures (<0 °C).76
 | ||
| Fig. 7 SEM images of poly(o-toluidine) (POT) hollow microspheres synthesized at different [APS]/[o-toluidine] ratios of: (a) 0.8:1 and (b) 0.6:1. (c) and (d) SEM images of POT hollow microspheres synthesized at different temperatures: (c) 25 °C and (d) −10 °C. The insets show the corresponding TEMs image of the POT hollow structures. Reproduced from ref. 76 with permission from John Wiley and Sons. | ||
2.3 Stimuli-responsive HPPs
Stimuli-responsive HPPs are smart materials that are capable of responding to environmental stimuli such as temperature, pH, light, and magnetic field, which allows for the controlled release and site-specific targeting of the loaded drugs modulated by environmental stimuli.77,78 The best technique to prepare stimuli-responsive HPPs is polymerization from templates. This technique allows the employment of a variety of functional monomers, which contributes to better stimuli-responsive performance than other techniques.79 The functionality of stimuli-responsive HPPs can be increased by introducing more than one shell layer and the addition of inorganic/magnetic particles, such as silica, aurum, and ferric oxide, to the particles. The magnetic-targeting stimuli-responsive HPPs show unique performance as intelligent carriers for the site-specific controlled delivery of drugs or genes. The most used monomer to prepare stimuli-responsive HPPs is N-isopropylacrylamide (NIPAM), which exhibits a volume-phase transition temperature (VPTT) close to the temperature of the human body.80 Upon a slight change of temperature, poly(N-isopropylacrylamide) (PNIPAM) chains undergo macromolecular transition from a hydrophilic to a hydrophobic structure.81Li et al. prepared silica-polymer double-shell HPPs with independent pH and temperature sensitivity via a sol–gel process. The poly(methacrylic acid) (PMAA) inner shell and the PNIPAM outer shell could respond independently to pH and temperature of the environment, respectively. Monodispersed double-walled hollow PMAA-PNIPAM microspheres were obtained by selective removal of the inorganic silica core and interlayer by HF treatment for 48 h.82 Du et al. designed double-shell HPPs with independent pH-responsive cross-linked PMAA inner shells, temperature-responsive cross-linked PNIPAM outer shells, and movable superparamagnetic cores from Fe3O4/SiO2/PMAA/SiO2/PNIPAM penta-layer onion-shaped core–shell microspheres based on polymerization on template approaches.79 Pan et al. synthesized magnetic/hollow double-shell-imprinted polymers (MH–MIPs) by Pickering emulsion polymerization. In this preparation process, ATP particles were used as stabilizers for oil-in-water Pickering emulsions, and a few hydrophilic Fe3O4 nanoparticles in the water phase were used as magnetic separation carriers. Then, the interfacial imprinted shells of double-shelled MIPs were fabricated in the oil phase, and ATP particles were attached to the surfaces of the interfacial imprinted shells to form permeable outer shells.83 Macková et al. designed hollow particles with a triple functionality by inverse emulsion polymerization. These hybrid particles have potential in magnetic and biomedical applications for temperature-driven preparative separations of biological media.84 Dubbert et al. prepared hollow double-shell microgels consisting of two thermoresponsive polymer shells made of PNIPAM and poly(N-isopropylmethacrylamide) (PNIPMAM). The hollow particles were derived from silica–PNIPAM–PNIPMAM core–shell–shell (CSS) particles by dissolution of the silica core. In the first synthesis step, the PNIPAM shell was attached to a functionalized silica core; this resulted in particles with a single shell. In the second step, a shell made of PNIPMAM was added. This led to the formation of doubly temperature-sensitive CSS particles, as shown in Fig. 8.51
 | ||
| Fig. 8 Schematic of the synthesis process for doubly temperature-responsive core–double-shell microgels and hollow spheres. Reproduced from ref. 51 with permission from John Wiley and Sons. | ||
3. Preparation methods of HPPs
Hollow polymer particles (HPPs) are typically prepared by template, self-assembly, and acid/alkali swelling using different synthesis techniques such as suspension, dispersion, emulsion, and precipitation polymerization. One of the earliest processes for making hollow emulsion particles was developed by Rohm and Haas. Their process involved emulsion polymerization and alkali swelling methods.56 Among the various approaches reported, template-directed synthesis is the most widely used method for the formation of polymer hollow particles.21,85,86 However, solid-shell HPPs are mostly prepared by acid/alkali swelling method.13,35,52–54 Table 2 lists the advantages and disadvantages of methods used for the preparation of hollow polymer particles (HPPs).| Method | Advantages | Disadvantages |
|---|---|---|
| (A) Template method | ||
| Hard-templates | Void size and shape of the particles are determined by the size and shape of the template | The templates are removed completely without any contribution to the composition of the final products |
| Hard-templating method in principle can be used to prepare any kind of HPPs and therefore is considered as the most popular method | In some cases, toxic etchants or solvents are required to remove the template core, which seriously limits its application | |
| Easy to control size, morphology, and surface functionality of the HPPs | ||
| Soft-templates | In most soft-templating approaches, only the coating process is needed, because it is unnecessary to remove the soft templates | Coating process usually requires an additional surface modification procedure, which could potentially cause low reproducibility and high cost |
| Self-templates | Self-templating strategy is preferred in practical applications because of the significantly reduced production cost, simplified synthesis procedures, and the ease of scaling up | Self-templating is only suitable for the synthesis of hollow structures with specific compositions, limiting its application field |
| Practical application of the soft-templating method has been mainly limited by the size and uniformity because it produces materials mainly in the micrometer range with high polydispersity | ||
| (B) Self-assembly | Produces HPPs in nanometer scale with high spherical particles | Shell cross-linking must be conducted at very low concentration of micelles solution; this restricts the self-assembly method for industrial application |
| Electrostatic interaction as a driving force is only suitable for layer-by-layer (LbL) assembly of polymer electrolytes | ||
| Micelle formation of block copolymer involves UV cross-linking of the shell-layer and subsequent tedious ozonolysis of the core | ||
| Compositions and structures of the block copolymers are very important for the preparations of polymer vesicles as seen in the limited successful examples | ||
| (C) Acid/alkali swelling | Formation of hollow particles with a definite morphology (particles containing one central hollow) | Complicated process controlled by a number of thermodynamic and kinetic factors |
3.1 Template method
The template method is known to be one of the most effective approaches to achieve hollow nanostructures.87 The method can be divided into four categories: (1) conventional hard-templating synthesis, (2) sacrificial-templating synthesis, (3) soft-templating synthesis, and (4) template-free methods. The most commonly employed hard templates are monodisperse silica particles and polymer latex colloids. The sacrificial template is the template itself, which is involved as a reactant in the synthetic process of the shell material (or its intermediate). The most commonly used soft (liquid or gaseous) templates are emulsion droplets, surfactants and other supramolecular micelles, polymer aggregates/vesicles, and gas bubbles.88 The template-free method/self-templated method is based on novel mechanisms, such as Ostwald ripening (a spontaneous process, first described by Wilhelm Ostwald in 1896).89 Templates are selected, and their diameters can be controlled easily to predetermine the sizes of HPPs;90 to ensure that the hollow particles retain integrity after the template synthesis, a cross-linking strategy is employed.91 Polymer nanoparticles, such as emulsion and polymeric micelles, have been increasingly used to fabricate HPPs because it is easy to control the size, morphology, and surface functionality of these nanoparticles. Emulsion particles are employed to fabricate hollow particles with larger diameters in the range from the submicron to micrometer, whereas polymeric micelles are used for smaller hollow particles with diameters less than 100 nm.92,93MAH/VAc (maleic anhydride/vinyl acetate) (PMV) particles have been used as hard templates for the synthesis of maleic anhydride/divinylbenzene (MAH/DVB) hollow polymer particles. The approaches consist of three major steps, as shown in Fig. 9: the synthesis of template particles based on MAH/VAc; the formation of core/shell particles via precipitation polymerization using MAH/DVB as comonomers and PMV as templates; and the removal of the core by dissolving it with an organic solvent, i.e., n-butyl acetate. The shell thickness of the hollow spheres and the volume of their hollow core could be controlled simply by varying the size of the template, the monomer concentration, and the ratio of the monomer to template.94
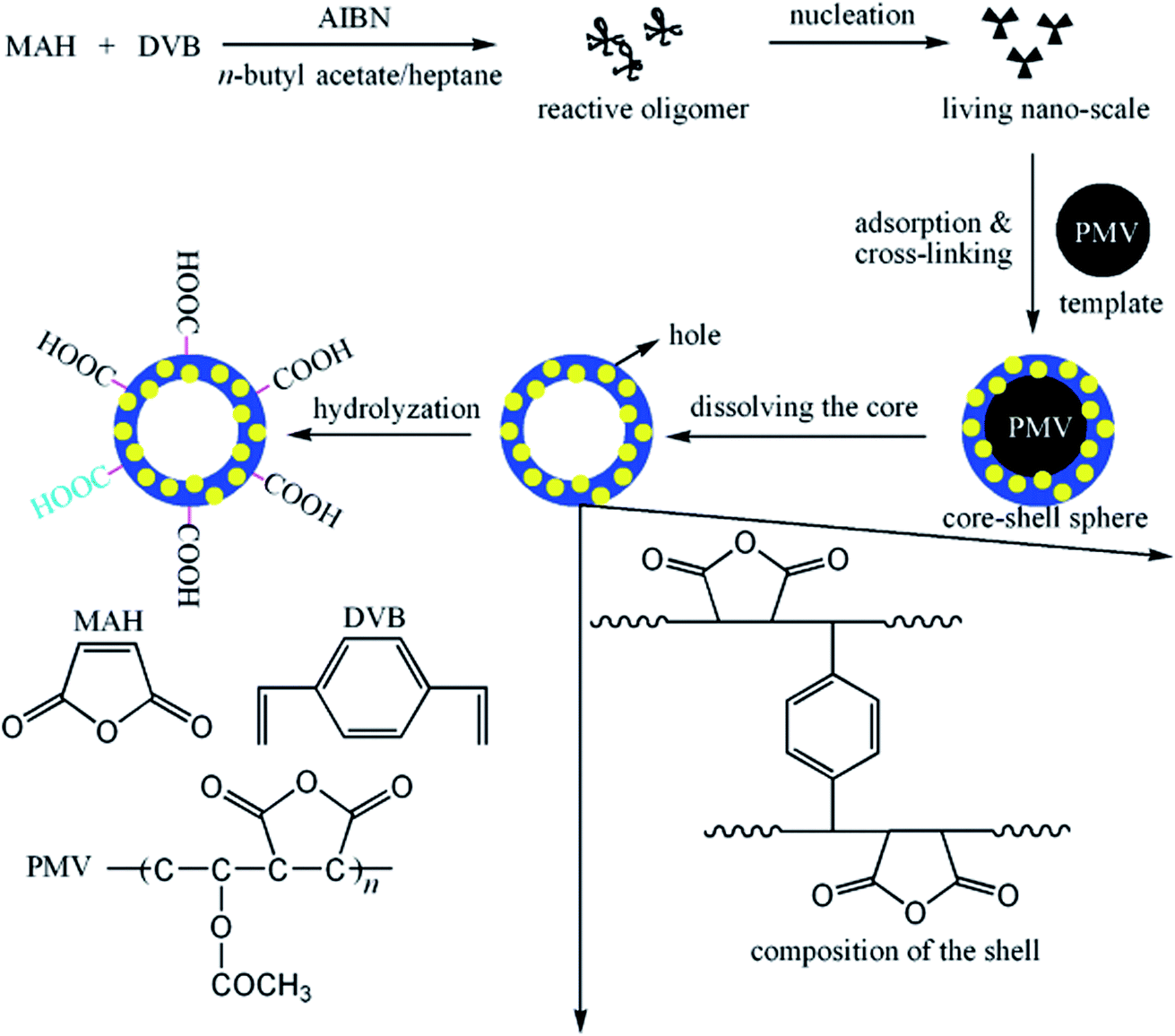 | ||
| Fig. 9 Schematic of the strategy employed in the preparation of hollow spheres. Reproduced from ref. 94 with permission from the American Chemical Society. | ||
In another study reported by Li et al., poly(divinylbenzene-co-acrylic acid) (P(DVB-co-AA)) microspheres were used as templates to synthesize hollow microspheres with movable P(DVB-co-AA) cores. Tri-layered-structure microspheres were prepared by three-stage distillation precipitation polymerization. The hollow P(DVB-co-AA) microspheres with a 48 nm shell thickness were formed by selective dissolution of the noncross-linked PAA segment.95 In another study, polypyrrole-chitosan (PPy-CS) hollow nanospheres with movable Ag nanoparticles (Ag@PPy-CS) cores were prepared using a photoreduction method. This method does not require the preparation of polymer layers as compared to the template-assisted approach. In this method, hollow nanospheres were prepared first before the introduction of the core material.4
Boyer et al. prepared hollow polymer nanocapsules of styrene (St) and maleic anhydride (MAH), (St-alt-MAH), exploiting gold nanoparticles (GNP) as sacrificial templates for polymers obtained by RAFT polymerization, as illustrated in Fig. 10. Block polymers (made by RAFT polymerization) were assembled onto the GNP surfaces using a grafting process. The polymers were then cross-linked using a small diamine molecule. Finally, the gold cores were removed using aqua regia; this resulted in the formation of a stable, biocompatible, low cytotoxic, and antifouling hollow polymer.96
 | ||
| Fig. 10 Synthesis of nanocapsules using gold nanoparticles as sacrificial templates: (1) nanoparticle assembly; (2) cross-linking; and (3) GNP core removal. Reproduced from ref. 96 with permission from John Wiley and Sons. | ||
McKelvey and co-workers reported a catanionic equilibrium vesicles templating method for producing hollow polymeric spheres of styrene and divinylbenzene. The catanionic equilibrium vesicles were formed by cetyltrimethylammonium tosylate (CTAT) and sodium dodecylbenzenesulfonate (SDBS) or cetyltrimethylammonium bromide (CTAB) and sodium octyl sulfate (SOS). The hydrophobic monomers were added to swell the bilayers of a vesicle solution and subsequently polymerized. The goal was to capture or template the vesicle structure to form hollow polymer spheres. The particles had an average radius of ca. 60 nm and a membrane shell less than 10 nm thick.97 In another study, a surfactant bilayer of dimethyldioctadecylammonium chloride (DODAC) was used as a vesicle template to allow the preparation of poly(butyl methacrylate) (PBMA) hollow spheres with dimensions ranging from several nanometers up to several hundred micrometers. After extraction of the surfactant matrix, the polymer particles contracted considerably; thus, this led to the formation of hollow spheres.98 Wang et al. prepared hollow ferrocenyl coordination polymer microspheres with micropores in the shells by Ostwald ripening. The formation of hollow microspheres with microporous shells resulted from the mass transfer and crystallization of small crystallites from the Ostwald ripening process. The detailed formation mechanism of hollow microspheres is depicted in Fig. 11 involving: (a) dissolution of the small particles; (b) diffusion and redeposition of the dissolved species; and (c) dissolution and outmigration of the inner crystallites.66
 | ||
| Fig. 11 Proposed formation process of hollow iron-based ferrocenyl coordination polymer microspheres (Fe–Fc–HCPS) by the Ostwald ripening mechanism, together with the corresponding TEM images. Reproduced from ref. 66 with permission from the American Chemical Society. | ||
Gao et al. prepared polymer nanocapsules embedded with noble metal nanoparticles by a template-free method, as shown in Fig. 12. Compared with other methods for fabricating nanocapsules, this method is performed in an aqueous solution without any organic solvent or template core. By this technique, it is not necessary to prepare any uniform spherical templates, regular amphiphilic polymers, complex dendrimers, or hyperbranched polymers, and repeated absorption and washing processes are also not required.99
 | ||
| Fig. 12 Proposed schematic for the formation of nanocapsules. Reproduced from ref. 99 with permission from the Royal Society of Chemistry. | ||
3.2 Self-assembly
The self-assembly approach involves a careful selection of the experimental parameters, such as the copolymer composition, block length, ionic content, solvent, and polymer concentration, to control the morphology.100 HPPs have been prepared via the self-assembly of amphiphilic block copolymers, rod–coil block copolymers, polymer pairs, and polymer–peptide conjugates.48 Owing to their amphiphilic nature and molecular geometry, lipid molecules can aggregate in a dilute aqueous solution into spherically closed bilayer structures, the so-called vesicles or liposomes. It is quite reasonable that the hollow sphere morphology of these aggregates should render them suitable as precursors for the preparation of more stable nanocapsules. This can be realized using different concepts.6 Amphiphilic diblock copolymers possess similar properties as surfactants. They can undergo a self-assembly process to obtain diverse morphologies.46 The self-assembly method produces HPPs in the nanometer scale with high spherical particles. This method involves shell cross-linking reactions and then removal of their core to form stable HPPs. The shell cross-linking must be conducted at a very low concentration of micelle solution to prevent the polymer micelles from aggregating; this restricts the use of the self-assembly method in industrial applications.101 Polymer aggregates exhibit a higher stability and durability as compared to small-molecule aggregates due to their mechanical and physical properties. Therefore, polymer self-assembly has attracted significant attention because of its potential applications in many fields, such as biomedicine, biomaterials, and microelectronics, and in the synthesis of photoelectric materials, catalysts, etc.100Jang and Ha reported the preparation of P(MMA)/cross-linked PS core/shell nanospheres via emulsion polymerization. The self-assembly of the triblock copolymers poly(oxyethylene)-poly(oxypropylene)-poly(oxyethylene) was used to form hollow polystyrene nanospheres. PS hollow nanospheres were obtained by a polymer-core etching technique using methylene chloride. Fig. 13 shows the schematic of polystyrene hollow nanosphere fabrication.33
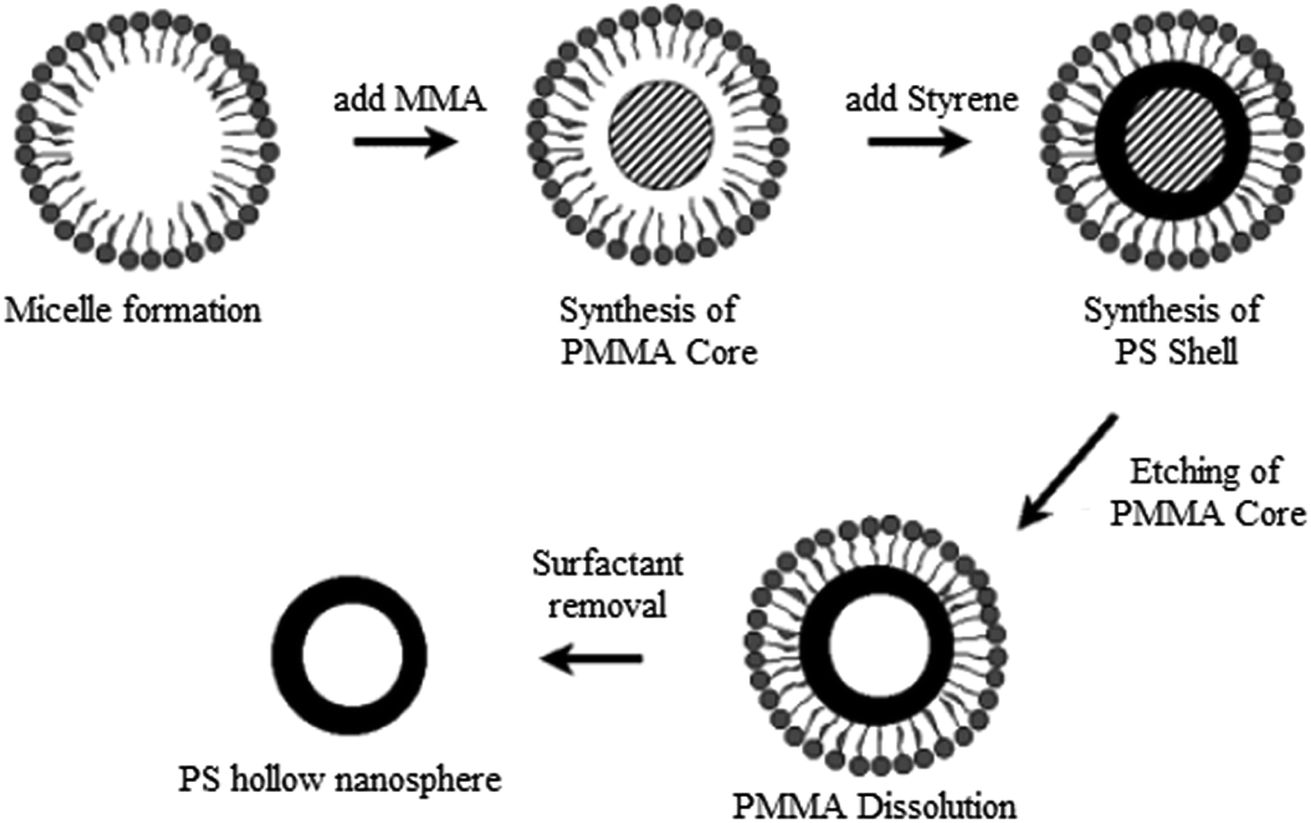 | ||
| Fig. 13 Schematic for polystyrene hollow nanosphere fabrication. Reproduced from ref. 33 with permission from the American Chemical Society. | ||
In another study reported by He and co-workers, cage-like polymer microspheres with hollow core/porous-shell structures were prepared by the self-assembly of PS particles at the emulsion droplet interface. The swelling of the PS particles by the oil phase provides a driving force to develop hollow cores. The PS particles also serve as a porogen that will disengage automatically during polymerization, leaving behind a uniform porous shell.73,102 In another contribution, He and co-workers prepared a polystyrene/melamine–formaldehyde (PS/MF) hollow polymer based on a similar approach. The reserving time of emulsion after the addition of the MF prepolymer will affect the morphology of the polymer layer. It was found that many pores appeared on the particle surface with the shortening of the reserving time and finally formed a porous structure.71
3.3 Acid/alkali swelling
Acid/alkali swelling/osmotic swelling is the earliest method and dominant for obtaining submicron-sized HPPs. This method was invented by Kowalski and colleagues and commercialized by Rohm & Haas Company.103 The typical concept is to prepare a core/shell polymer with a soft carboxyl core and rigid shell, followed by neutralizing the carboxyl group to generate a polyelectrolyte inside and then expanding the core by an osmotic swelling mechanism.35 This method was subsequently improved by Vanderhoff and co-workers104 and other researchers.105,106 The advantage of these methods comprises the formation of hollow particles with a definite morphology (particles containing one central hollow) as compared to other techniques that provide particles containing several hollows separated by polymer walls.13 A large number of emulsion polymerization techniques are available to produce hollow particles by acid/alkali swelling. The unsaturated acid monomer plays an important role in the formation of the hollow structure.49 At first, seed latex with carboxylic acid groups is prepared; then, a hard permeable polymer shell is formed on the outer surfaces of the carboxylic core polymer particles; after this, at around the glass-transition temperature (Tg) of the shell, particles containing water and polyelectrolyte are prepared by neutralizing the carboxylic acid groups of the core polymer particles using a volatile alkali.101Yuan and co-workers prepared monodispersed HPPs with a core/shell monomer ratio of 1:8 and 1% DVB cross-linker in the shell. At first, MMA–BA–MAA copolymer emulsions were prepared as seeds; after this, the seed emulsion polymerization of MMA–MAA–DVB was carried out to prepare carboxylated core particles. Then, the hydrophobic shell of St-ACN-DVB was synthesized onto the core. Finally, the HPP particles were obtained by alkalization treatment of the core–shell latex particles using NaOH.52 Based on a similar approach, Zhang and co-workers prepared HPPs with an MAA/BA/MMA core and BA/St shell, followed by a stepwise alkalization treatment with ammonia (NH4OH). The results showed that the optimum content of the cross-linker diallyl maleate (DAM) in the shell was 0.5–1.0 wt%. Monodispersed HPPs were obtained at a core/shell monomer ratio of 1:7.53 This concept was utilized by Lee and co-workers to produce P(NIPAM-co-MAA)/Fe3O4 thermosensitive magnetic HPPs.54,107
3.4 Advanced techniques to prepare hollow polymer particles
Various physical and chemical methods have been reported for the preparation of HPPs. Preparation of HPPs by emulsion polymerization via the encapsulation of a hydrocarbon non-solvent is a complicated process controlled by a number of thermodynamic and kinetic factors.108 Electrostatic interaction, as a driving force, is only suitable for the layer-by-layer (LbL) assembly of polymer electrolytes. Micelle formation of a block copolymer involved UV cross-linking of the shell layer and a subsequent tedious ozonolysis of the core.109 However, although the abovementioned pioneering works are very interesting, the preparation processes seem to be time-consuming; for example, multistep processes are needed for the synthesis of core–shell composite particles, and then, removal of the core particles by selective dissolution or by calcination is required. Therefore, development of facile and feasible methods to prepare HPPs continues to be a great challenge to material scientists. The fabrication of monodisperse HPPs with size in the range from the micrometers to nanometers and a tailored structural shape and surface properties remains a great challenge in material science.110 Accordingly, in this review, advanced techniques such as electrohydrodynamic atomization (CEHDA), inverse mini-emulsion periphery RAFT polymerization (IMEPP) process, urea treatment, and a four-step approach for the preparation of HPPs have been reported.Chang et al. prepared a hollow microsphere with a single hole in its shell by a CEHDA technique. Fig. 14 shows the experimental setup of the CEHDA technique consisting of a pair of concentric needles. The inner stainless steel needles with diameters in the range between 300 and 150 μm were supplied with liquid perfluorohexane (PFH), and outer needles with diameters in the range between 1100 and 685 μm were supplied with a polymethylsilsesquioxane (PMSQ) solution. The mean diameter of the hollow microsphere could be varied in the range from 310 to 1000 nm by adjusting the flow rate, polymer concentration, or applied voltage. The shell thickness could be controlled by the polymer concentration. Compared with other methods, this strategy has two advantages. First, it enables the preparation of microspheres with a single surface hole at ambient temperature and pressure. Second, this green process does not require the use of surfactants or other additives and does not entail a template removal step.40,111
 | ||
| Fig. 14 Experimental setup for the electrohydrodynamic atomization (CEHDA) technique, consisting of a pair of concentric needles. Reproduced from ref. 111 with permission from the American Chemical Society. | ||
Utama et al. synthesized HPPs with a hydrophilic liquid core using a one-pot approach via a novel IMEPP process. An inverse mini-emulsion was obtained by dissolving the stabilizer, monomer, cross-linker, and initiator in the organic phase and the lipophobe in the aqueous phase. The combined mixture was then ultrasonicated, followed by polymerization in sealed glass ampoules under vacuum at 60 °C. The size and stability of the droplets (prior to polymerization) were monitored using varying amounts of dispersed phase, stabilizer, lipophobe, and the ultrasonication period.112 Further developments by Utama et al. showed that the shell thickness could be conveniently tuned by adjusting the stoichiometry and monomer conversion.39 Wang et al. prepared porous and hollow polymer particles by urea treatment. In this study, core/shell particles were synthesized by multistep seed polymerization using methyl methacrylate (MMA), acrylic acid (AA), divinylbenzene (DVB), and styrene (St), and then, a certain amount of ethanol and urea were added to the particles. The advantage of this approach as compared to that of the conventional methods is the creation of polymers with mesoporous shells. Fig. 15 shows the morphology of the particles (A) without urea treatment, and the particle has a smooth surface, and (B) after urea treatment, the particle displays a rough surface. The porous shells were created by carbon dioxide produced by the decomposition of urea and was released out from the particles.36
 | ||
| Fig. 15 TEM images of the multistage polymer particle (A) without and (B) with urea treatment. Reproduced from ref. 36 with permission from Springer. | ||
Shi et al. reported a novel route to prepare HPPs decorated with gold nanoparticles (AuNPs) in the shell. The approach needed no additional reducing agent in the formation process of the AuNPs. As shown in Fig. 16, the method mainly involves four steps: (1) pretreatment of the surface of silica particles with polyvinylpyrrolidone (PVP), and PVP-treated silica particles are labeled as SiO2/PVP; (2) seeded polymerization of ethylene glycol dimethacrylate (EGDMA) and methacrylic acid (MAA) on the SiO2/PVP particles, and the resultant particles are labeled as SiO2/PVP/PEM, where PEM represents the poly(EGDMA-co-MAA) copolymer; (3) selective etching of SiO2 from SiO2/PVP/PEM particles using an aqueous HF solution to generate PVP/PEM hollow particles; and (4) AuNP formation in the shell of the PVP/PEM hollow particles to yield PVP/PEM-Au hybrid hollow particles.38
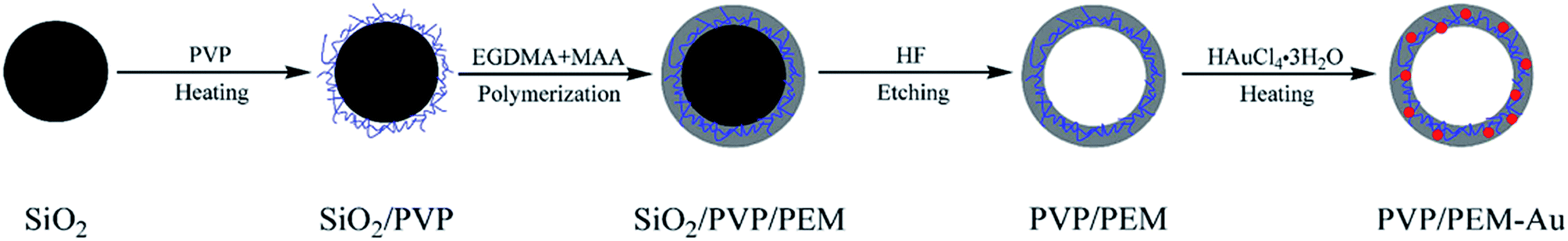 | ||
| Fig. 16 Schematic for the synthesis process of PVP/PEM-Au hybrid hollow particles. Reproduced from ref. 38 with permission from Springer. | ||
4. Properties
The features, such as encapsulation property, surface functionality, low density, high specific area, good flow ability, optical properties, surface permeability, and opacity,35,101,113,114 of HPPs have been actively investigated due to their possible role in technological applications in various areas. The functionalization of HPPs could be achieved by encapsulating guest species into the hollow core, which endowed them with diverse properties.115 Consequently, incorporation of functional groups (e.g., –COOH) comprising biological units by surface modification could specifically deliver the loaded guest molecules to target areas and ideally suited for in vivo applications.116 Shi et al. developed a charged hollow core/shell lipid–polymer–lipid hybrid nanostructure constructed with three functional features. An outer PEG–lipid layer was incorporated into the system and could enable the particles to escape immunological recognition, and improve the particle stability during circulation and slow polymer degradation and drug release. The middle poly(lactic-co-glycolic acid) (PLGA) forms a barrier through the sustained release of encapsulated small interfering RNA (siRNA) and additionally serves to carry water-insoluble drugs. Moreover, the inner positively charged hollow core, composed of cationic lipids, could encapsulate siRNA much more efficiently than PLGA alone.117Nanoparticles, such as tin, gold, Fe3O4, or silica NPs, could be incorporated into the interior and exterior of HPPs.4 The inclusion of stimuli-responsive properties into these microstructures has allowed their unique application in biomaterials, such as in anticancer drug delivery systems, because of their increased functionality in comparison with that of conventional inert structures.118 Kim et al. prepared monodispersed HPPs embedded with magnetic Fe3O4 nanoparticles. The resultant magnetic hollow polymer showed excellent colloidal stability. The interior void size could be controlled by adjusting the particle size of the core polymer, and the shell thickness could be adjusted by varying the weight ratio of the iron oxide nanoparticles to the styrene monomer. These magnetic polystyrene microspheres were superparamagnetic and highly magnetized as much as 4.69 emu g−1 microsphere.119 Shi et al. prepared HPPs decorated with gold nanoparticles in the shell. By introducing the PVP polymer into the shell of the hollow particles, AuNPs were easily formed within the polymer shell just in the presence of a gold precursor under heating. This method will open up the possibility for the generation of a series of HPPS that can incorporate various noble metal nanoparticles (Au, Ag, Pt, etc.) into the shell.38 Lee et al. synthesized P(NIPAM-MAA)/Fe3O4 thermosensitive magnetic HPPs. They found that an increase in the amount of Fe3O4 particles would increase the saturation magnetic flux density of the magnetic HPPs.54
Opacity or the hiding power is an important optical property of white paint coating and results from the multiple scattering of incident light due to interactions with pigment particles.120 HPPs with excellent optical properties, such as opacity and whiteness, are utilized as an opacifying agent or a white pigment for industrial coatings. The needed structure and properties of this type of HPPs can be obtained by changing the composition of the shell, the size of particles, and the holes or kinds of internal substances.53 Huang et al. prepared novel HPPs with both opaque and self-film forming properties. The HPPs of four-layered polymers consisted of a high carboxyl-containing soft core, a transition layer, a rigid supporting layer, and an outermost film-forming layer. The optimized recipe with a BA/St mass ratio of 2/1, DVB content of 1 wt%, and core/film forming layer mass ratio of 1/6 was utilized in the film forming layer preparation, and the self-film forming hollow latex with the best opaque property was obtained. Accordingly, this kind of hollow polymers might have prospective applications in various fields such as in waterborne coatings, inks, paints, and biomedicine. Borthakur et al. prepared a series of core–shell particles containing a copolymer of P(n-BA-co-MAA-co-EGDMA) as the core and a copolymer of P(St-co-MMA) as the shell. In their studies, newly synthesized core–shell particles were used as a pigment in the emulsion paint along with titanium dioxide (TiO2). Particles with a St:MMA ratio of 60:40 by percentage showed the best result in these studies. It was found that the optical properties of the paint did not change with an up to 17% reduction of TiO2 in the pigment volume. The gloss, rock hardness, and washability of the emulsion paint also remained unchanged.9
Furthermore, creation of a single hole in polymer shells offers a large number of binding sites generated in the proximity of the interior surface. The target species can easily diffuse into the interior sites of hollow spheres through the open holes; this leads to a higher capacity and faster kinetics for the uptake of target species. Hollow core–porous-shells have promising application as high-loading drug carriers. This is due to the large cavity inside the HPPs and the numerous mesopores present in the shell.64 A permeable shell can allow small molecules to penetrate the core for effective catalytic reactions and drug-loading capacity.64,65 Several interesting materials have been reported such as inorganic silica, in which both macropores and micro- or mesopores are incorporated into the structure.71 Guan et al. reported HPPs with a mesoporous shell containing small silica particles on the surface. The surface roughness of the hollow particles has technical importance in controlling the surface-associated adsorption process. The resultant hollow particle had a hydrophobic cavity and an amphiphilic surface, thus serving as a good colloidal collector (high loading) for hydrophobic contaminants in water. The particles were also found to be good particle emulsifiers probably because of the amphiphilicity of the silicate and polymeric moieties on the surface.70
5. Recent study on hollow polymer particles
Hollow polymer particles (HPPs) have attracted wide research interest from fundamental science to prospective applications. The unique properties of HPPs have attracted attention from scientists, who are re-investigating the older systems to develop a new system using advanced techniques and methods. To simplify the complex processes of fabrication of HPPs, a one-step method was proposed by Yang et al.121 Other facile methods to prepare HPPs have been developed by a number of researchers recently.122–124 In this section, the recent studies have been reviewed.Nakamura et al. prepared poly(ionic liquid) (PIL) particles with a single-hollow structure by suspension polymerization from monomer droplets. The obtained PIL hollow particle shells had switchable properties, which could be changed from hydrophobic to hydrophilic by anion exchange using a LiBr/ethanol solution. These properties are useful for preserving encapsulated materials in the hollow part by changing the hydrophobicity of the shell particle.125 Qin et al. designed and synthesized a novel type of ammonium-functionalized HPPs (HPP–NH3+). Due to the presence of dual functional groups (ammonium and carboxyl groups), HPP–NH3+ showed a significant pH-dependent equilibrium adsorption capacity, which increased dramatically from 59 mg g−1 to 449 mg g−1 as the solution pH was decreased from 9 to 2. Because of its high adsorption capacity, pH-sensitivity, easy regeneration, and good reusability, the HPP–NH3+ has great potential for application in the field of water treatment, controlled drug release, and pH-responsive delivery.126 Kitayama et al. studied an efficient pathway for the preparation of HPPs. In this approach, site-specific a posteriori photocrosslinking of homogeneous spherical polymer particles and subsequent removal of the particle core – the self-templating strategy – has been developed as an efficient pathway for hollow particle formation. This facile route via a self-templating strategy has great potential to be used as an alternative route because this route has high mass productivity and high simplicity as a result of the non-use of additional sacrificial template particles and highly toxic solvents.122 Zhang et al. prepared a novel amphiphilic diblock copolymer, PHEMAPOSS-b-P(DMAEMA-co-CMA), via reversible addition–fragmentation chain transfer (RAFT) polymerization. The hollow polymeric capsules were obtained via etching the POSS core in the solution of hydrofluoric acid (HF). The tetraphenylporphyrin tetrasulfonic acid hydrate (TPPS)-loaded polymeric capsules demonstrated efficient photodynamic therapy (PDT) efficacy and a low dark toxicity towards the MCF-7 cells. Thus, the TPPS-loaded polymeric capsules presented potential application in PDT.127 Nakayama et al. fabricated HPPs from the solvent treatment of aqueous bubble-stabilized polystyrene (PS) emulsion particles. The aqueous bubbles stabilized with PS particles could be transformed into polymeric hollow microspheres containing air by exposure to a solvent. The polymeric hollow microspheres maintained their shapes even after complete water evaporation; this confirmed that a rigid polymeric film was successfully formed.128 Wichaita et al. prepared natural rubber (NR)-based hollow latex (HL) particles by a seeded emulsion polymerization of methyl methacrylate/divinyl benzene/acrylic acid (MMA/DVB/AA) monomers on the NR seed. A void was subsequently formed at an MMA/DVB molar ratio of 2.7/1 without the necessity of any residual core removal. The void cavity was enlarged when the monomer to seed (M/S) weight ratio was increased to 4/1. The large void cavity (155 ± 37 nm) of these NR-based HL particles makes them particularly suited as delivery vehicle systems.129 Yang et al. developed an innovative one-step synthesis of hollow polymer particles by microsuspension polymerization of styrene (St) and methyl acrylate (MMA) with Mg(OH)2 as the dispersant. Due to difference in the reactive activity of St and MMA, hollow polymer particles were generated at an appropriate pH and polymerization temperature.130 Okubo et al. proposed an innovative easy method to synthesize hydrophobic hollow PS particles by emulsion polymerization with an polyoxyethylene nonylphenyl ether nonionic emulsifier (Emulgen 910). The vol% (relative to the particles) of water was increased to 46% by heat treatment at 90 °C for 24 h, which was based on further water absorption and resulted in spherical hollow particles.131 Other contributions relating to HPPs have been described by a number of researchers.132–138
6. Applications
Hollow polymer particles (HPPs) have attracted extensive attention in recent years due to their potential applications in the fields of coating, papermaking, and cosmetics because of their excellent characteristics of light scattering and low density;139 moreover, they have potential applications in catalysis, biochemistry, and biomedicine due to their capacity of accommodating objects in their hollow structure.3,140 The application of HPPs as fillers that decrease the density of polymeric composites is a well-known technique. Another and more complicated area of HPPs is their use as white pigments in coatings and paint materials. This approach almost eliminates the possibility for using many of the conventional microspheres.13,141 Stimuli-responsive HPPs are very promising candidates as drug delivery systems because of their high stability for prolonged circulation in the blood stream and their ability to encapsulate bioactive drugs.142 In particular, their application provides an interesting opportunity for drug delivery in which the delivery system becomes an active participant, rather than a passive carrier, in the optimization of disease therapy.143 When used as drug carriers, stimuli-responsive HPPs have shown a much larger drug-loading capacity than ordinary HPPs as stimuli-responsive HPPs are easier to load with drugs, particularly biomacromolecular drugs, or to release the entrapped drugs on demand at the desired target because the pores of their shells can be gated upon phase transition triggered by external stimuli.143 However, porous-shell HPPs are also well known for a superhigh capacity of drugs due to their porous shell structures.Yang et al. prepared monodisperse pH-sensitive hollow P(MBAAm-co-MAA)microspheres with a diameter below 200 nm and a shell thickness in the range of 8–26 nm. The in vitro loading and release behavior studied demonstrated that the hollow P(MBAAm-co-MAA) microspheres toward the DOX drug possessed a good loading capacity (as high as 357 mg mg−1) with a high encapsulation efficiency (as high as 83%). The loading and release behavior of DOX drug with P(MBAAm-co-MAA) HPPS as a reservoir was highly dependent on the pH values in the environment, which could be well interpreted by the difference in strength of the hydrogen–bonding interaction between the host P(MBAAm-co-MAA) HPPs and the guest DOX drug under different pH conditions. Furthermore, these HPPs were improved to enhance the effect of targeted delivery by adding Fe3O4 nanoparticles to P(MBAAm-co-MAA) HPPs. These results imply that P(MBAAm-co-MAA) hollow microspheres may have potential application as a reservoir for controlled release. The drug loading of the dual-targeting hollow P(MBAAm-co-MAA) microspheres toward the DOX drug possessed a good loading capacity (as high as 176 mg mg−1) and a high encapsulation efficiency (as high as 61%) in the case of an initial DOX concentration of 230 mg mL−1, whereas the releasing behavior with dual-targeting hollow microspheres as reservoirs was highly dependent on the pH values in the environment. All these results may offer a suitable way for the preparation of multi-functionalized drug carriers for tumor combination therapy.140 Chiang et al. prepared polymeric hollow nanogels designed as carriers for intracellular triggered drug release.144 Another contribution reported by Chiang et al. involved the use of superparamagnetic hollow nanogels as a potential guidable vehicle system for stimuli-mediated magnetic resonance imaging (MRI) and multiple cancer therapeutics. The system works by magnetic transport guidance toward the target and subsequent exposure to an alternating magnetic field. Then, the system exhibited accelerated drug release in response to both pH reduction and temperature increase as a result of a disruption of the interactions between the drug molecules and nanogels structures. Fig. 17 shows a schematic of the hollow nanogels serving as a multifunctional anticancer theranostic platform.145
 | ||
| Fig. 17 Schematic hollow nanogels serving as a multifunctional anticancer theranostic platform. Reproduced from ref. 145 with permission from the American Chemical Society. | ||
Ke et al. reported a novel smart system based on PLGA hollow microspheres (HMs) that could deliver an anticancer drug into tumor cells and quickly release the drug in an acidic organelle such as a lysosome.146 Nuasen et al. prepared hollow latex (HL) particles functionalized with chitosan for the removal of formaldehyde from indoor air. Chitosan and polyethyleneimine (PEI)-functionalized HL particles were conveniently fabricated by coating (P(MMA/DVB/AA)) HPPs with 5 wt% chitosan or 14 wt% PEI. Besides providing opacity and whiteness, the multilayer HL-chitosan particles could effectively remove indoor air pollutants, i.e., formaldehyde gas, and, hence, would be useful in special coating applications.10
7. Conclusions and outlook
This review presents a general overview of hollow polymer particles (HPPs). The formation of single pore structures in the particles endows a high potential to encapsulate large guest molecules. Core–shell polymer particles with hollow core structures are the most developed HPPs due to their interesting morphologies, which lead to them achieve superior properties. These advanced morphologies have accelerated their exploration in a growing list of applications such as in drug delivery, cosmetics, inks, pigments, chemical reagents, the protection of biologically active species, and the removal of pollutants. The highest loading capacity of functional materials can be achieved by porous-shell structures, and the fastest loading capacity can be achieved by creating a single hole on the polymer particle surface. Magnetic-targeting stimuli-responsive HPPs have received significant attention from scientists due to their capability to work as intelligent carriers. Template-directed synthesis is the most widely used method for the formation of HPPs. However, with solid-shell HPPs, the acid/alkali swelling method is mostly used to obtain the hollow core. Pioneering works show that the preparation processes is time-consuming; therefore, development of a facile method with high-quality (e.g., nonagglomerated, uniform, controlled size) HPPs is still a great challenge to material scientists. The developed methods also need to be environmentally friendly, clean, and versatile to meet the demands of modern technologies. The best example is the opaque polymers developed by Rohm and Haas (bought by Dow chemical in 2009) for filler and coating applications. However, the development of opaque polymers took 30 years before they could be commercialized. For applications, especially biomedical applications, careful studies should be conducted to reveal the toxicity of the HPPs to the human body.A number of interesting developments have been focused in principle; however, more studies are necessary to optimize the experiments, to improve results reproducibility, and to demonstrate applications testing. Scaling-up this product to produce industrial-scale quantities for applications is expected to face critical challenges, such as uniformity, size, stability, synthesis scale, and production cost. To meet the requirements for industrial applications, future research needs to overcome the critical challenges. These aspects of HPPs synthesis are still in an early stage; this offers exciting opportunities for newcomers to this area. Table 3 summarizes the polymers that form HPPs and classifications and the preparation methods of HPPs.
| Polymer | Classification | Preparation | Ref. | |
|---|---|---|---|---|
| Technique | Method | |||
| MMA/MAA/DVB core, St/ACN/DVB shell | Solid-shell | Seeded emulsion | Acid/alkali swelling | 52 |
| P(St-co-MMA-co-BA-co-MAA) | Solid-shell | Soap-free emulsion | Acid/alkali swelling | 55 |
| PBzMA | Solid-shell | LRP | Core dissolution | 56 |
| PS | Solid-shell | Dispersion | HF etching | 21 |
| PS/DVB | Solid-shell | Seeded dispersion | SaPSeP | 57 |
| P(AA-EGDMA) | Solid-shell | 2-Steps polymerization | Core dissolution | 3 |
| PVAc, PMMA, PBA, PBMA | Porous-shell | Emulsion | Self-assembly | 73 and 102 |
| PAA | Porous-shell | Suspension | Core-removal | 64 |
| PEA core, silica shell | Porous-shell | Pickering emulsion | Solvent evaporation | 70 |
| Au-POMA | Porous-shell | Precipitation | Solvent evaporation | 65 |
| PS, PMMA | Single particles | Pre-synthesis, emulsion | Solvent evaporation | 74 |
| PS | Single particles | 3-Steps approach | Solvent evaporation | 75 |
| POT | Single particles | Chemical polymerization | Droplet template | 76 |
| PMAA-PNIPAM | Stimuli responsive | Sol–gel process | Core removal/HF treatment | 82 |
| Fe3O4/SiO2/PMAA/SiO2/PNIPAM | Stimuli responsive | Precipitation | Template | 79 |
| MH-MIPs | Stimuli responsive | Pickering emulsion | Soft-templates | 83 |
| P(NIPAAm-co-MBAAm-co-GA) | Stimuli responsive | Inverse emulsion | Phase separation | 84 |
| Silica–PNIPAM–PNIPMAM | Stimuli responsive | 2-Steps approach | Core dissolution | 51 |
| P(MAH/DVB) | Solid-shell | Distillation precipitation | Hard templates | 94 |
| P(DVB-co-AA) | Solid-shell | Precipitation | Hard templates | 95 |
| PPy-CS | Solid-shell | Photoreduction | Sacrificial templates | 4 |
| St-alt-MA | Solid-shell | RAFT | Sacrificial templates | 96 |
| St, DVB | Solid-shell | Vesicle polymerization | Vesicle templates | 97 and 98 |
| Fe–Fc–HCPS | Porous-shell | Ostwald ripening | Self-templates | 66 |
| AG-VP | Single-particles | FRP | Template-free | 99 |
| PS | Solid-shell | Emulsion | Self-assembly/Core etching | 33 |
| PS | Porous-shell | Emulsion | Self-assembly/Emulsion droplet | 73 and 102 |
| PS/MF | Porous-shell | Emulsion | Self-assembly/Emulsion droplet | 71 |
| MMA–MAA–DVB | Solid-shell | Emulsion | Acid/alkali swelling | 52 |
| MAA/BA/MMA core, BA/St shell | Solid-shell | Emulsion | Acid/alkali swelling | 53 |
| P(NIPAAm–MAA)/Fe3O4 | Solid-shell | Emulsion | Acid/alkali swelling | 54 and 107 |
| PMSQ | Solid-shell | CEHDA | 40 and 111 | |
| PMMA | Single particles | Inverse mini-emulsion | IMEPP | 39 and 112 |
| P(MMA/AA/St) | Porous-shell | Emulsion | Urea treatment | 36 |
| PVP/PEM-Au | Solid-shell | Emulsion | HF etching | 38 |
| Lipid–polymer–lipid | Solid-shell | Emulsion | Self-assembly | 117 |
| PS-Fe3O4 | Solid-shell | Emulsion | Acid/alkali swelling | 119 |
| St/BA | Solid-shell | Emulsion | Acid/alkali swelling | 35 |
| P(BA-co-MAA-co-EGDMA) core, P(St-co-MMA) shell | Solid-shell | Emulsion | Acid/alkali swelling | 9 |
| PIL | Solid-shell | Suspension | SaPSeP | 125 |
| PHEMA-b-P(DMAEMA-co-CMA) | Solid-shell | RAFT polymerization | HF etching | 127 |
| PS | Single particles | Emulsion | Phase separated | 128 |
| NR-based P(MMA/DVB/AA) | Solid-shell | Emulsion | Core removal | 129 |
| St/MMA | Single particles | Microsuspension | Phase separation | 130 |
| P(MBAAm-co-MAA) | Solid-shell | Distillation precipitation | Dissolution of core | 140 |
| P(NIPAM-co-AA) | Stimuli responsive | FRP | Dissolution of core | 144 |
| P(MMA/DVB/AA)/CS@PEI | Solid-shell | Emulsion | Phase separated | 10 |
Conflicts of interest
There are no conflicts to declare.Abbreviations
| AA | Acrylic acid |
| AAm | Acrylamide |
| ACN | Acrylonitrile |
| Ag | Argentum |
| AG | Alginic acid |
| Au | Aurum |
| 4-AP | 4-Aminophenol |
| 4-NP | 4-Nitrophenol |
| VP | 4-Vinyl pyridine |
| BA | Butyl acrylate |
| BMA | Butyl methacrylate |
| CMA | Coumarin methacrylate |
| CS | Chitosan |
| CSS | Core–shell–shell |
| CTAB | Cetyltrimethylammonium bromide |
| CTAT | Cetyltrimethylammonium tosylate |
| CEHDA | Coaxial electrohydrodynamic atomization |
| DAM | Diallyl maleate |
| DMAEMA | 2-(Dimethylamino)ethyl methacrylate |
| DODAC | Dimethyldioctadecylammonium chloride |
| DOX | Doxorubicin |
| DVB | Divinylbenzene |
| EGDMA | Ethyleneglycol dimethacrylate |
| EHDA | Electrohydrodynamic atomization |
| Fc | Ferrocenyl |
| Fe | Ferum |
| Fe3O4 | Ferric oxide |
| FRP | Free radical polymerization |
| GA | Glycidyl acrylate |
| GNP | Gold nanoparticles |
| HCPS | Hollow coordination polymer microspheres |
| HEMAPOSS | Polyhedral oligomeric silsesquioxane |
| HF | Hydrofluoric acid |
| HFMF | High-frequency magnetic field |
| HMs | Hollow microspheres |
| HPC | Hydroxypropyl cellulose |
| HP | Hollow polymer |
| HPMIPs | Hollow porous molecularly imprinted polymers |
| HPPs | Hollow polymer particles |
| IMEPP | Inverse mini-emulsion periphery RAFT polymerization |
| LBL | Layer-by-layer |
| LiBr | Lithium bromide |
| LRP | Living radical polymerization |
| MAA | Methacrylic acid |
| MAH | Maleic anhydride |
| MBAAm | N,N-methylenebisacrylamide |
| MC | Methylene chloride |
| MCF-7 | Michigan Cancer Foundation-7 |
| MIP | Molecularly imprinted polymers |
| MA | Methyl methacrylate |
| MF | Melamine–formaldehyde |
| Mg(OH)2 | Magnesium hydroxide |
| Mn | Number average molecular weight |
| Mw | Weight average molecular weight |
| MRI | Magnetic resonance imaging |
| NaBH4 | Sodium borohydride |
| NaOH | Sodium hydroxide |
| NH3+ | Ammonium |
| NIPAM | N-isopropylacrylamide |
| NPs | Nanoparticles |
| PBA | Poly(butyl acrylate) |
| PBMA | Poly(butyl methacrylate) |
| PBzMA | Poly(benzyl methacrylate) |
| PCL | Polycaprolactone |
| PDT | Photodynamic therapy |
| PEA | Isopentyl acetate |
| PEG | Polyethylene glycol |
| PEI | Polyethyleneimine |
| PEM | P(EGDMA-co-MAA) |
| PFH | Liquid perfluorohexane |
| PIL | Poly(ionic liquid) |
| PLGA | Poly(lactic-co-glycolic acid) |
| PLLA | Poly (L-lactide) |
| PMMA | Poly(methyl methacrylate) |
| PMSQ | Polymethylsilsesquioxane |
| PNIPAM | Poly(N-isopropylacrylamide) |
| PNIPMAM | Poly(N-isopropylmethacrylamide) |
| POT | Poly(o-toluidine) |
| POMA | Poly(o-methoxyaniline) |
| PMV | MAH/VAc particles |
| PPy | Polypyrrole |
| PS | Poly(styrene) |
| Pt | Platinum |
| PVA | Poly(vinyl acetate) |
| PVP | Polyvinylpyrrolidone |
| RAFT | Reversible addition–fragmentation chain transfer |
| SaPSeP | Self-assembling of phase-separated polymer |
| SDBS | Sodium dodecylbenzenesulfonate |
| SDS | Sodium dodecyl sulfate |
| SEM | Scanning electron microscope |
| SiO2 | Silicone dioxide |
| siRNA | Small interfering RNA |
| St | Styrene |
| SOS | Sodium octyl sulfate |
| TEM | Transmission electron microscope |
| TiO2 | Titanium dioxide |
| TPPS | Tetraphenylporphyrin tetrasulfonic acid hydrate |
| TPM | 3-(Trimethoxysilyl)propyl methacrylate |
| Tg | Glass-transition temperature |
| VAc | Vinyl acetate |
Acknowledgements
The author would like to thank Universiti Malaysia Pahang for the financial support under University Grant (No. RDU1703286).References
- J. Han, G. Song and R. Guo, Chem. Mater., 2007, 19, 973–975 CrossRef CAS.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- G. Guan, Z. Zhang, Z. Wang, B. Liu, D. Gao and C. Xie, Adv. Mater., 2007, 19, 2370–2374 CrossRef CAS.
- D. Cheng, X. Zhou, H. Xia and H. S. O. Chan, Chem. Mater., 2005, 17, 3578–3581 CrossRef CAS.
- D. G. Shchukin, G. B. Sukhorukov and H. Möhwald, Angew. Chem., Int. Ed., 2003, 42, 4472–4475 CrossRef CAS PubMed.
- W. Meier, Chem. Soc. Rev., 2000, 29, 295–303 RSC.
- H. Bamnolker, B. Nitzan, S. Gura and S. Margel, J. Mater. Sci. Lett., 1997, 16, 1412–1415 CrossRef CAS.
- A. Khan, B. Ray, J. Maiti and S. Dolui, Pigm. Resin Technol., 2009, 38, 159–164 CrossRef CAS.
- L. J. Borthakur, T. Jana and S. Dolui, J. Coat. Technol. Res., 2010, 7, 765–772 CrossRef CAS.
- S. Nuasaen and P. Tangboriboonrat, Prog. Org. Coat., 2015, 79, 83–89 CrossRef CAS.
- A. Khan, B. Ray and S. Dolui, Prog. Org. Coat., 2008, 62, 65–70 CrossRef CAS.
- R. F. Brown, C. Carr and M. E. Taylor, Prog. Org. Coat., 1997, 30, 185–194 CrossRef CAS.
- V. Pavlyuchenko, O. Sorochinskaya, S. Ivanchev, V. Klubin, G. Kreichman, V. Budtov, M. Skrifvars, E. Halme and J. Koskinen, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1435–1449 CrossRef CAS.
- X. Yang, L. Chen, B. Han, X. Yang and H. Duan, Polymer, 2010, 51, 2533–2539 CrossRef CAS.
- X. Kang, D. Yang, P. A. Ma, Y. Dai, M. Shang, D. Geng, Z. Cheng and J. Lin, Langmuir, 2013, 29, 1286–1294 CrossRef CAS PubMed.
- C. Mangeney, S. Bousalem, C. Connan, M.-J. Vaulay, S. Bernard and M. M. Chehimi, Langmuir, 2006, 22, 10163–10169 CrossRef CAS PubMed.
- J.-H. Kim, R. D. Burnett and A. Gabriel, J. Biomed. Nanotechnol., 2012, 8, 432–438 CrossRef CAS PubMed.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. A. Davis and H. Möhwald, Angew. Chem., Int. Ed., 1998, 37, 2201–2205 CrossRef.
- S. Miao, C. Zhang, Z. Liu, B. Han, Y. Xie, S. Ding and Z. Yang, J. Phys. Chem. C, 2008, 112, 774–780 CAS.
- X. Wang, H. Ji, X. Zhang, H. Zhang and X. Yang, J. Mater. Sci., 2010, 45, 3981–3989 CrossRef CAS.
- X. Xu and S. A. Asher, J. Am. Chem. Soc., 2004, 126, 7940–7945 CrossRef CAS PubMed.
- B. Debord, M. Alharbi, T. Bradley, C. Fourcade-Dutin, Y. Wang, L. Vincetti, F. Gérôme and F. Benabid, Opt. Express, 2013, 21, 28597–28608 CrossRef CAS PubMed.
- C. Markos, G. Antonopoulos and G. Kakarantzas, IEEE Photonics Technol. Lett., 2013, 25, 2003–2006 CrossRef.
- G. Li, Q. Shi, S. Yuan, K. Neoh, E. Kang and X. Yang, Chem. Mater., 2010, 22, 1309–1317 CrossRef CAS.
- R. A. Ramli, W. A. Laftah and S. Hashim, RSC Adv., 2013, 3, 15543–15565 RSC.
- F. Caruso, R. A. Caruso and H. Möhwald, Chem. Mater., 1999, 11, 3309–3314 CrossRef CAS.
- D. L. Wilcox, Hollow and Solid Spheres and Microspheres: Science and Technology Associated with their Fabrication and Application: Symposium held November 3-December 1, 1994, Mater. Res. Soc., Boston, Massachusetts, USA, 1995 Search PubMed.
- A. Kowalski, M. Vogel and R. M. Blankenship, US Pat., 4, 468, 498, 1984.
- R. M. Blankenship and A. Kowalski, US. Pat., 4, 594, 363, 1986.
- Z. Zhang, J. Deng, J. Sui, L. Yu, M. Wan and Y. Wei, Macromol. Chem. Phys., 2006, 207, 763–769 CrossRef CAS.
- J. Du and Y. Chen, Angew. Chem., Int. Ed., 2004, 43, 5084–5087 CrossRef CAS PubMed.
- O. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374–2384 CrossRef CAS.
- J. Jang and H. Ha, Langmuir, 2002, 18, 5613–5618 CrossRef CAS.
- M. S. Wong, J. N. Cha, K.-S. Choi, T. J. Deming and G. D. Stucky, Nano Lett., 2002, 2, 583–587 CrossRef CAS.
- X. Huang, L. Li, M. Xu, J. Xu, Y. Jiang and C. Kan, J. Appl. Polym. Sci., 2015, 132, 42541 Search PubMed.
- Z. Wang, Q. Hong, Y. Shi, J. Zhang, Y. Song, S. Wu and B. Yang, Polym. Sci., Ser. B, 2015, 57, 600–607 CrossRef CAS.
- D. Hu, X. Zhang, C. Zhang, S. Ding and Z. Yang, Polymer, 2012, 53, 3802–3806 CrossRef CAS.
- S. Shi, C. Chang, T. Wang, S. Ren, Y. Gao and N. Wang, J. Polym. Res., 2015, 22, 632 CrossRef.
- R. H. Utama, M. H. Stenzel and P. B. Zetterlund, Macromolecules, 2013, 46, 2118–2127 CrossRef CAS.
- M.-W. Chang, E. Stride and M. Edirisinghe, Langmuir, 2010, 26, 5115–5121 CrossRef CAS PubMed.
- C. F. Lee, M. L. Hsu, C. H. Chu and T. Y. Wu, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3441–3451 CrossRef CAS.
- M. Sasidharan and K. Nakashima, Acc. Chem. Res., 2013, 47, 157–167 CrossRef PubMed.
- P. Panahian, M. Salami-Kalajahi and M. Salami Hosseini, Ind. Eng. Chem. Res., 2014, 53, 8079–8086 CrossRef CAS.
- M. Fuji, T. Shin, H. Watanabe and T. Takei, Adv. Powder Technol., 2012, 23, 562–565 CrossRef CAS.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- G. L. Li, H. Möhwald and D. G. Shchukin, Chem. Soc. Rev., 2013, 42, 3628–3646 RSC.
- F. Caruso, Adv. Mater., 2001, 13, 11–22 CrossRef CAS.
- G.-D. Fu, G. L. Li, K. Neoh and E. Kang, Prog. Polym. Sci., 2011, 36, 127–167 CrossRef CAS.
- W. Deng, W. Ji, Y. Jiang and C. Kan, J. Appl. Polym. Sci., 2013, 127, 651–658 CrossRef CAS.
- X. Huang and B. Voit, Polym. Chem., 2013, 4, 435–443 RSC.
- J. Dubbert, K. Nothdurft, M. Karg and W. Richtering, Macromol. Rapid Commun., 2015, 36, 159–164 CrossRef CAS PubMed.
- C. D. Yuan, A. H. Miao, J. W. Cao, Y. S. Xu and T. Y. Cao, J. Appl. Polym. Sci., 2005, 98, 1505–1510 CrossRef CAS.
- Q. Zhang, Z. Yang, X. Zhan and F. Chen, J. Appl. Polym. Sci., 2009, 113, 207–215 CrossRef CAS.
- C. F. Lee, M. L. Lin, Y. C. Wang and W. Y. Chiu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2626–2634 CrossRef CAS.
- T. Tan, S. Wang, X. Li, C. Wang and Y. An, Curr. Appl. Phys., 2009, 9, 989–992 CrossRef.
- T. K. Mandal, M. S. Fleming and D. R. Walt, Chem. Mater., 2000, 12, 3481–3487 CrossRef CAS.
- H. Minami, H. Kobayashi and M. Okubo, Langmuir, 2005, 21, 5655–5658 CrossRef CAS PubMed.
- D. Chen, J. Deng, J. Liang, J. Xie, C. Hu and K. Huang, Sens. Actuators, B, 2013, 183, 594–600 CrossRef CAS.
- W. L. Zhang, Y. D. Liu and H. J. Choi, J. Mater. Chem., 2011, 21, 6916–6921 RSC.
- W. Lu, C. Wang, W. Yue and L. Chen, Nanoscale, 2011, 3, 3631–3634 RSC.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- R. Liu, Y. Guo, G. Odusote, F. Qu and R. D. Priestley, ACS Appl. Mater. Interfaces, 2013, 5, 9167–9171 CAS.
- X. Xia, D. Chao, X. Qi, Q. Xiong, Y. Zhang, J. Tu, H. Zhang and H. J. Fan, Nano Lett., 2013, 13, 4562–4568 CrossRef CAS PubMed.
- Y. Chen, X. Zheng, H. Qian, Z. Mao, D. Ding and X. Jiang, ACS Appl. Mater. Interfaces, 2010, 2, 3532–3538 CAS.
- J. Han, M. Wang, R. Chen, N. Han and R. Guo, Chem. Commun., 2014, 50, 8295–8298 RSC.
- J. Huo, L. Wang, E. Irran, H. Yu, J. Gao, D. Fan, B. Li, J. Wang, W. Ding and A. M. Amin, Angew. Chem., Int. Ed., 2010, 49, 9237–9241 CrossRef CAS PubMed.
- J. Liu, Q. Yang, L. Zhang, H. Yang, J. Gao and C. Li, Chem. Mater., 2008, 20, 4268–4275 CAS.
- S. W. Choi, Y. Zhang and Y. Xia, Adv. Funct. Mater., 2009, 19, 2943–2949 CrossRef CAS PubMed.
- T. Chen, P. J. Colver and S. A. Bon, Adv. Mater., 2007, 19, 2286–2289 CrossRef CAS.
- Y. Guan, X. Meng and D. Qiu, Langmuir, 2014, 30, 3681–3686 CrossRef CAS PubMed.
- X. D. He, X. W. Ge, M. Z. Wang and Z. C. Zhang, J. Colloid Interface Sci., 2006, 299, 791–796 CrossRef CAS PubMed.
- H. Li, X. Hu, Y. Zhang, S. Shi, X. Jiang and X. Chen, J. Chromatogr. A, 2015, 1404, 21–27 CrossRef CAS PubMed.
- X. He, X. Ge, H. Liu, M. Wang and Z. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 933–941 CrossRef CAS.
- U. Jeong, S. H. Im, P. H. Camargo, J. H. Kim and Y. Xia, Langmuir, 2007, 23, 10968–10975 CrossRef CAS PubMed.
- S. H. Im, U. Jeong and Y. Xia, Nat. Mater., 2005, 4, 671–675 CrossRef PubMed.
- J. Han, G. Song and R. Guo, Adv. Mater., 2006, 18, 3140–3144 CrossRef CAS.
- L. Zha, B. Banik and F. Alexis, Soft Matter, 2011, 7, 5908–5916 RSC.
- R. A. Ramli, S. Hashim and W. A. Laftah, J. Colloid Interface Sci., 2013, 391, 86–94 CrossRef CAS PubMed.
- P. Du, T. Wang and P. Liu, Colloids Surf., B, 2013, 102, 1–8 CrossRef CAS PubMed.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441–1455 CrossRef CAS.
- L.-H. Wang, T. Wu, Z. Zhang and Y.-Z. You, Macromolecules, 2015, 49, 362–366 CrossRef.
- G. Li, C. Lei, C. Wang, K. Neoh, E. Kang and X. Yang, Macromolecules, 2008, 41, 9487–9490 CrossRef CAS.
- J. Pan, L. Li, H. Hang, R. Wu, X. Dai, W. Shi and Y. Yan, Langmuir, 2013, 29, 8170–8178 CrossRef CAS PubMed.
- H. Macková, D. Horák, E. Petrovský and J. Kovářová, Colloid Polym. Sci., 2013, 291, 205–213 Search PubMed.
- G. Réthoré and A. Pandit, Small, 2010, 6, 488–498 CrossRef PubMed.
- Y. Yang, Y. Chu, F. Yang and Y. Zhang, Mater. Chem. Phys., 2005, 92, 164–171 CrossRef CAS.
- C. Wang, J. Yan, X. Cui, D. Cong and H. Wang, Colloids Surf., A, 2010, 363, 71–77 CrossRef CAS.
- X. W. D. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- W. Ostwald, Lehrbuch der Allgemeinen Chemie, Engelmann, Leipzig, Germany, vol. 2, 1896, part 1 Search PubMed.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- A. Khanal, Y. Inoue, M. Yada and K. Nakashima, J. Am. Chem. Soc., 2007, 129, 1534–1535 CrossRef CAS PubMed.
- D. E. Bergbreiter, Angew. Chem., Int. Ed., 1999, 38, 2870–2872 CrossRef CAS PubMed.
- J. Deng, Y. Yu, S. Dun and W. Yang, J. Phys. Chem. B, 2010, 114, 2593–2601 CrossRef CAS PubMed.
- G. Li and X. Yang, J. Phys. Chem. B, 2007, 111, 12781–12786 CrossRef CAS PubMed.
- C. Boyer, M. R. Whittaker, C. Nouvel and T. P. Davis, Macromolecules, 2010, 43, 1792–1799 CrossRef CAS.
- C. McKelvey, E. Kaler, J. Zasadzinski, B. Coldren and H.-T. Jung, Langmuir, 2000, 16, 8285–8290 CrossRef CAS.
- J. Hotz and W. Meier, Langmuir, 1998, 14, 1031–1036 CrossRef CAS.
- Y. Gao, X. Ding, Z. Zheng, X. Cheng and Y. Peng, Chem. Commun., 2007, 3720–3722 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- B. Wei, S. Wang, H. Song, H. Liu, J. Li and N. Liu, Pet. Sci., 2009, 6, 306–312 CrossRef CAS.
- X. He, X. Ge, H. Liu, M. Wang and Z. Zhang, Chem. Mater., 2005, 17, 5891–5892 CrossRef CAS.
- A. Kowalski and M. Vogel, US. Pat., 4, 469, 825, 1984.
- E. S. Daniels, E. D. Sudol and M. S. El-Aasser, Polymer Latexes: Preparation, Characterization, and Applications, ACS Publications, 1992 Search PubMed.
- D. I. Lee, M. R. Mulders, D. J. Nicholson and A. N. Leadbetter, US. Pat., 5, 521, 253, 1996.
- G. K. Chip and A. Rudin, US. Pat., 4, 863, 973, 1989.
- C. F. Lee, Y. C. Wang and W. Y. Chiu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2880–2891 CrossRef CAS.
- M.-K. Park, K. Onishi, J. Locklin, F. Caruso and R. C. Advincula, Langmuir, 2003, 19, 8550–8554 CrossRef CAS.
- S. Stewart and G. Liu, Chem. Mater., 1999, 11, 1048–1054 CrossRef CAS.
- Z. Deng, M. Chen, S. Zhou, B. You and L. Wu, Langmuir, 2006, 22, 6403–6407 CrossRef CAS PubMed.
- M.-W. Chang, E. Stride and M. Edirisinghe, J. R. Soc., Interface, 2010, 7, S451–S460 CrossRef CAS PubMed.
- R. H. Utama, Y. Guo, P. B. Zetterlund and M. H. Stenzel, Chem. Commun., 2012, 48, 11103–11105 RSC.
- G. Liu, H. Wang and X. Yang, Polymer, 2009, 50, 2578–2586 CrossRef CAS.
- S. Yang and H. Liu, J. Mater. Chem., 2006, 16, 4480–4487 RSC.
- S. Malathi and S. Balasubramanian, J. Biomed. Nanotechnol., 2011, 7, 150–151 CrossRef CAS PubMed.
- H. Liu, N. Finn and M. Yates, Langmuir, 2005, 21, 379–385 CrossRef CAS PubMed.
- J. Shi, Z. Xiao, A. R. Votruba, C. Vilos and O. C. Farokhzad, Angew. Chem., 2011, 123, 7165–7169 CrossRef.
- L. Zhang, R. Guo, M. Yang, X. Jiang and B. Liu, Adv. Mater., 2007, 19, 2988–2992 CrossRef CAS.
- N. S. Kim and J.-D. Kim, J. Ind. Eng. Chem., 2012, 18, 1721–1729 CrossRef CAS.
- C. J. McDonald and M. J. Devon, Adv. Colloid Interface Sci., 2002, 99, 181–213 CrossRef CAS PubMed.
- J. Shen, J. Xu, Y. Hu, J. Li and C. Kan, Colloid Polym. Sci., 2017, 295, 679–688 CAS.
- Y. Kitayama, K. Yoshikawa and T. Takeuchi, Langmuir, 2016, 32, 9245–9253 CrossRef CAS PubMed.
- S. H. Park, J. Kim, W.-E. Lee, D.-J. Byun and M. H. Kim, Langmuir, 2017, 33, 2275–2282 CrossRef CAS PubMed.
- S.-J. Park, H.-S. Lim, Y. M. Lee and K.-D. Suh, RSC Adv., 2015, 5, 10081–10088 RSC.
- R. Nakamura, M. Tokuda, T. Suzuki and H. Minami, Langmuir, 2016, 32, 2331–2337 CrossRef CAS PubMed.
- Y. Qin, L. Wang, C. Zhao, D. Chen, Y. Ma and W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 16690–16698 CAS.
- Z. Zhang, Y. Xue, P. Zhang, A. H. Müller and W. Zhang, Macromolecules, 2016, 49, 8440–8448 CrossRef CAS.
- S. Nakayama, K. Fukuhara, Y. Nakamura and S. Fujii, Chem. Lett., 2015, 44, 773–775 CrossRef CAS.
- W. Wichaita, D. Polpanich, T. Suteewong and P. Tangboriboonrat, Polymer, 2016, 99, 324–331 CrossRef CAS.
- Z.-k. Yang, Z.-l. Wang, Z.-m. Mao, W.-s. Li, Y.-j. Zhou, X. Liu and M. Okubo, Colloid Polym. Sci., 2017, 295, 565–572 CAS.
- M. Okubo, H. Kobayashi, C. Huang, E. Miyanaga and T. Suzuki, Langmuir, 2017, 33, 3468–3475 CrossRef CAS PubMed.
- H. Minami, A. Kojima and T. Suzuki, Langmuir, 2017, 33, 1541–1546 CrossRef CAS PubMed.
- N. S. J. Williams, S. A. Wheeler and R. D. Bradley, US Pat., 9,546,297, 2017.
- Y. Zhang, S. Costeux and T. H. Kalantar, US Pat., 9, 650, 500, 2017.
- N. Sudjaipraparat, C. Kaewsaneha, S. Nuasaen and P. Tangboriboonrat, Polymer, 2017, 121, 165–172 CrossRef CAS.
- W. Deng, H.-C. Guo, G.-A. Li and C.-Y. Kan, Chin. Chem. Lett., 2017, 28, 367–371 CrossRef CAS.
- M. Kano, N. Yanagisawa, Y. Takahashi and Y. Kondo, J. Fluorine Chem., 2017, 197, 34–41 CrossRef CAS.
- D. Chen, Y. Liu, Y. Qin, L. Wang, Y. Ma and W. Yang, Chin. J. Chem., 2017, 35, 596–604 CrossRef CAS.
- S. Freiberg and X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS PubMed.
- X. Yang, L. Chen, B. Huang, F. Bai and X. Yang, Polymer, 2009, 50, 3556–3563 CrossRef CAS.
- V. Pavlyuchenko, N. Byrdina, S. Ivanchev, M. Skrifvars, E. Halme, H. Laamanen and J. Koskinen, US. Pat., 6, 235, 810, 2001.
- S. V. Vinogradov, Nanomedicine, 2010, 5, 165–168 CrossRef CAS PubMed.
- Z. Xing, C. Wang, J. Yan, L. Zhang, L. Li and L. Zha, Soft Matter, 2011, 7, 7992–7997 RSC.
- W.-H. Chiang, V. T. Ho, W.-C. Huang, Y.-F. Huang, C.-S. Chern and H.-C. Chiu, Langmuir, 2012, 28, 15056–15064 CrossRef CAS PubMed.
- W.-H. Chiang, V. T. Ho, H.-H. Chen, W.-C. Huang, Y.-F. Huang, S.-C. Lin, C.-S. Chern and H.-C. Chiu, Langmuir, 2013, 29, 6434–6443 CrossRef CAS PubMed.
- C. J. Ke, T. Y. Su, H. L. Chen, H. L. Liu, W. L. Chiang, P. C. Chu, Y. Xia and H. W. Sung, Angew. Chem., 2011, 123, 8236–8239 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |