
Keratin: dissolution, extraction and biomedical application
Amin
Shavandi
 *a,
Tiago H.
Silva
b,
Adnan A.
Bekhit
c and
Alaa El-Din A.
Bekhit
*d
*a,
Tiago H.
Silva
b,
Adnan A.
Bekhit
c and
Alaa El-Din A.
Bekhit
*d
aCenter for Materials Science and Technology, University of Otago, Dunedin, New Zealand. E-mail: amin.shavandi@otago.ac.nz
b3B's Research Group – Biomaterials, Biodegradables and Biomimetics, University of Minho, Headquarters of the European Institute of Excellence on Tissue Engineering and Regenerative Medicine, AvePark-Parque de Ciência e Tecnologia, 4805-017 Barco, Guimarães, Portugal
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Alexandria, Alexandria, Egypt
dDepartment of Food Science, University of Otago, Dunedin, New Zealand. E-mail: aladin.bekhit@otago.ac.nz
First published on 31st May 2017
Abstract
Keratinous materials such as wool, feathers and hooves are tough unique biological co-products that usually have high sulfur and protein contents. A high cystine content (7–13%) differentiates keratins from other structural proteins, such as collagen and elastin. Dissolution and extraction of keratin is a difficult process compared to other natural polymers, such as chitosan, starch, collagen, and a large-scale use of keratin depends on employing a relatively fast, cost-effective and time efficient extraction method. Keratin has some inherent ability to facilitate cell adhesion, proliferation, and regeneration of the tissue, therefore keratin biomaterials can provide a biocompatible matrix for regrowth and regeneration of the defective tissue. Additionally, due to its amino acid constituents, keratin can be tailored and finely tuned to meet the exact requirement of degradation, drug release or incorporation of different hydrophobic or hydrophilic tails. This review discusses the various methods available for the dissolution and extraction of keratin with emphasis on their advantages and limitations. The impacts of various methods and chemicals used on the structure and the properties of keratin are discussed with the aim of highlighting options available toward commercial keratin production. This review also reports the properties of various keratin-based biomaterials and critically examines how these materials are influenced by the keratin extraction procedure, discussing the features that make them effective as biomedical applications, as well as some of the mechanisms of action and physiological roles of keratin. Particular attention is given to the practical application of keratin biomaterials, namely addressing the advantages and limitations on the use of keratin films, 3D composite scaffolds and keratin hydrogels for tissue engineering, wound healing, hemostatic and controlled drug release.
 Amin Shavandi | Amin Shavandi is a Postdoctoral fellow at the Centre for Materials Science and Technology at the University of Otago, Dunedin, New Zealand. He received a master's degree in Chemical Engineering from the University of Putra Malaysia and obtained his Ph.D. in Medicine/Food Science from the University of Otago. His current research interest includes extraction and characterization of natural polymers, development of biomaterials based on natural materials and application of marine-derived polymers for biomaterial application. |
 Tiago H. Silva | Tiago H. Silva, PhD (male, born: 1979), is an Assistant Researcher at 3B's Research Group, being coordinator of the Marine Inspired Biomaterials research area. He graduated in Chemistry (2001) and PhD in Chemistry (2006), both at the Faculty of Sciences – University of Porto (Portugal) and was a visiting researcher at the Swiss Federal Institute of Technology in Lausanne (EPFL, Switzerland) in 2003. He has more than 11 years of experience in the valorization of marine resources and development of marine inspired biomaterials for biomedical applications, namely tissue engineering, and about 15 years of experience in surface modification and electrostatic self-assembly of polyelectrolytes, with applications in (bio)sensors and nanomedicine. |
 Adnan A. Bekhit | Adnan A. Bekhit obtained his PhD in Pharmaceutical Chemistry (1993) from the Faculty of Pharmacy, University of Alexandria (UoA), Egypt. He is currently a professor of Pharmaceutical Chemistry at UoA and a visiting professor at Kyoto Pharmaceutical University, Japan. His research focuses on the synthesis and evaluation of new compounds as well as the analysis of pharmaceutical, environmental and food compounds. Over the last 30 years, he has been involved in vigorous drug development programs aimed at the synthesis of anti-inflammatory-antimicrobial, anticancer, antimalarial, and antileishmanial compounds, and HIV protease and MAO inhibitors and other bioactive agents. Also, his research focuses on drug delivery systems using functional biomaterials. |
 Alaa El-Din A. Bekhit | Alaa El-Din A. Bekhit received his MSc (1994) from the School of Chemistry, Food & Pharmacy, at the University of Reading, UK. He obtained his Ph.D. in Biochemistry (2004) from Lincoln University. He is currently a senior lecturer at the Food Science department, Sciences Division at the University of Otago, an adjunct senior lecturer at Lincoln University and a distinguished professor at the Chinese Academy of Agricultural Sciences and Xinjiang Agricultural University. His research includes bioprocessing of waste materials and generation of functional biomaterials. |
1 Introduction
Keratin, as the major component of wool, hair, nails, hooves, feathers and horns, is one of the most abundant and underexploited protein sources. With the exception of good quality wool that is used in garments and rugs, there are challenges associated with the disposal and management of these materials. Wool fibre is composed of a multi-cell structure that is normally high in sulfur content. More than 2.5 million tonnes of wool are produced annually worldwide1–3 with Australia, China, New Zealand, Iran and Argentina being the top five wool producers. While wool has important commercial value for the textile industry, several low grades and trimmings obtained from slaughterhouses cannot be used in the wool industry and end up as a waste stream. On the other hand, there are over 65 million tonnes of feathers produced worldwide, with many of these by-products normally being disposed through incineration or landfill4,5 or utilized in small scale/low value applications such as fertilizers or biodegradable surfactants. The sulfur content of keratin makes it undesirable for burning and as a fuel source.The present review aims to critically examine the various methods used for keratin solubilisation and extraction, assessing the impact of these methods, and the chemical compounds used on the properties of extracted keratin and on its functional use in various biotechnological and medical applications. Moreover, this review will discuss the influence of different plasticizer and cross-linking agents on the mechanical properties of keratin materials to explain the impact of incorporation of these agents on the film processing and electrospinning ability of keratin. This will be followed by a discussion on the incorporation of natural and synthetic materials into the keratin matrix to fabricate keratin films or 3D scaffolds for biomedical applications. Since calcium phosphate (CaP) materials, such as hydroxyapatite (HA), play an important role in the field of biomaterials, this review will also cover those studies that fabricated biomimetic matrices using CaP materials and keratin. In the last section of this article, concluding remarks are offered on the current and future role of keratin in biomedical applications. For information on disadvantages and advantages of keratin compared with other natural polymers, the reader should consult the excellent book by Neves and Reis.6
1.1 Overview of keratin properties
Wool is constituted largely by a three-dimensional mesh structure of keratin – about 95% keratin proteins – which contain 7–20 mol% cystine residue7,8 and small amounts of lipid (0.1%) and minerals (0.5%). Keratin is a polypeptide made of different amino acids that have inter-molecular bonding of the disulfide cysteine amino acid and inter- and intra-molecular bonding (Fig. 1) of polar and nonpolar acids.9,10 Wool proteins are resistant to the majority of chemical and physical environmental factors. These proteins are insoluble in water and in many weak acids, alkali solutions or organic solvents, as well as resistant to common protein-digesting enzymes such as pepsin or trypsin.11 Keratin has high contents of cystine, glycine, proline and serine, but it is low in lysine, histidine and methionine, and tryptophan is barely present.11 Cystine has an important role in determining the physicochemical properties of wool keratin. Compared to most of the proteins, keratin has higher stability and lower solubility due to inter and intra-chain cross-links of cysteine disulfide bonds. However, hydrogen, hydrophobic and ionic bonds also play an influential role in the stability and properties of the wool keratin. The presence of ionic bonds is pH-dependent and is highest at the isoelectric point of pH = 4.9 when the protein is in the form of zwitterions (+H3N–CHR–COO–), while under extreme acidic or basic conditions, the ionic bonds are at their lowest level. The ionic bond occurs between carboxylic anions and ammonium cations. Therefore, these bonds are reduced by protonation of the carboxylic group at low pH and deprotonation of the amine group at high pH.12 The disulfide bonds give keratin a compact three-dimensional structure and provide high resistance to chemical or enzymatic reactions. Wool keratin with 4–8 wt% sulfur is known as hard keratin whereas keratin found in the epidermal tissue of skin has 2% sulfur and 50–75% moisture and is considered as soft keratin.13 Wool solubilisation occurs by disruption of the complex keratin structure. Keratin obtained from the wool fibre can be classified into four different molecular weight groups: a low sulfur content (LS) with a MW of 45–60 kDa and a fraction with a high sulfur content with a MW of 11–28 kDa, and fractions with a high glycine or high tyrosine content with a MW of 9–12 kDa.12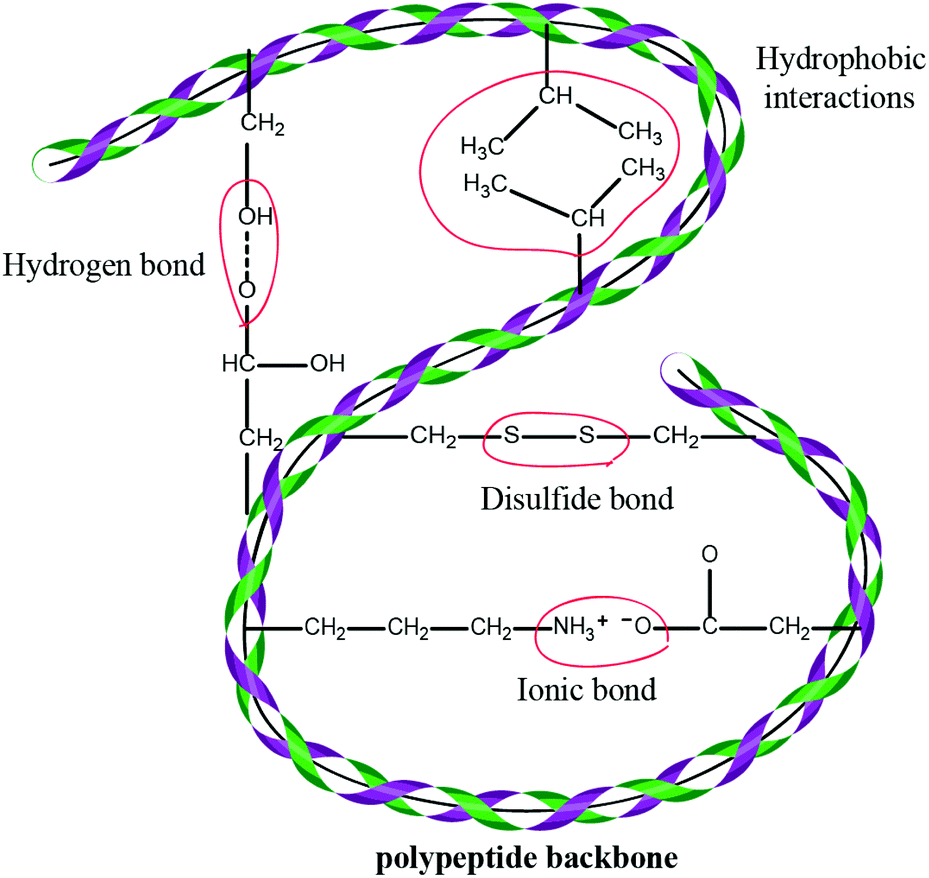 | ||
| Fig. 1 A diagram showing inter- and intra-molecular bonding in keratin. Various chemical bonds, e.g. hydrogen, ionic and disulfide bonds, which result in increased strength and stability of the protein, determine the structure of the keratin. | ||
1.2 Available methods for keratin extraction
The major methods used to solubilise and isolate keratin from keratin-rich materials are reduction,14 oxidation,15 microwave irradiation,16 alkali extraction,17 steam explosion,18 sulfitolysis19 and ionic liquids20 (Fig. 2). The alkaline extraction method requires significant amounts of alkaline chemicals for hydrolysis and acids for neutralization. The primary chain of keratin is damaged and its structure is disrupted in the hydrolysis method. Isolation of keratin from wool by the reduction method using reducing agents, such as thiols (e.g. mercaptoethanol), has been the most reported technique to break the cystine disulfide bonds (R–S–S–R), and the formation of cysteine (R–S–H). Despite the fact that the keratin chain structure is preserved in this method, the use of mercaptoethanol has the disadvantages of being expensive and can be toxic and harmful. Sodium sulfide was used as a cheaper chemical replacement of mercaptoethanol and was widely utilized for the extraction of keratin from wool through the sulfitolysis step with the formation of cysteine (R–S–H) and cysteine-sulfonate (R–S–S–O3H)8 as the stable sites for solubilized keratin. Both methods require the use of large amounts of urea as a protein denaturant (Table 1), which can change the physicochemical properties of the final keratin. Ionic liquids are relatively newer green solvents that have attracted great attention and have been used for the regeneration of keratin from wool.21 However, this process needs to be carried out under nitrogen, requires a precise temperature control, the raw material needs to be added in small portions to the hot liquid, and the obtained keratin is not water soluble. Oxidation methods have been reported in the literature for decades22 with oxidizing materials such as formic or peracetic acids being the most frequently used acids to form a sulfonic acid (RSO3H).16,23 The process is generally a time-consuming process with more than 24 h of reaction time required to obtain a reasonable yield. Depending on the presence or absence of disulfide bonds in the keratin structure, several subfractions can be obtained that can have different physical properties.24 This part of the review will discuss various keratin dissolution and extraction methods. The advantages and limitations of these methods will be highlighted in relation to the physicochemical properties of the obtained keratin.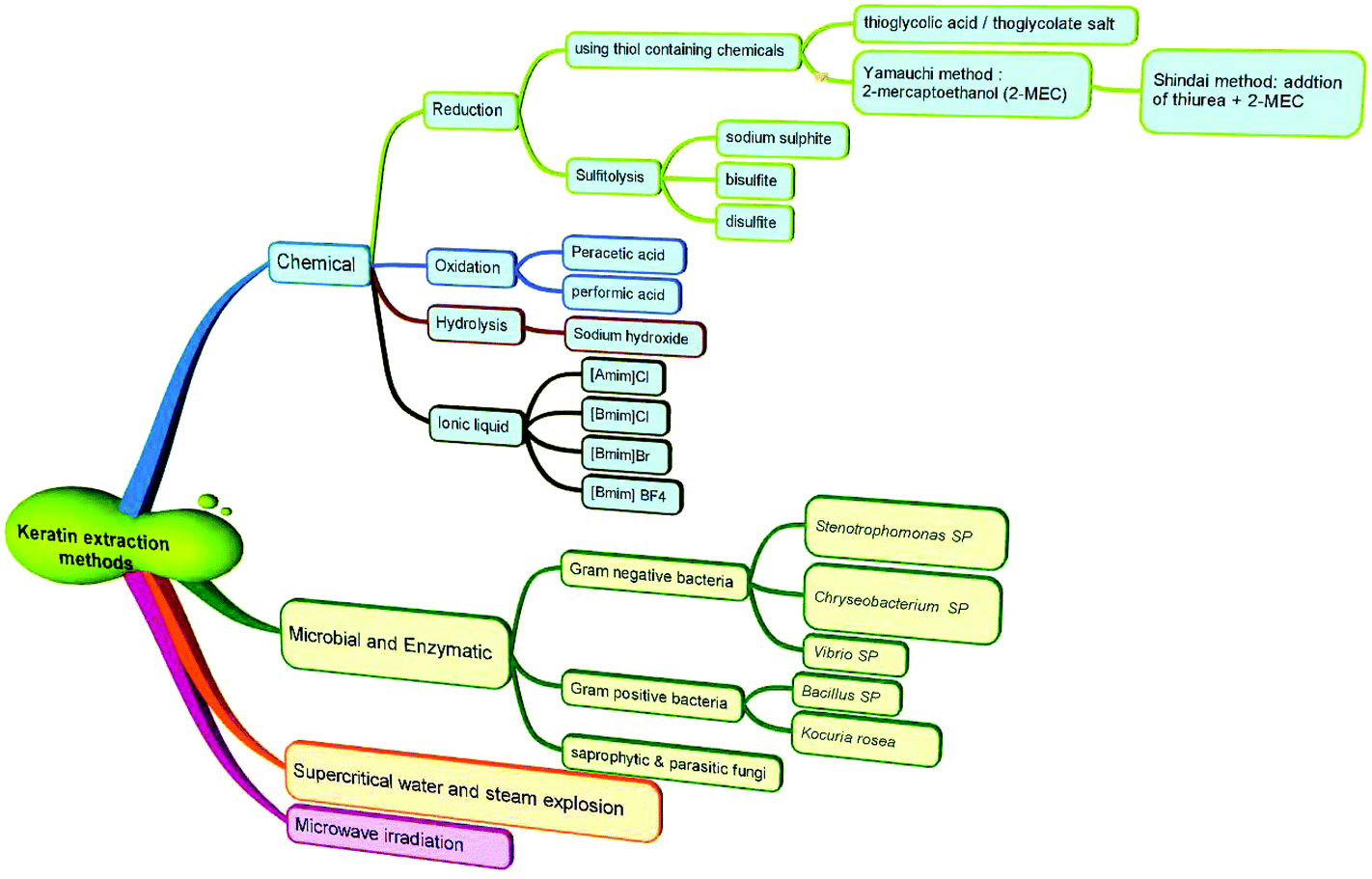 | ||
| Fig. 2 Classification of various methods used for the extraction of keratin from keratin-rich materials, such as wool, feathers and hooves. | ||
| Thiol | Conditions | Temperature and time | %Yield | Ref. | ||
|---|---|---|---|---|---|---|
| Urea (M) | Surfactant/buffering | pH | ||||
| MEC = mercaptoethanol. | ||||||
| MEC (5% v/v) | 7 | 2% SDS (w/v) | Neutral | 50 °C for 24 h | 60 | 14 |
| MEC (5% v/v) | 7 | No SDS | Neutral | 50 °C for 24 h | 45 | 14 |
| Thiourea (2.5 M), 5% thioglycolic acid | 5 | 25 mM Tris-HCl | 9.5 | 50 °C for 3 d | 235 | |
| Thioglycolic acid (0.2 M) | 6 | 10.5 | 40 °C, for 3 h | 85 | 236 | |
| Thioglycolic acid (0.2 M) | 6–10 | 11 | 40 °C for 2 h | 91 | 237 | |
| MEC (0.6 M) | 8 | 1 M Tris/0.25 M EDTA | 11 | 20 °C for 16 h | 73 | 23 |
| MEC (4 M) | — | 5 | 20 °C for 24 h | 75 | 31 | |
| MEC (0.14 M) | 8 | 10.5 | 20 °C for 3 h | 80 | 238 | |
| MEC (1.4 M) | 6 | 1.4 g SDS | 9 | 40 °C for 1 h | 72 | 33 |
| 3 mM EDTA, (0.2 M) KCl–NaOH NaHCO3, Tris | ||||||
| MEC (1.4 M) | 6 | No SDS | 9 | 40 °C for 1 h | 77 | 33 |
| 3 mM EDTA, (0.2 M) KCl–NaOH NaHCO3, Tris | ||||||
| MEC (125 mM) | 8 | 3 mM EDTA, 200 mM Tris | 9.0 | 40 °C, 30 min | 75 | 239 |
| Thiourea (2.4 M) | 15 | 15% DTT, 25 mM Tris | 8.5 | 50 °C for 2 d | 67 | 35 |
| 2-ME (5%) | 8 | 25 mM Tris-HCl | 9.5 | 50 °C for 1–3 d | 27 | 35 |
2 Methods for keratin extraction
2.1 Reduction method
The stable structure of keratin is associated with the disulfide bonds in the polypeptide chain. This disulfide linkage can be reduced using thiol containing chemicals. Several reducing agents under various processing conditions, i.e. in the presence of protein denaturing agents and various pH levels, have been reported.25–27 The following section will examine the effects of various chemicals used in the reduction method.In 1962, Thompson and O'Donnell31 compared mercaptoethanol (MEC) and thioglycolate as reducing agents at pH 5 for the reduction of wool and observed that both chemicals were very similar in their reduction extent when the thiol concentration was low, but at high thiol concentrations, neutral thiol was more effective at pH 7, and by increasing the thiol concentration the reduction process was driven to completion. The authors reported a maximum extractability of 75% and suggested that about 96% of the wool cystine can be reduced using 4 M MEC. Generally, MEC was suggested to be more effective as a reducing agent than potassium thioglycolate.31 The differences between these two thiols were more obvious at high concentrations when a higher ionic strength was exhibited by the ionized carboxyl groups of potassium thioglycolate.
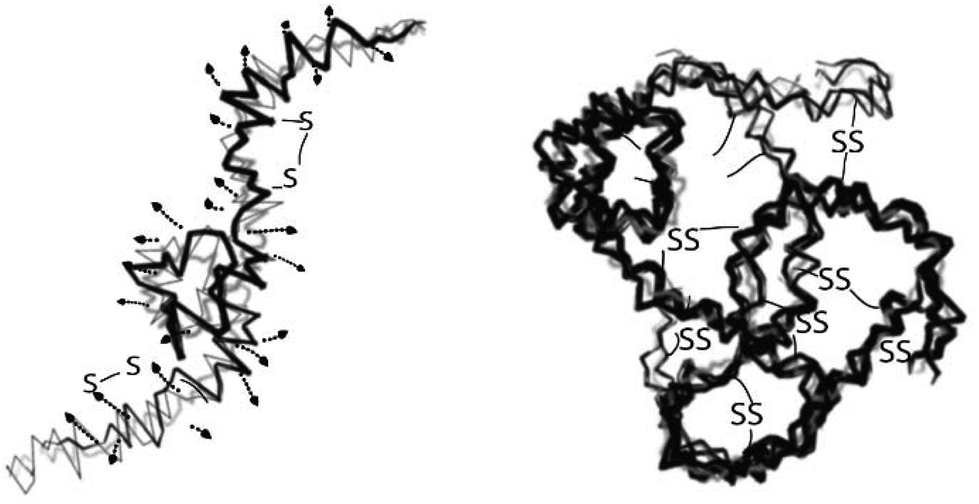 | ||
| Fig. 3 Schematic representation of the keratin complex in the presence of a high amount of SDS, which prevents the protein chain from agglomeration and has high intramolecular disulfide bonds, (B) keratin complex with a low or no amount of SDS which results in agglomeration and high intermolecular disulfide formation. The concept was adopted from ref. 33, reproduced with permission from Elsevier (license no. 4118920283618). | ||
Recently, Xu et al.32 replaced MEC with cysteine as an environmentally friendly reducing agent. The authors suggested that a controlled breakdown of disulfide bonds was achieved using cysteine. The authors also reported that the final product had good mechanical and fibre spinning properties indicating the possibility of using this method for successful development of films, sponges and other mechanically stable forms.
2.2 Alkaline method
It has been known for many years that strong and hot alkali solutions can solubilize wool.37 Upon treating wool with an alkali solution the sulfur nucleus begins to split off and degradation of the cystine residues occurs.37 High concentration alkali solution dissociates the hydrogen from sulfate and carboxylic groups and facilitates solubilisation, although followed by damage occurring in the peptide chains.38 The breakdown of these bonds can lead to the formation of the alkaline sulfide odour during the treatment process, which has a very objectionable odour.39 The damage and dissociation of the protein backbone, consumption of high amounts of alkali reagents and consequently, high amounts of acid required to neutralize and precipitate the protein are the main factors hindering the commercialization and scale-up of the alkali method.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol/mol). Despite the drastic effect of sodium hydroxide, the peptide chain was not damaged and no lanthionine was formed during the process. More importantly, the cystine residue was not damaged but was converted to a cysteic acid residue in the obtained protein. The authors believed that the Schweitzer's reagent acted as a catalyst to facilitate the oxidation of cystine and its subsequent conversion to the cystine residue. The final product was a protein–copper complex and copper was not eliminated through ordinary dialysis due to the strong bond between copper and the protein. Therefore, the final keratin product might not be suitable for feed and pharmaceutical applications due to the presence of copper, which hinders the use of this method commercially. In a recent study by Jiang-tao et al.,45 a two-step alkali-reduction process was used to solubilize hair samples. First, the hair samples were treated with 0.1 mol L−1 of NaOH and then processed with a solution mixture of NaSO3, urea and SDS at 80 °C for 5 h. A dissolution rate of 55% was observed and the authors reported that the α-helix and β-sheet structures of the product were preserved and the obtained keratin had a molecular weight of 25–37 kDa. In practical sense, the harsh effects of alkaline treatment on the keratin protein limit its application and commercialization.43
:1 mol/mol). Despite the drastic effect of sodium hydroxide, the peptide chain was not damaged and no lanthionine was formed during the process. More importantly, the cystine residue was not damaged but was converted to a cysteic acid residue in the obtained protein. The authors believed that the Schweitzer's reagent acted as a catalyst to facilitate the oxidation of cystine and its subsequent conversion to the cystine residue. The final product was a protein–copper complex and copper was not eliminated through ordinary dialysis due to the strong bond between copper and the protein. Therefore, the final keratin product might not be suitable for feed and pharmaceutical applications due to the presence of copper, which hinders the use of this method commercially. In a recent study by Jiang-tao et al.,45 a two-step alkali-reduction process was used to solubilize hair samples. First, the hair samples were treated with 0.1 mol L−1 of NaOH and then processed with a solution mixture of NaSO3, urea and SDS at 80 °C for 5 h. A dissolution rate of 55% was observed and the authors reported that the α-helix and β-sheet structures of the product were preserved and the obtained keratin had a molecular weight of 25–37 kDa. In practical sense, the harsh effects of alkaline treatment on the keratin protein limit its application and commercialization.43
2.3 Sulfitolysis method
The reduction of disulfide bonds using MEC has been the standard method for keratin extraction with a good yield of keratin that has a maintained structure. However, MEC is a toxic chemical and is undesirable commercially and environmentally due to its high cost and issues related to its unpleasant odour and for being naturally toxic to the environment. Sodium sulfite can be a good alternative to break down the sulfide bonds and extract keratin. This method has major industrial and analytical impacts on wool processing. Sulfitolysis (Fig. 4) of a cystine residue by sulfite gives a cysteine thiol and S-sulfonated residue (reaction (1)). Sodium sulfite (SO32−), bisulfite (HSO3−), and disulfite (S2O52−) are major sulfite compounds that exist in aqueous solution and can be used for sulfitolysis. However, the reaction rate of sulfite ions with cystine is faster than that of bisulfide ions46 and the concentration of these sulfite ions increases by increasing the pH up to 9, which consequently make the sulfitolysis reaction faster. HSO3− is dominant under acidic conditions while SO32− is the main species when pH > 7. At pH above 9, cystine sulfitolysis is a reversible reaction (reaction (1)), and the rate of sulfitolysis decreases due to the repulsion force of carboxylic anions on the sulfite ions. While at pH < 9 the reaction is more complex with the formation of bisulfite thiol and S-sulfonate anions.47 Therefore, the optimum pH should be considered to have a maximum rate and equilibrium constant. It has been suggested that bisulfite ions might be the species responsible for sulfitolysis.25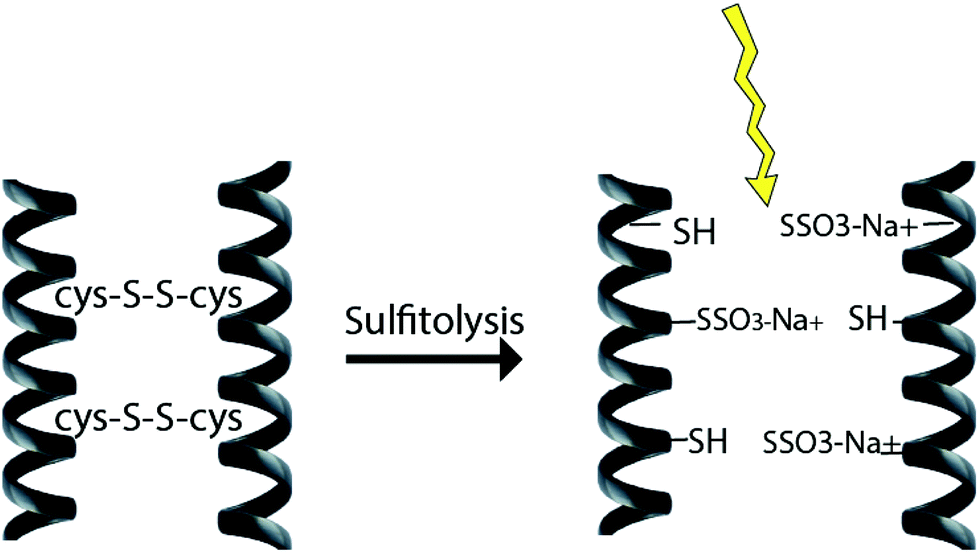 | ||
| Fig. 4 Schematic diagram of the sulfitolysis reaction that breaks the strong disulfide bonds of the keratin fibre. | ||
Sulfitolysis of a cystine residue by sulfite:
 | (1) |
| Material | Chemical used | Conditions | Remarks | Ref. | |
|---|---|---|---|---|---|
| Feathers | Na2S (10 g L−1) | 130 rpm, 30 °C | Flush with N2 gas | Keratin yield of 62% after 24 h | 10 |
| Feathers | Na2S (10 g L−1) + SDS (10 g L−1) + urea (9 M) | 130 rpm, 30 °C | Flush with N2 gas | Addition of urea and SDS enhanced the yield | 10 |
| Feathers | 7 g in 250 ml, urea (8 M), 0.6 g SDS per g of feather/0–0.5 M Na2S | 65 °C for 7 h, pH 6.5 | — | 87.6% at 0.2 M Na2S solution | 7 |
| pH adjusted to 6.5 | |||||
| Wool | 150 g in 1.5 L, urea (8 M), 75 g SDS, 150 g sodium disulfite | 100 °C for 30 min | — | 19 | |
| Wool | 5 g in 100 ml, urea (8 M), sodium metabisulfite (0.5 M) | Shaking for 2 h at 65 °C | — | pH adjusted to 6.5 using 5 M NaOH, extraction yield of 38% | 8 and 149 |
| Wool | 0.5 mol L−1 LiBr, 0.1 mol L−1, SDS (0.02 mol L−1) | 90 °C for 4 h, pH = 12 | — | 94% wool dissolution rate (WDR) and 50.2% keratin extraction rate (KER) | 240 |
| Wool | 1 g in 10 ml, 0.125 mol l−1 Na2S2O5, 0.05 mol l−1 SDS, 2.0 mol l−1 urea | 30–100 °C | Dissolution yield of 48.6% | 50 | |
| 15–45 min | Regeneration yield 76.7% | ||||
| Wool | 5 g per 100 mL: urea (8 M), sodium metabisulfite (0.5 M) (SDS, 0.1 M) | 65 °C overnight | — | — | 152 |
Formation of hydrosulfite and hydroxyl ions from the reaction of sodium sulfite with water:
| Na2S + H2O → 2Na+ + HS− + OH−. | (2) |
In a following step, the hydroxyl ions break the disulfite bonds and form dehydroalanine and perthiocysteine as proposed in reaction (3)46 which are then dissociated to cysteine and sulfur.
Hydroxyl ions break disulfite bonds and form dehydroalanine:
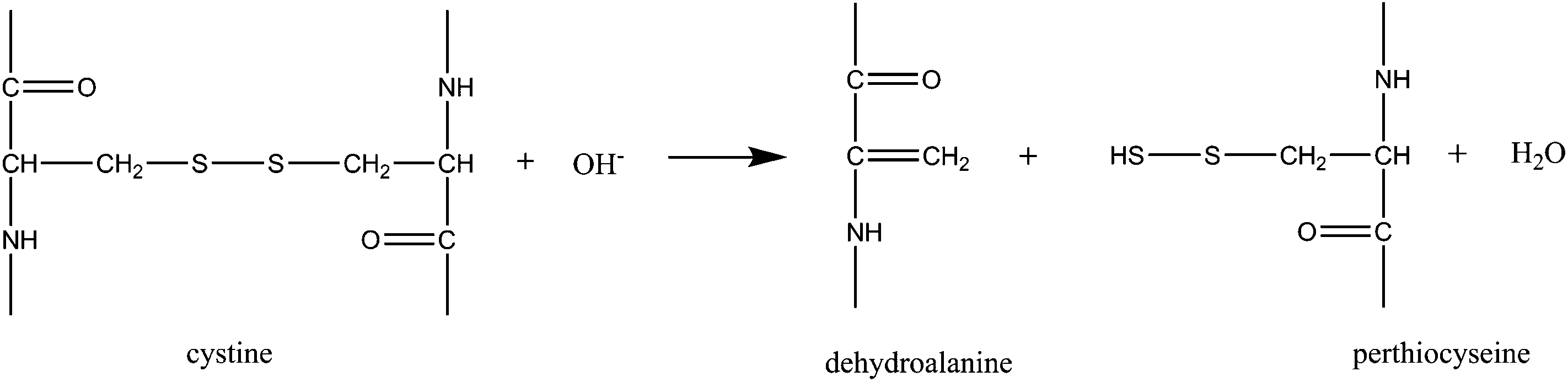 | (3) |
Dehydroalanines that are formed from reaction (3) are very reactive and form lanthionine and lysinoalanine via cross-linking with cysteine and lysine (reactions (4) and (5)). These crosslinking reactions can improve the mechanical properties of the final regenerated product, nevertheless, sodium sulfite treatment can damage the protein backbone, and therefore, optimizing the extraction conditions is an important step to preserve the keratin structure.
Formation of lanthioalanine by the addition of cysteine to dehydroalanine:
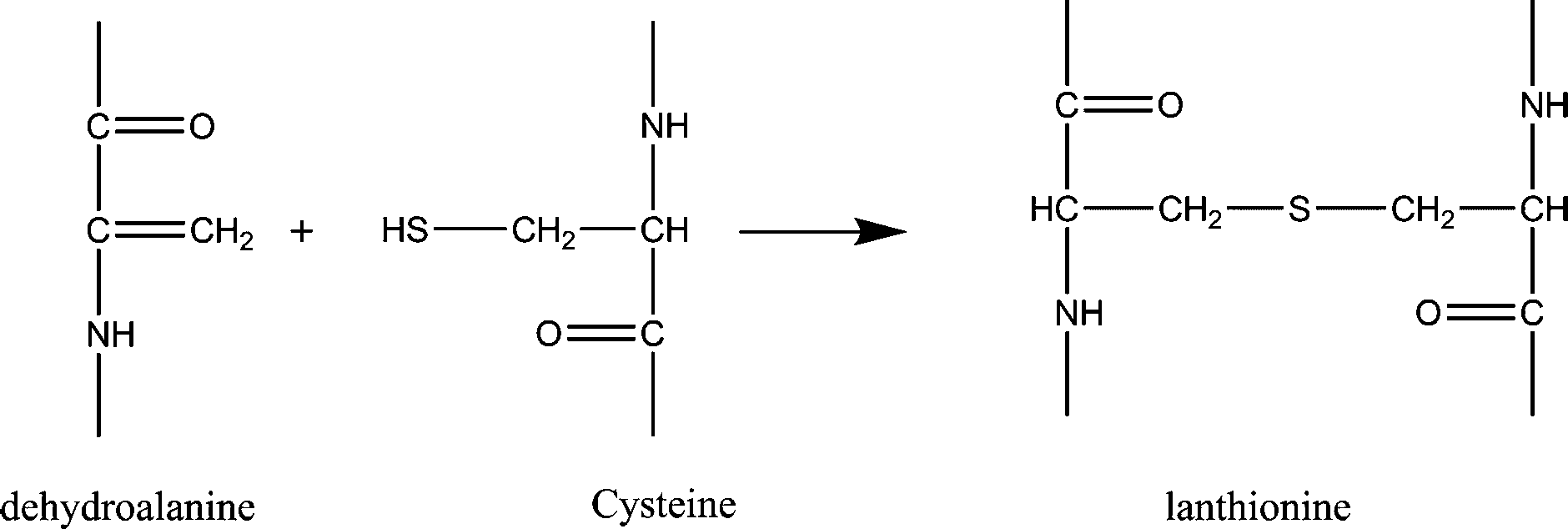 | (4) |
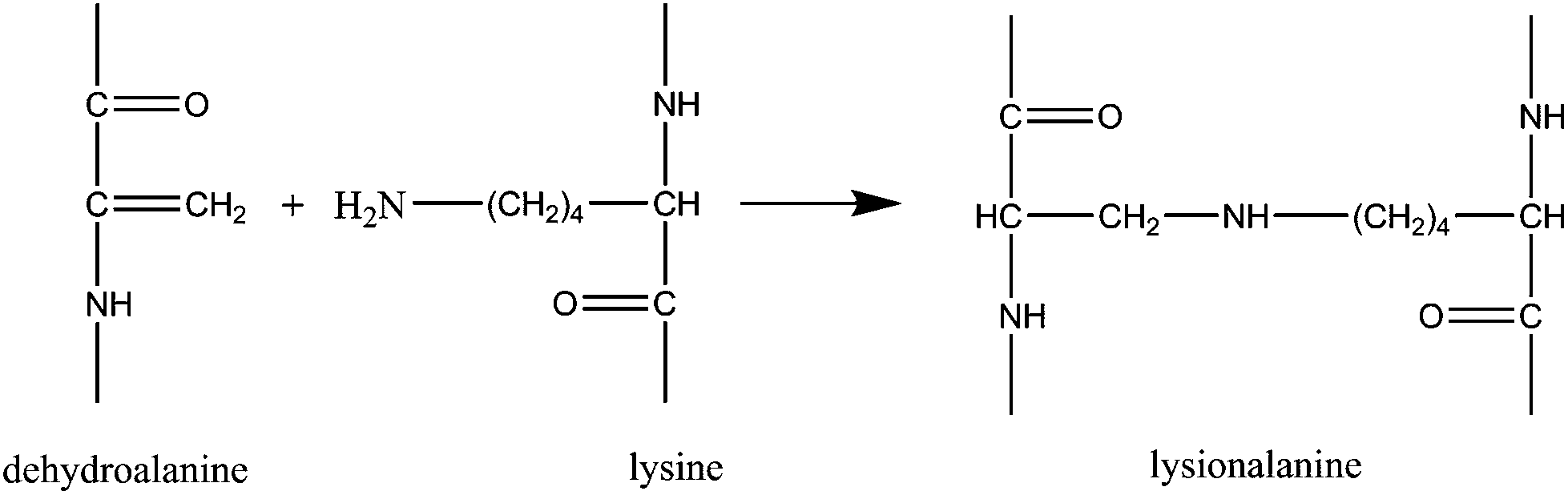 | (5) |
2.4 Ionic liquids
Ionic liquids (ILs) are salts composed of an organic cation and a number of different organic and inorganic anions that melt at temperatures below 100 °C.51,52 Ionic liquids have some unique physicochemical properties including low vapour pressure, high ion conductivity, non-flammability, high thermal stability, high solvation for specific solutes and non-volatility.53–55 Due to these properties, and for also being recognised as a green liquid, ILs have been widely used for a variety of applications such as extraction of biomass or in organic synthesis56 and electrochemistry59 and as ion conductive media57 and catalysts,58 and for use under vacuum where there is a limitation for other solvents due to evaporation.60 ILs have also been widely used as polymer solvents for solubilisation of materials such as silk, wool, cellulose and chitin.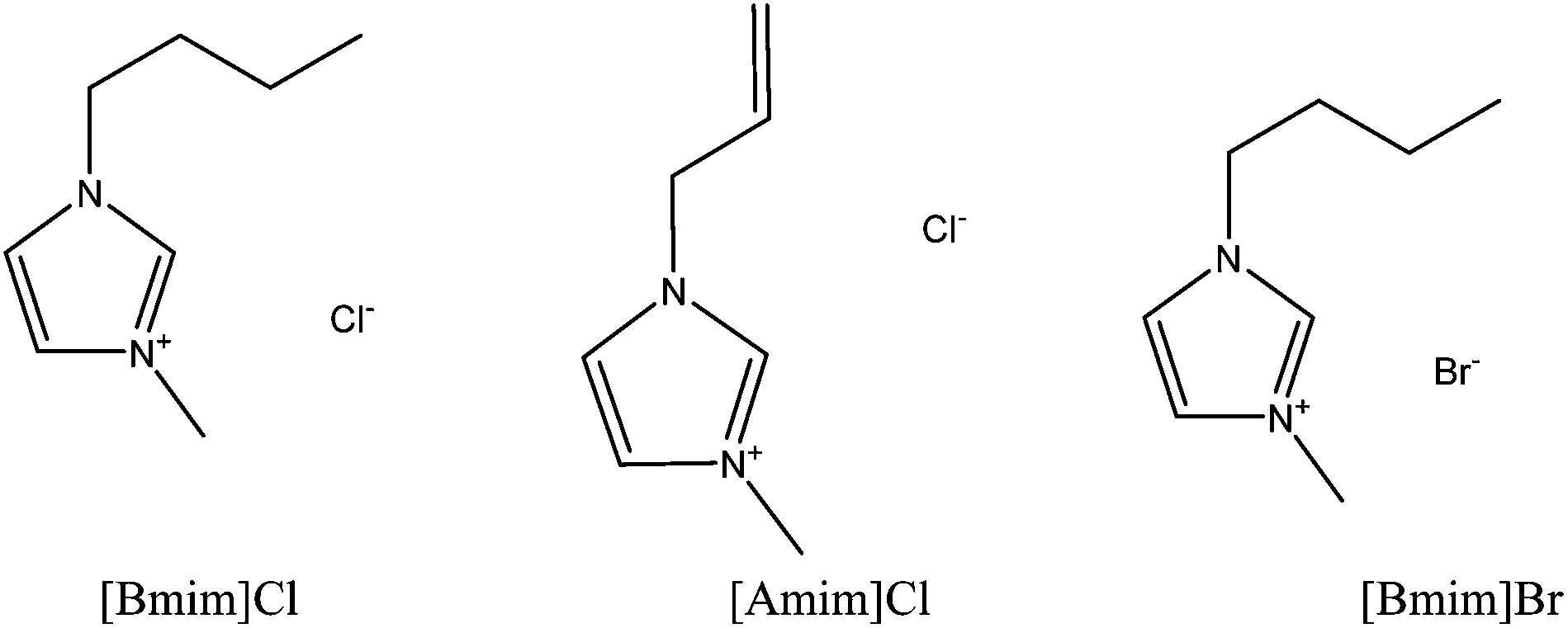 | ||
| Fig. 5 Schematic structures of three major ionic liquids that have been widely used for the dissolution of keratin fibres. 1-Butyl-3-methylimidazolium chloride [Bmim]Cl, 1-allyl-3-methylimidazolium chloride [Amim]Cl, and 1-butyl-3-methylimidazolium bromide [Bmim]Br. | ||
| Material | Ionic liquids and additives | Conditions | Yield of keratin | Ref. | |||
|---|---|---|---|---|---|---|---|
| Temperature (°C) | Solid: liquid ratio |
Time | Solubility (wt%) | ||||
| Feathers | [Amim]Cl + 10 wt% Na2SO3 | 90 | 1:20 |
1 h | 4.8% | — | 62 |
| Feathers | [Bmim]Cl + 10 wt% Na2SO3 | 90 | 1:20 |
1 h | 4.8% | — | 62 |
| Feathers | [Bmim]Br + 10 wt% Na2SO3 | 90 | 1:20 |
1 h | 4.2% | — | 62 |
| Feathers | [Bmim]NO3 + 10 wt% Na2SO3 | 90 | 1:20 |
1 h | 4.2% | — | 62 |
| Feathers | [Hmim]CF3SO3 + 10 wt% Na2SO3 | 90 | 1:20 |
1 h | 0.2% | — | 62 |
| Feathers | [Bmim]HSO4 + 10 wt% Na2SO3 | 90 | 1:20 |
1 h | 4.1% | — | 62 |
| Wool | [Bmim]Br | 130 | — | 10 h | 2% | — | 20 |
| Wool | [Bmim]Cl | 100 | — | 10 h | 4% | — | 20 |
| Wool | [Bmim]Cl | 130 | — | 10 h | 11% | — | 20 |
| Wool | [Amim]Cl | 130 | — | 10 h | 8% | — | 20 |
| Wool | [Bmim]BF4 | 130 | — | 24 h | Insoluble | — | 20 |
| Wool | [Bmim]PF6 | 130 | — | 24 h | Insoluble | — | 20 |
| Wool | [Amim]Cl | 130 | — | 640 min | 21% | — | 61 |
| Wool | [Bmim]Cl | 130 | — | 535 min | 15% | — | 61 |
| Wool | [Bmim]Cl | 120 | 1:6 |
30 min | — | 57% | 21 |
| Wool | [Bmim]Cl | 150 | 1:6 |
30 min | — | 35% | 21 |
| Wool | [Bmim]Cl | 180 | 1:6 |
30 min | — | 18% | 21 |
| Feathers | [Bmim]Cl | 130 | 1:2 |
10 h | 50% | 60% | 68 |
| Feathers | [Amim]Cl | 130 | 1:2 |
10 h | 50% | 60% | 68 |
| Feathers | Choline thioglycolate | 130 | 1:2 |
10 h | 45% | 55% | 68 |
| Feathers | [Bmim]Cl | 100 | — | 48 h | 23% | — | 64 |
| Wool | [Amim][dca] | 130 | — | — | 23% | — | 71 |
| Wool | [Bmim]Cl | 130 | — | — | 12% | — | 71 |
| Wool | [Amim]Cl | 130 | — | — | 10% | — | 71 |
| Wool | Choline thioglycolate | 130 | — | — | 11% | — | 71 |
| Feathers | [HOEMIm][NTf2] + 1.0 g NaHSO3 | 80 | 1:45 |
4 h | — | 21.75% | 67 |
Formation of R-SSO3Na and breakage of disulfite bonds:
| RSSR′ + SO32− → RSSO3− → R′S− | (6) |
Ji et al.62 reported a keratin yield of 75.1% under an extraction time of 1 hour at 90 °C with a liquid/feather weight ratio of 20 with 10% Na2SO3. A processing time of 1 h at 90 °C temperature used in this study was much lower than the recommended time by Xie et al.20 (10 h at 130 °C) where only 4% of wool was solubilized in [Bmim]Cl after 10 hours. This large difference may be related to the addition of Na2SO3 in the study by Ji et al.62 or the looser structure of feathers compared to wool. No amino acid composition or protein profiles were reported in the above studies, which make the comparison difficult between both the methods. A study on the effect of temperature of the solution was carried out by Ghosh et al.21 to investigate the disordered structure of keratin regenerated from IL extraction and to elucidate more information on the mechanism of extraction. Different temperatures of 120 °C, 150 °C and 180 °C were studied for the dissolution of wool in [Bmim]Cl with a keratinous material/liquid ratio of 1:6 for 30 minutes. A maximum yield of 57% was obtained at 120 °C whereas at 150 °C and 180 °C, a yield of 35 and 18% (wt%) was achieved, respectively. The authors reported that the tested wool and feather contained more than 90 and 70% of protein, respectively. However, these low yields can be due to the loss of the keratin water-soluble protein that was not precipitated and remained in the IL solution.21 This is actually one of the main drawbacks of this method that affects the rate of regeneration and the quality of the final product due to the loss of valuable water soluble amino acids such as cysteine.18,63 The extraction of keratin using ILs needs to be performed under an inert atmosphere (e.g. N2) due to the hygroscopic nature of ionic liquids,20,64 which might need expensive specialized equipment.
20,61 until it dissolved completely. In this way, it is problematic to treat a large amount of wool in a short time. Furthermore, there is a limitation on the maximum concentration of fibre that can be solved in the solution. Xie et al.20 dissolved up to 11% of wool in [Bmim]Cl at 130 °C during a 10-hour process, while a maximum concentration of 15% was obtained by Li and Wang61 using the same IL over a 9 h processing time. The same group used [Amim]Cl and found that about 21% of wool was dissolved in 10.7 h at 130 °C.61 Higher dissolution rates were reported for feathers. Idris et al.68 reported 50% of dissolution using both IL solutions of [Bmim]Cl and [Amim]Cl at 130 °C, but the processing time was not reported. This higher dissolution rate could be due to the loose structure of feathers compared to wool. In parallel to the IL salt, Idris et al.68 synthesized a series of thioglycolate ionic liquids to evaluate their efficacy in wool solubilisation. Choline thioglycolate was able to solubilize feathers at a slightly lower concentration compared to ILs. Reduction and partial cleavage of the disulfite bonds can be the mechanism involved in the dissolution of wool using choline thioglycolate.69 No dissolution was observed using [bis(2-ethylhexyl)ammonium][thioglycolate], which could be related to its high viscosity and long cationic chain that hinder the penetration of liquids into the keratin network. A thermogravimetric analysis (TGA) showed that the regenerated keratin had slightly lower thermal stability compared to natural wool,61 which could be due to the high crystallinity of natural wool along with its higher molecular weight compared to the regenerated keratin. These findings were in accordance with the DSC results of Ghosh et al.21 who concluded that the high temperature dissolution process resulted in the disruption of the secondary structure and therefore, denaturation and transition of the regenerated keratin. As a result, the regenerated keratin shifted to a lower temperature in the thermogram compared to natural wool.18,70 Contradictory to these findings, Xie et al.20 reported that the thermal stability of the regenerated keratin using ILs was slightly superior to the natural wool. Different sources of keratin used in this study may explain the conflicting results. Different arrangements of the wool structure have been reported for different wools. For example, Merino wool has a bilateral arrangement, Lincoln's wool shows a cylindrical arrangement and Mohair consists of predominantly the ortho-cortex.70 These differences in the wool structure could result in different thermal properties of the final keratin product. Using ILs, wool cannot dissolute at a temperature lower than 90 °C. Idris et al.68 considered 65 °C for dissolution and only partial dissolution was achieved, while, similar to other studies, at 130 °C, a complete dissolution was achieved. High temperature is required for the unfolding of the protein structure and to open up the structure for ILs to react. Additionally, a higher temperature accelerates the physical and chemical changes that occur during the process and therefore, enhances the dissolution of keratin.67 The required temperature for wool and feather dissolution can be different. Wang and Cao67 observed that when the temperature was over 90 °C, the yield of keratin from feathers decreased markedly which could be due to the scission of the peptide bond at the higher temperature. Therefore, 80 °C was reported in their study for optimum yield. A hydrophobic ionic liquid 1-hydroxyethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide ([HOEMIm][NTf2]) was used by Wang and Cao67 to dissolve chicken feathers to obtain keratin. The authors investigated the addition of NaHSO3 at different mass ratios to feathers (1:0, 1:0.3, 1:0.5, 1:0.75, 1:1, 1:1.25 and 1:1.5) to reduce the disulfite bonds. It was clear that more disulfite bonds of feathers’ keratin were broken with a higher mass ratio of NaHSO3 to the feather. In this study, a maximum yield of 21.5% was obtained after 4 h extraction time using a feather to the liquid mass ratio of 1:40. The obtained keratin, in contrast to the majority of other related reports, was soluble in water and the immiscibility of [HOEMIm][NTf2] in water helped to separate keratin easily from the IL. The authors were able to reuse and recycle the IL for five cycles without the loss of extraction power. This ability to separate the IL from the final solution could be a major advantage of this IL compared to other reported ILs for the dissolution of keratin.67 Regarding the MW distribution of the regenerated keratin from the IL, Idris et al.68,71 did not observe a clear distinction in the molecular weight distribution of the protein bands, while the majority of the proteins were between 10 and 40 kDa. These results are in agreement with those reported by Ghosh et al.21 who suggested partial fragmentation of the proteins as a result of extraction using ILs. Therefore, the hydrolysis of the proteins results in a mixture of different proteins with heterogeneous molecular weights.21,71
:20 ratio and obtained a film that had enhanced thermal stability and mechanical properties compared to the individual component, due to the intermolecular hydrogen bonding between the components.72 In another study by Tran and Mututuvari,73 keratin, cellulose and chitosan were blended together using [BMIM]Cl to produce a film for drug release. The result showed that the incorporation of keratin into the mixture slowed down the drug release regardless of the concentration of chitosan and cellulose. Therefore, the release rate can be controlled by using various amounts of keratin in the blend. These properties might be related to the compact and denser structure of keratin compared to two other polymers.73
2.5 Oxidation methods
Oxidation methods have been reported in the literature for decades with the early work by Earland et al.22 describing the extraction of keratin with 2% peracetic acid for 30 h followed by mild ammonia (0.2 N) treatment and finally a precipitation step using HCl. Buchanan et al.15 used 2% performic acid and obtained 6.6% of keratin. The oxidation method has been relatively the same over the years and it was used for the extraction of keratin from wool and hair. The wool does not behave as a chemically homogeneous material during the extraction with this method.22 Therefore, the bulk of the wool is solubilized, but an insolubilized keratin residue was found in all studies, which is believed to be mainly β-keratin. The keratin solution obtained in this method is mainly α-keratin, which is obtained from the cortex and has a crystalline structure before the extraction process. The insoluble keratin residue forms a thick jelly-like material and mainly consists of β-keratin, which is primarily found in the hair cuticle. It has been reported that the folded α form of keratin is more soluble than the extended β form.22 Treatment with peracetic acid partially oxidizes the disulfite linkages of keratin and converts them to hydrophilic pendant sulfonic groups on the side chains of the cysteine amino acid that can complex with water. The Cys–Cys sequence occurs frequently in keratin, and as a result of disulfite breakage, the dipeptide cysteinylcysteine is released from wool15 and produces a protein with cysteic acid (–CH2SO3H) residues (Fig. 6). Buchanan et al.15 reported keratin which was rich in cysteine and cysteic acid and contained 30% peptides with low MWs. In a study by Weston,76 wool was treated for 30 h with 2% peracetic acid and he found that disulfite bonds were oxidized to sulfonate groups. These results were later confirmed by infrared analysis that was reported by Strasheim and Buijs.77 The latter study used mild oxidation conditions (2% peracetic acid for only 5 min) and reported the presence of cystine monoxide and dioxide in addition to sulfonate groups. Sulfonate was also reported as a major oxidation product when wool or keratin-rich material was treated with hydrogen peroxide, sodium hypochlorite, peracetic acid or potassium permanganate at various pH values.78 Therefore, the major reaction in all these oxidation processes was the transformation of disulfite to sulfonate.79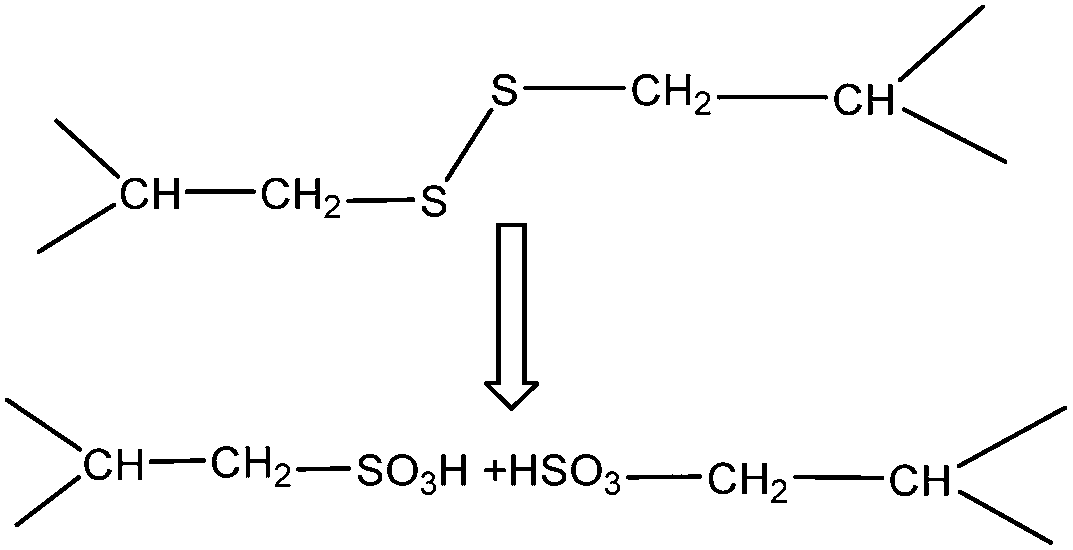 | ||
| Fig. 6 Oxidation of keratin results in the transformation of disulfite to sulfonate. | ||
:1 v/v) and observed low recovery for tyrosine (0.4%) and phenylalanine after oxidation, while other amino acids were essentially unaffected. In a patent by Blanchard et al.87 wool samples were treated with 32% peracetic acid at 4 °C for 24 h, then after vacuum drying the powder was re-suspended using 3 N ammonium hydroxide containing ammonium thioglycolate and the suspension was heated to 60 °C for 4 h. The inventors believed that in the first step disulfite linkages were partially oxidized to cysteic acid residues and remain as disulfite linkages, then in a second step, the remaining disulfite linkages are broken to produce cysteine residues in the protein structure. The keratin protein is believed to contain cysteic acid, cysteine and cysteine-thioglycolate disulfite residues.87
| Material | Pressure | Temperature (°C) | Time (min) | Sample properties | Ref. |
|---|---|---|---|---|---|
| Steam explosion | |||||
| Wool | 0.2–0.8 MPa | — | — | Scales on the fibre surface were damaged, sample moisture was regained, mechanical properties and the dissolving ability in caustic solution decreased as the explosion pressure increased | 99 |
| Wool | 0.2–0.6 MPa | 164.2 | 2–8 | Up to 80% digestion yield, 50% reduction in cystine content | 98 |
| Wool | — | 220 | 10 | The decrease of disulfite bonds, 62.36% of the dry solid, 18.66% of proteins dissolved in the supernatant, 1.12% of sediment, the presence of the wool structure in the treated sample | 18 |
| Feathers | 0–2.0 MPa | 50 | <3 | 93.2% pepsin digestibility, the wool structure disrupted completely in the treated sample | 63 |
| Feathers | 1.4–2.0 MPa | 60 | 0.5–5 | Extraction rate of feathers of 65.78% and a yield of keratin of 42.78% | 38 |
| Feathers | 0.5–2.5 MPa | — | 1 | 91% digestibility | 1 |
| Feathers | 2.2 MPa | 220 | 120 | Arginine diminishes | 102 |
| Microwave irradiation | |||||
| Wool | Microwave irradiation | 150–180 | Up to 60 | 60% extraction yield | 16 |
| Wool | Microwave superheated water | 180 | 30 | 31% extraction yield | 100 and 241 |
| Feathers | Microwave 1200 W | 160–200 | 20 | 71.83% yield | 103 |
| Material | Processing parameters | Properties of the hydrolysed product | Ref. |
|---|---|---|---|
| Wool | 30 h, 2% peracetic acid | Sulfonic acid formation | 76 |
| Wool | 5 min, 2% peracetic acid | Cystine monoxide and dioxide residues | 77 |
| Wool | 24% peracetic acid–10% H2O2 | Sulfonate and cystine monoxide | 77 |
| Wool | H2O2 (3.5 N), pH 11.5, 9.5, 4.5 | Formation of either sulfonate or sulfonic acid groups, the highest oxidation at pH 11.5, no oxidation at pH 4.5 | 78 |
| Wool | 2% peracetic acid for 10 h at 37 °C on a 150 rpm orbital shaker | 82 | |
| Wool | 24 h at 25 °C with 100 ml of 1.6% (w/v) peracetic acid, | 80 | |
| Wool | Performic acid [100-volume H2O2/98% formic acid (1:39, v/v)] for 18 h at 4 °C |
Peracetic acid oxidizes the disulfite bond (–S–S–) of the cystine dimeric amino acid into two cysteic acids containing the sulfonic acid (eSO3H) functional group >99% protein | 15 |
| Hair | 2% peracetic acid, boiled for 2 hours | 242 | |
| Hair | 30 grams of hair with 500 mL of 32% peracetic acid at 4 °C for 24 hours. | Partially oxidise the naturally occurring disulfite linkages to produce a protein with cysteic acid (–CH2SO3H) residues, and remaining disulfite linkages | 87 |
| Wool | Or 50% aqueous n-propanol for 4 h at 70 °C | Rich in glycine, tyrosine, phenylalanine, and serine, moderately rich in half-cystine, and low in lysine, histidine, methionine, and isoleucine, the molecular weight of approximately 6000–10000 | 243 |
| Wool | 98–100% formic acid at 20 °C for 1 h | Rich in glycine, tyrosine, phenylalanine, and serine with a large amount of glutamic acid and virtually no half-cystine, molecular weight of approximately 6000–10000 |
243 |
2.6 Supercritical water and steam explosion
| Pe = (ΔHs + ΔH1 + ΔHm)/(t × V) |
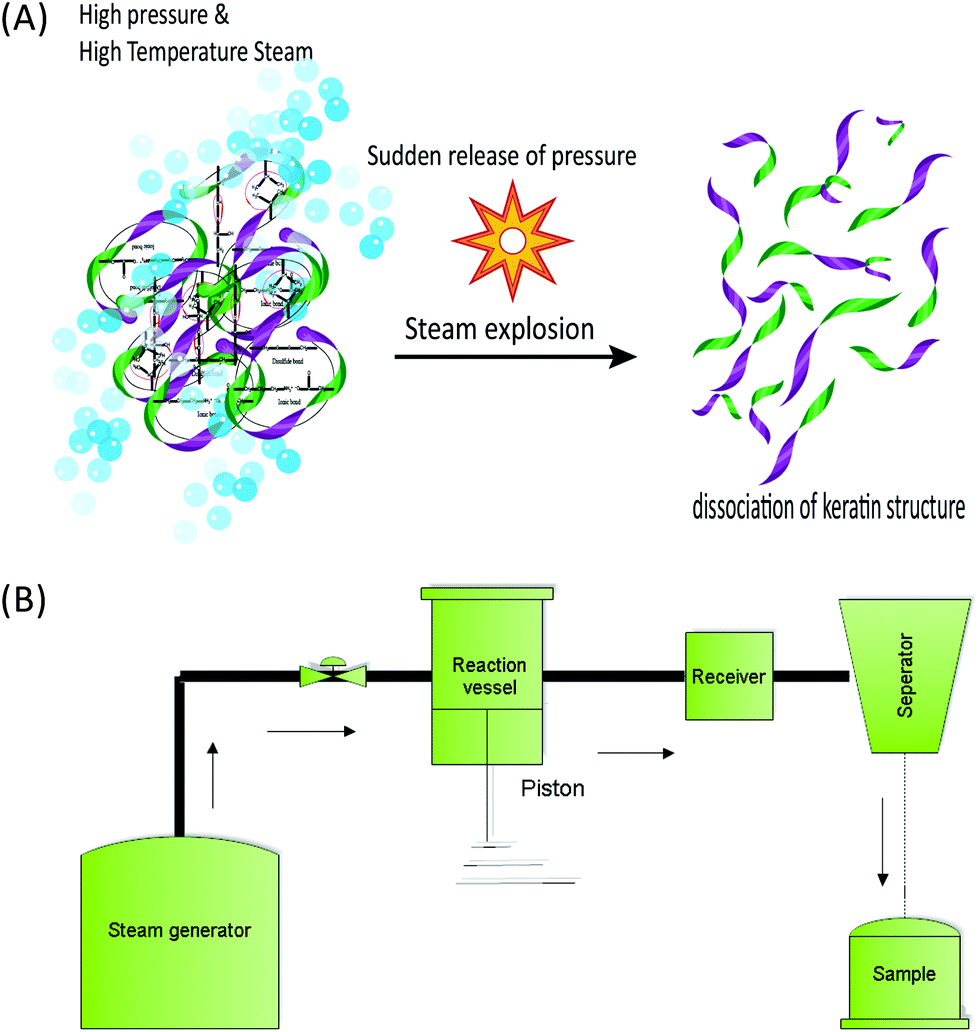 | ||
| Fig. 7 Schematic drawing (A) and schematic diagram (B) of the steam flash explosion system for the extraction of keratin from the fibre. | ||
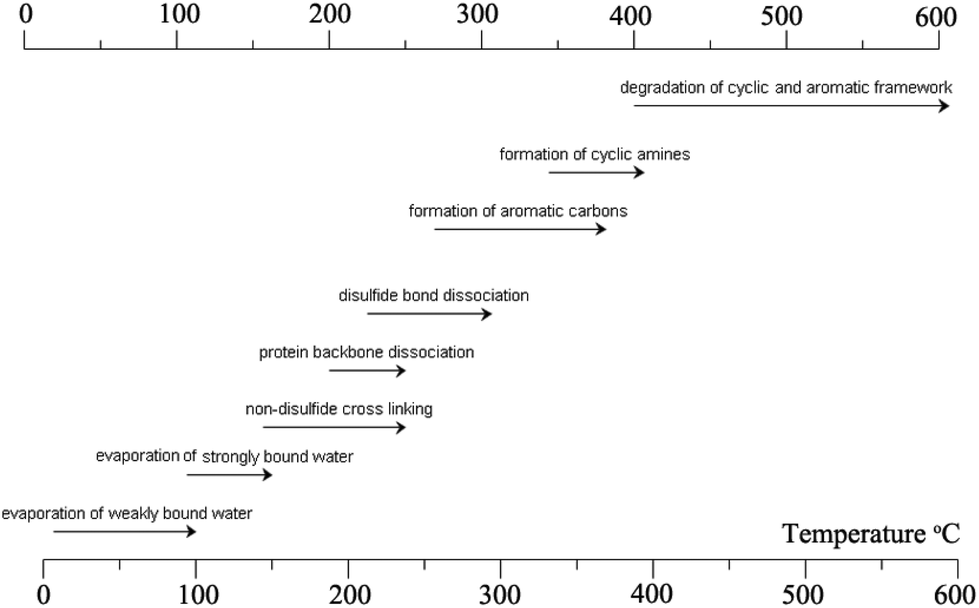 | ||
| Fig. 8 Major reactions that occur during the heat treatment of the keratin fibre. The image concept was adopted from ref. 278 and reproduced with permission from Elsevier (license no. 4118920599160). | ||
In this equation, ΔHs is the enthalpy drop of the steam and ΔH1 and ΔHm are the enthalpy drop of the liquid and water, respectively. t and V represent the duration and volume of the explosion, respectively, and the values of ΔH are measured from temperature changes before and after the reaction process, and time is normally neglected but it depends on the type and the design of the equipment and the pressure used.
:3 solid–liquid ratio) was treated with saturated steam at 220 °C for 10 min, and used a blow valve where the pressure was released and a dark yellow slurry was recovered. The process yield was 62.4% of a solid product, 18.7% for a water soluble fraction, and 1.1% of sediment. The authors reported that 17.9% of the initial wool mass was lost during the process, which can be due to the presence of non-proteinous materials in the wool and also the incomplete recovery of the process. Despite the intense processing conditions, wool fragments and the structure of the surface cuticle were still recognisable in scanning electron micrographs. The authors were able to produce pure keratin samples using centrifugation of the supernatant, similar to the study by Miyamoito et al.98 The amino acid analysis indicated that the cysteine content was very low and that this amino acid was destroyed during the strong heating process. A small cysteine residue was reported, which the authors suggested was related to the undamaged fraction of the wool and not to the extracted keratin. The protein profile using SDS-PAGE gel showed that bands related to high sulfur fractions of 67–43 kDa disappeared and the majority of proteins had low MWs in the broad range of 18–3 kDa without any recognisable band. This indicated that the chemical structure of keratin was strongly disintegrated during the high temperature and pressure treatment.18 This distribution of protein MWs was similar to that obtained by IL treatment (section 5). Similar to those reported for IL extraction, the thermal properties of SFE obtained keratin exhibited denaturation at low temperature compared to wool due to the low cysteine and reduced alpha helical structure.18 Zhao et al.63 argued that in CSE the use of valve blow mode suffers from a long processing time at a high temperature, which is not desirable for heat sensitive components such as cystine in wool and thus can lead to a decrease in the quality of the final protein product. The authors supported the use of the SFE system to extract keratin from wool. According to the authors, a temperature as low as 50 °C in a short processing time (<3 min) can produce enough energy to disrupt and unfold the compact structure of the fibrous protein. Additionally, the authors believed that the kinetic energy produced was the most important factor in the SFE systems compared to the conventional thermochemical reaction.63 The dissolution and pepsin digestibility of the keratin samples obtained from SFE increase with the increase of the reaction pressure. A maximum dissolution of 70% was found at the highest tested pressure of 2 MPa using phosphate buffer at pH 9 or 0.2% potassium hydroxide.63 Only 10% keratin was obtained under the same processing conditions using deionized water. Dissolution of 65% was obtained using phosphate buffer (pH 7.5) and 2% urea. Keratin obtained from SFE has a lower solubilisation rate in water compared to the chemical methods14,35 and the use of urea and high pH is required to have water soluble keratin or a higher dissolution rate. However, according to the authors, this method might be more eco-friendly due to the lower levels of chemicals required for solubilization compared to other chemical extraction methods. Zhao et al.63 also reported the pepsin digestibility of 93.2% of a keratin product that was obtained from feathers through SFE under a pressure of 2 MPa, which is higher than 80% as was previously reported by Miyamoito et al.98 for wool. According to the authors, the structure of the feather was changed from fibres to amorphous and the central axis of the feather was destroyed and was not identifiable compared with the original structure of the feather. These morphological observations and the fast processing time of less than a minute indicate a better performance to be obtained by the piston and cylinder method compared to the conventional blow valve method.
2.7 Microwave assisted extraction
The use of microwave irradiation has been used in a similar way for keratin extraction (Table 4). Zoccola et al.16 applied microwave radiation with variable power in the range of 150–570 W for up to 7 minutes and at temperatures of up to 180 °C, and the authors reported a 60% extraction yield. The suggested role of microwave irradiation was only heating up the solution and the key benefit of the technique was perceived as lowering the processing time due to the homogeneous heat distribution and internal heat generation. Comparing the conventional steam processing for feathers, at 200 °C for 10 and 120 minutes reported by Tonin et al.18 and Yin et al.,102 respectively, the microwave method appeared to have the advantage of being faster. However, it is hard to justify the efficacy of microwave irradiation when it is compared with the SFE process that is faster (2 min) and can be operated at a lower temperature (50 °C).38The use of microwave radiation to assist the extraction of keratin causes a significant loss in cysteine, from 9.41 mol% in wool to about 0.5 mol% in the extracted keratin sample16 after 90 minutes of microwave treatment time.
A cost analysis for energy consumption of microwave irradiation compared to SFE might provide some insights into the potential commercialization of these technologies and any economic benefits they might have. Chen et al.103 suggested that microwave heating significantly decreased the required activation energy for the extraction of keratin compared with traditional heating methods, due to the non-uniform, and irregular heating found in traditional heating. It is worth mentioning that the exact role of electromagnetic radiation and its interaction with the wool matrix is still largely unknown.104 Hydrolysis of the ester groups due to microwave irradiation is one of the possible mechanisms suggested for the lower activation energy requirement in microwave processing; however, the exact reason might be very complicated due to the complex structure of keratin. The presence of electrostatic, hydrophobic, hydrogen bonds and disulfite bonds and also α-helix and β-sheet structures that folded in a fatty oil layer make it very hard to extrapolate the exact reaction responsible for better hydrolysis using microwave irradiation.
2.8 Microbial and enzymatic methods for the digestion and hydrolysis of keratin
Whole keratin cannot be extracted or isolated using microbial and enzymatic treatments as the degradation of the protein is normally encountered. Using these techniques keratin rich materials can be degraded and hydrolysed to peptides that may be useful in other biotechnological or food applications. Keratin-rich materials such as wool and feathers are good sources of nutrients such as carbon, nitrogen and sulfur for microorganisms. Therefore, this method provides a completely different approach for the utilization of waste keratin rich materials compared to other methods that have been discussed earlier in this review. The above methods were mainly aimed at the isolation of the keratin protein from the materials. The following section provides information about the application of microbial methods for the conversion of keratin rich materials to hydrolysed keratin.Enzymes as catalysts have several advantages over chemicals and so they are widely utilized in many industrial and biotechnological processes. Around 80% of the enzymes used worldwide are produced via microbial pathways,105 and approximately 65% of the industrial enzymes are used for hydrolysis reactions.106 Proteases are widely used in the food processing, animal hide processing and detergent industries for the hydrolysis of peptide bonds.107 In addition to being environmentally safe, enzymatic hydrolysis of keratin has lower energy consumption and relatively mild treatment conditions compared to chemical and hydrothermal methods.108
Keratinases are microbial proteases that can hydrolyse keratin and they are produced by certain microorganisms. Keratinases have a wide range of applications such as cleaning and treatment of obstruction in sewage systems,109 cleaning of wool,110 finishing treatment of textiles, and for mild and gentle removal of hair from hide in the leather industry without the need to apply strong chemicals that affect the mechanical properties of the leather.111 The hydrolysis of keratin by microbial enzymes is a green and environmentally safe method, which does not damage the protein backbone and preserves the functional properties of keratin under certain processing conditions.108,112,113 However, commercial microbial enzymes have not been widely used for this purpose due to the limited enzymes that can have high efficiency and can be used on diverse substrates.
Keratinophilic fungi are normally mesophilic, however, some of them can tolerate higher temperature and a low number of them has thermophilic properties.11,121 The ability of the microorganisms to degrade keratin is measured by amino groups, mass loss of the keratin substrate, amino acid profile, substrate alkalinisation, release of ammonia/peptides and excretion of sulfate or sulfhydryl groups.122,123 In a study by Korniłłowicz-Kowalska,124 65–85% mass loss of the substrate (feather) and solubilisation of 50% of peptides were observed after 21 days of culture using a mixture of 16 different strains of keratinophilic fungi. Keratinolytic microorganisms are environmentally friendly and the process is probably more cost effective than using chemicals, however, the long processing time required for the microorganism to degrade the keratin is a major problem considering other methods like microwave irradiation or using ILs which can extract keratin within a few hours. In addition, as discussed earlier, the hydrolysed keratin product obtained through using microorganisms is different from the intact protein chain that can be generated through other thermochemical methods.
Around 49% of the chicken feather is carbon, 14% is nitrogen and about 4% is sulfur, considering that the microbial cell contains around 6% nitrogen and up to 1% sulfur, therefore, during keratin degradation the excess amount of these will be wasted into the environment.11 However, the chemical composition of the final product and the ratios between the nitrogen and sulfur and carbon depend on the structure of the substrate keratin and type of microorganism. It has been reported that the chemical compositions of the nitrogen products released from fungi and Actinomycetes such as Streptomyces fradiae were similar, while the profile of the sulfur products was different. Korniłłowicz-Kowalska124 reported that up to 75% of nitrogen was converted to the ammonium form after 21 days when Chrysosporium was used on the feather substrate; however, up to 60% of nitrogen was in the gas form due to the alkalinisation of the substrate by the released ammonia. Similar properties were reported for the degradation of the wool using a bacterial source (Streptomyces fradiae), and up to 75% of nitrogen was converted to ammonia.122 Korniłłowicz-Kowalska124 also observed that only 20% of nitrogen in the feather substrate is converted to peptides and amino acids which were dominated by >10 kDa molecular weight proteins. The authors reported that 10–20% of the lysate protein was cysteine/cystine and 20–30% was serine. In another work by Kunert,125 no more than 20% of the hair substrate were released as peptides with a molecular weight higher than 10 kDa, using Microsporum gypseum (geophilic dermatophyte) while the released product was mainly predominant with 1–2 kDa molecular weight peptides. A similar pattern of protein fractions was also reported for the wool lysate using S. fradiae.122 On the other hand, more than 55% of the nitrogen content of feather keratin were released as free amino acids and oligopeptides during the degradation of feathers using the thermophilic bacterium Meiothermus ruber H328.126 Nam et al. suggested that free amino acids were dominant in the protein lysate from feather degradation using the thermophilic anaerobic bacterium Fervidobacterium islandicum AW-1 and similar to Korniłłowicz-Kowalska, cysteine, serine, alanine and proline were the dominant amino acids.127 Keratinolytic microorganisms also produce some sulfur-containing products during keratin hydrolysis.124 Depending on the genus of fungi, up to 50% sulfur content of the raw material can be converted to a sulfite product.124 Noval and Nickerson (1959)122 reported that 25% of the cysteine content in the wool substrate was converted to sulfhydryl compounds during degradation using S. fradiae while up to 75% of cysteine conversion to sulfhydryl compounds was reported by Kunert125 using the same bacteria during wool degradation.125 However, it has been shown that the concentration and chemical composition of the resultant sulfur compounds differed from the actions of fungi and actinomycetes, and the fungi species. Additionally aerobic or anaerobic strains can behave differently in producing sulfur compounds from the keratin substrate. For example, Bacillus licheniformis produced higher sulfhydryl compounds when cultured under aerobic conditions compared to the anaerobic culture.128 The alkaline serine protease under the optimized conditions (2.6% v/v) was evaluated by Eslahi et al.129 for the degradation of wool and feathers. A keratin yield of 21.25% and 17.73% was obtained for wool and the feather, respectively, at 55 °C for 4 h. The authors also reported that the addition of an anionic surfactant enhanced the extraction regardless of the substrate type.129 The molecular weight of the hydrolysed samples was preserved and had a similar pattern to the original keratin.129 The amino acid content and availability of the hydrolysed sample were improved using Kocuria rosea on feather keratin.130 Additionally, using Kocuria rosea, carotenoid pigments (68 ppm) were produced during the microbial fermentation, which can enrich the feather meal and its application for animal feed.130 Various microorganisms; including Gram negative, Gram positive and fungi, have shown keratin degradation ability. Table 6 shows some of the important bacterial keratinases.
| Bacterial isolate(s)/enzyme | Substrate | Maximum degradation conditions | Ref. |
|---|---|---|---|
| Gram-negative bacteria | |||
| Chryseobacterium sp. P1-3 | Feather meal | Hydrolysed feather meal within 2 days and possesses a high level of keratinase activity (98 U mL−1). | 244 |
| Chryseobacterium sp. strain kr6 | Feathers | Complete degradation, optimum growth at pH 8.0 at 30 °C | 245 |
| Vibrio sp. strain kr2 | Feathers | Optimum at pH 6.0 and 30 °C | 246 |
| The hydrolysate was rich in serine, leucine, alanine and glutamate residues and contains minor amounts of histidine and methionine | |||
| Vibrio sp. kr2 | Feathers | pH ranging from 6.0 to 8.0, at 30 °C medium containing up to 60 g L−1 raw feathers, amounts of soluble protein, reaching maximum values around 2.5 g L−1 | 247 |
| Lysobacter NCIMB 9497 | Feathers | Optimum activity occurred at 50 °C, pH 7.5 | 248 |
| Stenotrophomonas maltophilia BBE11-1 | Feathers | pHs 7–11 and temperatures 40–50 °C, two days | 249 |
| Stenotrophomonas maltophilia R13 | Feathers | pH 7.0 at 30 °C, the | 250 |
| maximum yield of the enzyme was 82.3 ± 1.0 U ml−1, | |||
| Stenotrophomonas maltophilia L1 | Feathers | pH 7.8 at 40 °C | 251 |
| Gram-positive bacteria | |||
| Bacillus cereus Wu2 | Feathers | 30 °C and pH 7.0, B. cereus possessed disulfite reductase activity along with keratinolytic activity lysine, methionine and threonine | 252 |
| Bacillus subtilis | Feathers | 40 °C and pH 11–7 days | 253 |
| Bacillus sp. MTS | Capable of degrading whole chicken feathers | Bacteria produced extracellular alkaline keratinase and disulfite reductase, for keratinase at pH 8–12, and for disulfite reductase at pH 8–10. The optimum temperature for the extracellular keratinase was 40–70 °C, for disulfite reductase it was 35 °C. | 254 |
| Bacillus subtilis DB 100 (p5.2) | 37 °C, 700 rpm agitation, released soluble proteins 0.7 mg mL−1 | Amino acids such as phenylalanine, tyrosine, valine, leucine, isoleucine, serine, alanine, glycine and threonine | 255 |
| Kocuria rosea | Feather degradation up to 51% in 72 h was obtained with a conversion yield in the biomass of 0.32 g g−1 | At 40 °C, a specific growth rate of 0.17 h−1 was attained in basal medium with feathers as a fermentation substrate. Under these conditions, after 36 h of incubation, biomass and caseinolytic activity reached 3.2 g l−1 and 0.15 U ml−1, respectively | 256 |
| Kocuria rosea keratinolytic capacity | Aerobically on submerged feathers | Pepsin digestibility of the fermented product (88%), improved the content of amino acids lysine (3.46%), histidine (0.94%) and methionine (0.69%). | 130 |
| Bacillus pumilus | Bovine hair | pH 8 and 35 °C. nearly 60% of hair was solubilized after 16 days, and the maximum keratinase production was 54–57 kU ml−1, after 9 days | 257 |
| Bacillus safensis LAU 13 | Feathers | pH 7.5 and 40 °C, degraded whole chicken feathers after 6 days at 30 ± 2 °C, optimum activity at 50 °C and pH 8.0 | 258 |
| Bacillus amyloliquefaciens 6B | Feathers | pH 8.0. and 50 °C completely degrade native feathers in the shortest time period (24 h) | 259 |
| Saprophytic & parasitic fungi | |||
| Hrysosporium, Malbranchea, Scopulariopsis, Microascus, and Gliocladium | Human hair | All the test fungi could grow on keratin (human hair) and degrade it. | 260 |
| Maximum cysteine was released in the glucose supplemented medium by Chrysosporium tropicum (28 g ml−1). Maximum release of protein was by Scopulariopsis brevicaulis (65 g ml−1) | |||
| Chrysosporium species | Hair | 28 °C for 14 days | 261 |
| S. brevicaulis, Trichophyton mentagrophytes | Feathers | The highest keratinase activity was estimated by S. brevicaulis (3.2 kU mL−1) and Trichophyton mentagrophytes (2.7 kU mL−1) in the culture medium with chicken feathers and shows (79% and 72.2% of degrading ability, respectively) | 262 |
| Alternaria tenuissima | Feather powder | The highest keratinolytic activities were produced after 4–6 days of cultivation under submerged conditions: 53.8 ± 6.1 U mL−1 (Alternaria tenuissima), 51.2 ± 5.4 U mL−1 (Acremonium hyalinulum), 55.4 ± 5.2 U mL−1 (Curvularia brachyspora), and 62.8 ± 4.8 U mL−1 (Beauveria bassiana) | 263 |
| Acremonium hyalinulum | |||
| Doratomyces microsporus | Feathers | pH 8–9 and 50 °C | 264 |
| Aspergillus fumigatus | Feathers | pH 9 and 45 °C | 265 |
| A. niger 3T5B8 | Feathers | Keratinase activity (172.7 U ml−1) after seven days at pH 5.0 | 266 |
| Trichoderma atroviride strain F6 | Feathers | 5 days with rotary shaking (30 °C, 150 rev. min−1) pH 8·0–9·0 at 50 °C | 267 |
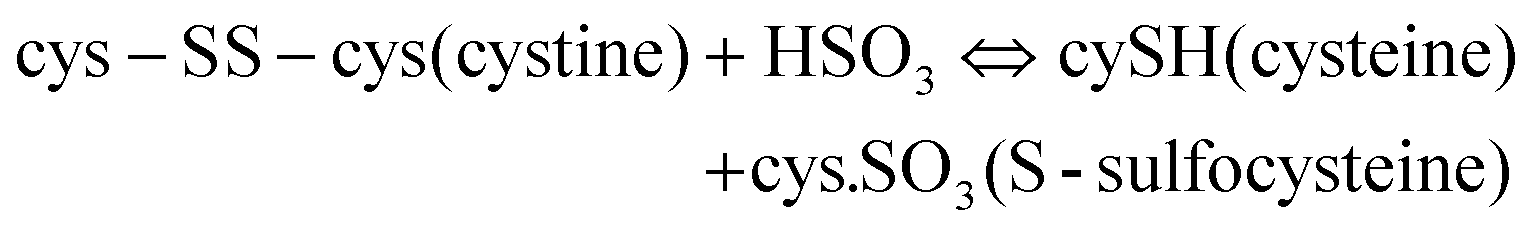
In addition to the production of the reducing agent sulfite, a dermatophyte also releases various endo-proteases like metalloproteases, therefore, these proteases can affect the structure of the protein since the denatured protein generated from the sulfitolysis process is accessible and susceptible to digestion by the proteases produced by fungi. Léchenne et al.132 in 2007 and Monod et al.133 in 2008 also proposed similar processes for the keratinolysis mechanism of Aspergillus fumigatus (AfuSSU1), dermatophytes Trichophyton rubrum and Arthroderma benhamiae. A significant amount of cysteic acid was detected in the reaction products, which was probably due to the air oxidation of sulfur amino acids.125 In contrast to these, Ruffin et al.134 hypothesized that sulfitolysis and proteolysis occur at the same time during the keratinolysis. For a detailed discussion on the mechanism of keratinolysis the reader should refer to the excellent review of Kowalska and Bohacz.11 However, sulfitolysis is probably a major step in the digestion of keratin that precedes the action of all proteases, and the efficiency of the hydrolysis process can be evaluated by measuring the enzyme activity, the concentration of the released thiol groups and soluble proteins, and weight loss.
3 Biomedical applications of keratin
Keratin based biomaterials have been widely produced and used in various biomedical applications. For example, keratin has the ability to function as a synthetic extracellular matrix (ECM) due to its biodegradability, biocompatibility and ability to create fibronectin-like cell binding domains that facilitate cell adhesion.135,136 It also has biological activities that facilitate and support the proliferation of cells. Moreover, keratin has an amino acid structure that can be fine-tuned and modified depending on the desired function. For example, it can be used to bind hydrophobic and hydrophilic therapeutic agents or adjust the degradation rate of the matrix.137,138 During the last decade, several mild and gentle techniques of keratin extraction from keratinous materials have been reported that offer the possibility of isolating different keratin fractions suitable for a broad spectrum of functions and applications. As a result, numerous studies have evaluated keratin for biomedical applications, such as bone tissue engineering,139,140 ocular regeneration,141 wound healing,142,143 nerve regeneration,144,145 skin replacement146 and controlled drug delivery.147,148 Products generated from keratin can generally be categorised as (1) films; (2) hydrogels; (3) scaffolds and composites (Fig. 9).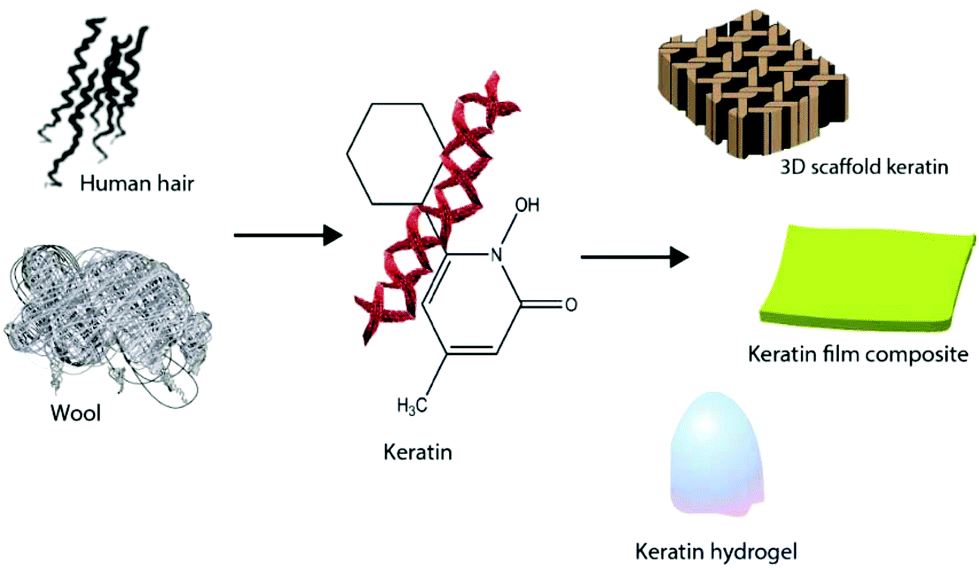 | ||
| Fig. 9 Different products such as keratin films, hydrogels, and composites can be generated from keratin. | ||
As a natural material, keratin has some limitations such as being brittle with poor mechanical and processing properties, therefore, the addition of plasticizers and cross linkers and incorporation of other synthetic or natural polymers/calcium phosphate particles into keratin are implemented to address these shortcomings.149–151
3.1 Keratin films and fibres
There are a number of methods for the production of keratin films (Table 7) such as solvent casting,141,152 thermal pressing2 and compression moulding,153 electrospinning147 and layer by layer (LbL) deposition.154 Given the interesting properties of electrospinning and LbL techniques, these methods are discussed in detail in section 3.1.4.| Film | Process conditions and composition | Results | Application and properties | Ref. |
|---|---|---|---|---|
| Wool cortical cells/chitosan | 30 wt% cortical cells | 29.6 ± 2.9 MPa for ultimate strength | Film with all improved mechanical properties compared to pure chitosan films | 167 |
| 5.6 ± 0.3% for ultimate elongation | ||||
| 35.3 ± 1.4 MPa Young's modulus | ||||
| Aqueous keratin dialysate with an alkaline keratin dialysate | 1% glycerol added as a softening agent. 90/10 (aqueous/alkaline keratin dialysate at a ratio of 90:10) cured at 100 °C for 2 h |
Facilitates corneal epithelial wound healing in vitro | 157 | |
| Photoactive keratin films | The film doped with different amounts of methylene blue | 99.9% killing rate against S. aureus upon irradiation with visible light | Tissue engineering, wound healing, antimicrobial photodynamic activity upon irradiation with visible light support for photodynamic therapy treatment | 152 |
| Keratin film cross linked by transglutaminase (TG) | Treatment with TGase (30 U g−1 keratin) for 18 h at 40 °C | The tensile strength of the film increased from 5.18 MPa to 6.22 MPa and decreased the elongation at break from 83.47% to 72.12 | Films with improved stability in PBS and in artificial gastric juice. Films with a lower drug release rate | 175 |
| Keratin film by compression molding of the S-sulfo keratin powder | The keratin powder mixed with water/ethanol (1:9) solution in a ratio of 1:1 (w/w). Moulding temperature up to 120 °C |
Good water tolerability, that is, it scarcely swelled in an acidic and neutral aqueous solution and fibroblast cell biocompatibility | The mechanical properties of the films can be modulated by controlling the moulding temperature and water content | 19 |
| Keratin/gelatin film (copolymerization of PHEMA on to keratin) | 10% keratin + 10% gelatin + 1 ml ethylene glycol + 0.35 ml glutaraldehyde | Keratin–gelatin–PHEMA film exhibited good mechanical properties and water absorption properties | Wound dressing materials | 164 |
| Keratin–chitosan film | 10–30% chitosan, 20% glycerol in 75% acetic acid | Ultimate strength: 27–34 MPa, ultimate elongation: 4–9% | Contact lens material | 179 |
| Keratin film | Shindai keratin + glycerol dried in a ventilated oven at 50 °C for 24 h | Good mechanical properties films provided a continuous release of loaded RB for up to 12 h. | Drug release (rhodamine B) | 147 |
| Max. ultimate strength: 7.56 MPa | ||||
| Max. elongation: 121.52% | ||||
| Max. Young's modulus: 27.61 MPa | ||||
| Keratin film | • Mixing the keratin dialysate with portions of the alkaline keratin dialysate at the ratios of 100, 90/10, 80/20, 70/30 and 50/50, respectively | Keratin films with the alkaline dialysate ≥30% were too fragile. | Ocular surface reconstruction | 141 |
| • Glycerol 1–3% | Max. ultimate strength (MPa): around 17 MPa for 100% keratin | |||
| • Cast on hydrophobic coated PET | Max. ultimate strength (MPa) wet: around 5 MPa | |||
| • Dry overnight in air | Max. E-modulus: around 350 MPa | |||
| Max. E-modulus wet: around 21 MPa | ||||
| Max. water absorption: 450% for 50/50 | ||||
| Keratin–chitosan | 250 mg of chitosan in 100 ml of 75% acetic acid | Max. swelling of 126% at the K:C ratio of 3:1 |
166 | |
| 100 mg of protein to give 75% acetic acid solution, | Ultimate strength: 27–34 MPa | |||
| chitosan 10–30% | Ultimate elongation: 4–9% | |||
| Glycerol 20% | ||||
| Keratin–HA films | Glycerol, 40% HA, 6% protein | Treated with ammonium thioglycolate, porosity: 63% | Full integration into the bone by 12 weeks | 191 |
| Kerateine disks | 5% kerateine stock solution into a 96 well tissue culture | Oxygen in the air was sufficient to catalyse the oxidative crosslinking of cystine to cysteine in these materials | Excellent compatibility with biological systems | 219 |
| PCL–keratin nanofiber | 10% PCL, 10% keratin in the ratios of 90:10, 80:20, 70:30, and 60:40 |
Max. Young's modulus (80:20): 5 (MPa) |
168 | |
| Max. breaking strength (100:0): 3 (MPa) |
||||
| Dried at 40 °C for 24 h | 1% (w/w) glycerol | Human nail plate model, especially for hydrophilic substances | 158 | |
| Keratin cell plate coating | Cell culture plates were coated using 0.03–1.0 mg per well | Keratin coating supports the attachment and proliferation of most cell types with advantages over the traditional polystyrene | Substrates for cell culture and tissue engineering | 155 |
| Film | • Mixing the aqueous keratin dialysate with portions of the alkaline keratin dialysate (at the ratios of 100, 90/10 and 80/20, respectively; e.g., a 90/10. | Low implant degradation which might be beneficial for certain applications such as transplantation of epithelial cell sheets | Good corneal biocompatibility of keratin films with minor host tissue reaction and preservation of corneal transparency | 141 |
| • 1% glycerol | ||||
| • Films were cured at 110 °C for 2 h | ||||
| Film | EGDE or GDE (7.5–30 mg) keratin aqueous solution containing 100 mg of protein | — | The crosslinked films showed excellent waterproof characteristics | 165 |
| Keratin film | Glycerol used as a plasticizer, the sample was sandwiched between aluminium foil and pressed into films at 160 °C for 2 to 8 min | — | Good physical properties | 2 |
| Keratin films | Plasticized with polyethylene glycol (PEG) with different molar weights (400, 1500, 4000, 6000), at the concentrations of 0.02, 0.05, 0.10, 0.20, and 0.30 g PEG = g keratin. | Films obtained with PEG400 were more hydrophilic than films obtained with higher molecular weight PEGs. | PEG causes an increase in the water vapour pressure of chicken feather keratin films | 178 |
| Keratin polyamide 6 film | Polyamide 6, electrospinning | — | Keratin improves the miscibility and hydrophilicity of the film | 183 |
| Keratin/ceramide | Unstable in organic solvents | Simple skin model | 159 |
Keratin films have been used for tissue engineering applications, with solvent casting becoming an attractive and common method for their production.155 Keratin film coated polystyrene cell culture plates supported and improved cell growth better than uncoated cell culture plates.155 Additionally, cell culture plates coated by normal drying of keratin solution on the surface performed better when compared to the precipitation of the protein on the plate surfaces. This enhanced performance can be due to the uniform distribution of the film as a result of the solvent casting method. Moreover, the authors suggested that keratin coating might be even superior to other tissue culture plastic coatings such as collagen and fibronectin, although the study lacks experimental data to support it.155 Reichl et al.156 proposed the keratin film for ocular surface reconstruction as an alternative for the human amniotic membrane (AM). The keratin film was cytocompatible toward the tested corneal epithelial cells and was more transparent, with better mechanical properties, when compared to AM. In a following study by the same researchers, epithelial wound healing properties of the keratin film were compared to the AM and polystyrene plates. The authors observed that the keratin film (KF) supported adhesion, migration, and proliferation of the epithelial HCE-T cell line. In spite of higher cell migration on the KF than the AM, the fastest cell migration was observed in the control polystyrene at all tested time points. Nevertheless, the authors suggested that the KF had improved transparency compared to the AM and polystyrene and therefore, the low cell migration can be modified by allowing a longer time.157 In a recent in vivo study,141 the same group evaluated the biocompatibility of the keratin film for ocular regeneration and observed good corneal biocompatibility of the films compared to the AM with minor host reaction and preservation of corneal transparency, although details on the behaviour of the film after sterilisation and its suturing ability during the surgical procedure are unknown.141 Keratin plates were also suggested as a suitable nail plate alternative to study drug release and permeation and probably its possible application as a human nail plate substitute.158 Keratin and ceramide were used to develop a human epidermis for in vitro studies, in order to avoid using human or animal skin.159 The membrane was not stable in organic solvent solutions such as water/ethanol for a long time, however the authors conclude that the membrane can be a simplified skin model to study small drug permeation. Keratin films have also been investigated as a drug carrier and means of controlled release of drugs. Alkaline phosphatase (ALP) was incorporated into the keratin film and it remained biologically active during the 14 days of the controlled release period.160 In this study, the authors used no surfactant agent for protein extraction, which enabled them to keep the ALP under the desired biologically active conditions during the testing period.160 In another study by Vasconcelos et al.161 the fabricated protein matrix for the delivery of the elastase inhibiting agent to wound by blending silk fibroin and wool keratin was investigated. The keratin concentration on the matrix played an important role in the rate of film degradation. It was concluded that the hydrolytic nature of the keratin enhanced the keratin dissolution and consequently the release rate of the elastase inhibiting agent. The release rate of the hydrophobic systems can therefore be adjusted by changing the keratin amount in the formula. The water soluble fraction of the keratin (keratose) was investigated as a bone morphogenic protein (BMP2) carrier, to enhance bone growth and regeneration in the rat femoral defect. The construct demonstrated a notable reduction of adipose tissues within the gap and enhanced bone regeneration.162 It is necessary to consider that the successful incorporation of the BMP2 was due to the positive charge of the BMP2 at the acidic and neutral pH level which enables its interaction with keratose through electrostatic interactions.163
Keratin solution has the ability to form a film in a self-assembled manner14 and can also enhance cell attachment and proliferation. However, a pure film of keratin is normally fragile and brittle. Therefore, the literature reported different approaches to resolve this issue such as the addition of plasticizers, e.g. glycerol, sorbitol and ethylene glycol147,157,164 or cross-linking agents.165 Incorporation of the natural166,167 or synthetic polymer168 into the matrix of the keratin structure has also been suggested to enhance the mechanical properties of the keratin films. These proposed solutions can have some limitations. For example, while plastisizers make the keratin film flexible and enhance its mechanical properties, some agents like glycerol can leach out of the film in an aqueous solution and be removed from the film.165 Additionally, when films that incorporate chitosan are to be used in acidic aqueous solution, swelling and dissolution of the chitosan content should be taken into consideration;165 this enhanced swelling can be considered as an advantage depending on the target application of the keratin film. There are a number of good reviews available on the biomedical applications of keratin,169–171 however very little attention has been paid to the impact of additives used in the process such as chemical cross-linkers and plasticizers and also the effect of incorporation of various synthetic and natural polymers into the keratin matrix. Therefore, the effects of cross-linking and plasticizing agents on the physiochemical properties of the keratin composites will be discussed in detail below.
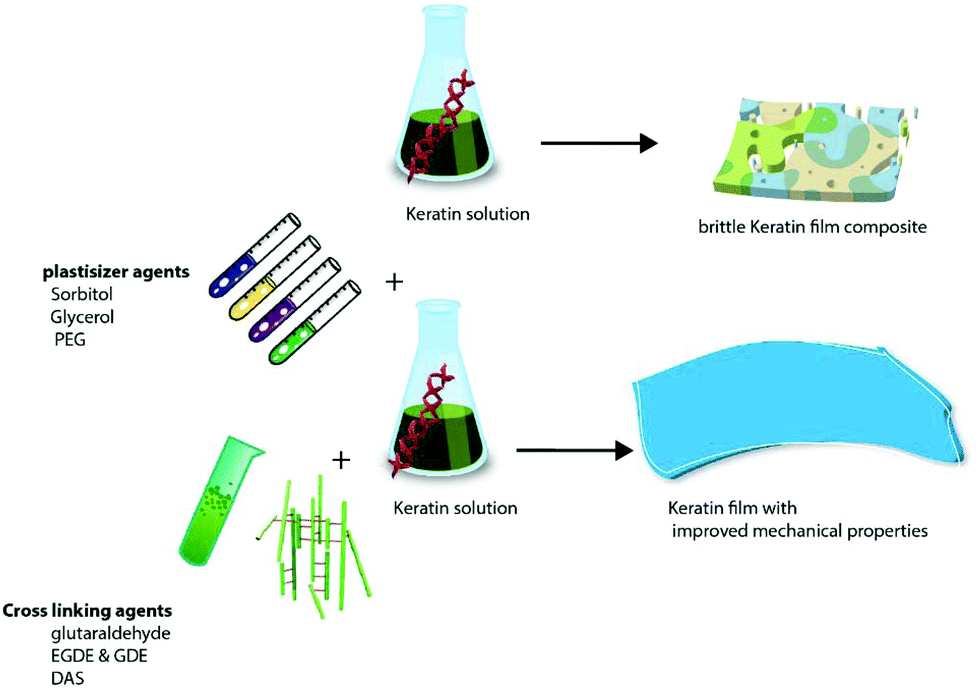 | ||
| Fig. 10 Effect of different plasticizers and crosslinking agents on the mechanical properties of the keratin film. | ||
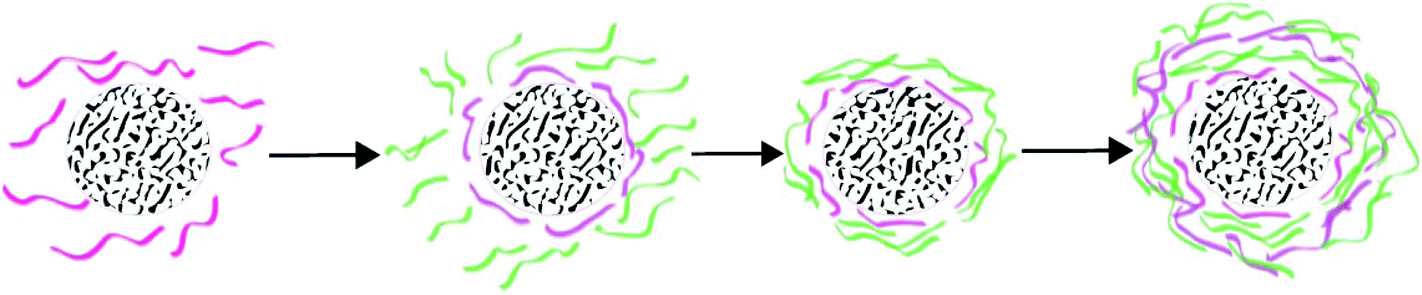 | ||
| Fig. 11 Layer by layer fabrication of keratin biomaterials. | ||
3.2 Keratin composite scaffolds
The self-assembling ability of keratin to form a porous 3-dimensional structure has made it an interesting material for the development of biocomposite scaffolds for biomaterial applications (Fig. 12). Freeze drying of frozen keratin solution, in general, has been the most common technique for the generation of the biocomposites. Additionally, the pH and concentration of the keratin solution, the presence of a cross-linker, plastisizer, or incorporation of other natural or synthetic polymers into the keratin matrix can also have an impact on the porous structure of the composite, in a way analogous to the described above for films. The use of a faster cooling/freezing rate during the freezing of the keratin solution mixture can affect the size of ice crystal formation and lead to pores with a smaller size during the sublimation process of the freeze dryer. | ||
| Fig. 12 Keratin-based calcium phosphate biomaterials. | ||
:1) and keratin/chitosan (2:1). The maximum porosity of the matrix was 31% and the authors observed improved mechanical properties in both composites and suggested that keratin/chitosan was superior to keratin/gelatin due to its slower degradation and antimicrobial properties. In a different study,198 a composite fabricated from keratin, gelatin, and chitosan at a ratio of 1:1:2 (w/w) and 86% porosity was reported, which had a much higher porosity than that reported by Balaji et al. (31%). Differences in the porosity may be due to the composition of the materials or their processing, although the effect of the method used for the porosity measurement cannot be neglected: Balaji et al.197 used the mercury intrusion porosimetry technique while a simple ethanol infiltration method was used in Kakkar's study.198
| Composition | Ratios | Process conditions | Properties | Ref. |
|---|---|---|---|---|
| Keratin–chitosan 2:1 (w/w) |
200 mg of chitosan | Frozen at −80 °C, slow degradation and antibacterial properties | Max. load (N) 6.30 ± 0.12 | 197 |
| 15 ml of 75% acetic acid | Max. extension (mm) 5.12 ± 0.15 | |||
| 10 ml of keratin solution (containing 420 mg keratin) | Elongation break (%) 21.63 ± 0.13 | |||
| Tensile strength (MPa) 1.58 ± 0.17 | ||||
| Pores in the range of 20–100 μm | ||||
| Porosity: 27% | ||||
| Water uptake: 850 ± 3 | ||||
| Keratin–gelatin (KG) 2:1 (w/w) |
200 mg of gelatin 10 ml water, 10 ml of keratin solution (containing 420 mg keratin) 10 ml of gelatin solution (containing 210 mg gelatin) | Frozen at −80 °C | Max. load (N) 7.15 ± 0.18 | 197 |
| Rapid degradation of gelatin in KG, pores: 20–100 μm | Max. extension (mm) 6.12 ± 0.12 | |||
| Elongation break (%) 18.65 ± 0.14 | ||||
| Tensile strength (MPa) 1.78 ± 0.16 | ||||
| Porosity 31% | ||||
| Pores in the range of 20–100 μm | ||||
| Water uptake 900 ± 3% | ||||
| Keratin, chitosan/gelatin 1:1:2 (w/w) |
Concentration of keratin and chitosan in solution was 2.5 mg ml−1 each gelatin was 5 mg ml−1 | Frozen at −40 °C | Ultimate tensile strength (kPa) | 198 |
| • Dry 95.69 ± 0.95 | ||||
| • Wet 10.06 ± 0.54 | ||||
| Compressive modulus (kPa) | ||||
| • Dry 8.58 ± 0.50 | ||||
| • Wet 5.27 ± 0.55 | ||||
| Water uptake (%): 1796.52 ± 23.1 | ||||
| Porosity (%): 86.86 ± 1.38 | ||||
| Keratin sponge | Keratin solution (250 ml) containing 15 mg of protein | Frozen at −20 °C treated with 10 ml of 0.1 M iodoacetic acid to produce the carboxyl-sponge | Pore size: 100 μm | 189 |
| Keratin sponge scaffold | The keratin solution (200 μl) containing 8 mg of protein was added to a flat-bottom tube | Frozen −20 °C | Pore size was 100 μm | 188 |
| Keratin–chitosan | 2% (w/v) CH solutions, 1.5 ml of ethylene glycol as a plasticizer | Max. tensile (MPa): K:CH (1:3) 21.14 |
196 | |
| Max. elongation at break: K:CH (1:1) 16.03 |
||||
| Max. Young's modulus (3:1) 3.14 |
||||
| Keratin–PEG | Carriers for doxorubicin hydrochloride salt (DOX·HCl) with a highest loading capacity of 18.1% (w/w) | 220 | ||
| Keratin hydrogel | Lyophilized material with PBS at a 15 wt%/vol% concentration | Keratin neuro conduit contains regulatory molecules capable of enhancing nerve tissue regeneration by inductive mechanisms | 268 | |
| PCL–keratin–HA | PCL and keratin 10 wt%, at a weight ratio of 7:3 |
Tensile strength (MPa) 16.53 ± 1.16 | 203 | |
| 1:10 HA |
Strain at break (%) 152.78 ± 19.86 | |||
| Young's modulus (MPa) 25.92 ± 0.96 | ||||
| Pore size (μm) 2.66 ± 0.41 | ||||
| Keratin hydrogel | 20% (weight per volume, w/v) hydrogels | Drug release over 3 weeks | 24 | |
| PLA/chitosan/keratin composites | A111: 70% PLA and 30% chitosan; A121: 68% PLA, 30% chitosan and 2% keratin; A131: 66% PLA, 30% chitosan and 4% keratin | A111 highest Young's modulus almost 3000 MPa and max tensile strength at break of about 50 MPa | PLA, chitosan and keratin composites support osteoblast attachment and proliferation during short-term culture | 269 |
| Cortical cells/chitosan | Chitosan 2 g was stirred in 200 mL of 50% acetic acid | The chitosan composite film with 30 wt% cortical cells: values of 29.6 ± 2.9 MPa for ultimate strength, 5.6 ± 0.3% for ultimate elongation 35.3 ± 1.4 MPa for Young's modulus, all higher than that of the pure chitosan film | Higher chitosan, higher thermal stability and higher crystallinity | 167 |
| Cortical of 0, 5, 10, 20 and 30 wt%, dispersed in solution respectively | ||||
| Keratin–chitosan | 3% solution of chitosan in acetic acid | Tenacity – 13.5 cN/tex, elongation – 33%, wetting angle – 33° and, in comparison to pure chitosan fibers, were less prone to biodegradation | 194 | |
| Keratin hydrogel | 15% keratin hydrogel | Nerve regeneration | Neuromuscular recovery with keratin was greater than with empty conduits in most outcome measures | 144 |
| Keratin/poly(vinyl alcohol) composite | 10% | Cross linked with glyoxal 10% | Keratin/PVA NFs | 270 |
| Young's modulus: 272.8 MPa | ||||
| Tensile strength: 19 MPa, strain: 175.6% | ||||
| Keratin sponge scaffolds | Keratin/NaCl as a porogen with different ratios | The S-sulfo keratin sponges with the regulated sizes of pores (100, 100–300 and 300–500 mm) more than 90% of the porosity for all | Weight ratios of the NaCl particulates to S-sulfo keratin were adjusted to 0, 5, 9, 15 and 20, respectively. Max. water uptake: 1206% for a ratio of 20 | 19 |
| Keratin–PVA | water uptake >26 g g−1 within 2000 min | Fibrous and highly porous morphologies | 271 | |
| The highest gel fraction and degree of swelling achieved at the dose of 40 kGy. | ||||
| Irradiated with an electron beam at a dose of 10 kGy–100 kGy |
To improve the mechanical and thermal stabilities of the composite, a chitosan/starch matrix with keratin was proposed.199 Using scanning electron microscopy and FTIR, Flores-Hernández199 showed that chitosan and keratin have good compatibility with each other and keratin up to 20 wt% was dispersed uniformly within the matrix of the chitosan matrix. Agar200 and calcium alginate beads (Fig. 13)201 were also blended with keratin solution in order to make keratin composites through the leaching method and the lyophilisation technique. The porosity values of around 94 and 98% were reported for agar and alginate–keratin composites respectively. Additionally, both sponges were biocompatible and had improved mechanical properties.
 | ||
| Fig. 13 The alginate incorporated keratin composite is flexible compared to the brittle structure of the pure keratin composite. Adopted from ref. 201 and reproduced with permission from Elsevier (license no. 4074120287302). | ||
:50 ratio and 7–10% polymer concentration can be easily electrospun. They suggested that this process results in a system with a less complex protein conformation, by preventing the self-assembly of S-sulfo keratin. In a following study, they improved the processability of keratin/PEO by using 2.7 wt% keratin with PEO (0.25, 0.20, 0.15 and 0.10 wt%). In this study a bead like nanofiber was observed at high concentrations of keratin (70/30) and the maximum strain was obtained at the lowest keratin ratio (keratin/PEO ratio 10/90). Keratin can improve the cell adhesion and proliferation of synthetic polymeric materials; and several studies by Yuan206 and Li et al.207 showed that the proliferation and attraction of the cells to electrospun (hydroxy butyrate-co-hydroxy valerate) PHBV fibre and poly(L-lactic acid) PLLA were increased when the polymer was blended with keratin to make wound dressing materials.206 Edwards et al.168 electrospun a PCL/keratin fibre with PCL keratin at the ratios of 90:10, 80:20, 70:30, and 60:40. Similar to the results reported by Aluigi,204 the mechanical strength of the fibre decreased by increasing the ratio of the keratin in the mixture. The maximum Young's modulus reported for the keratin–PEO fibre (12 ± 3 MPa) by Aluigi is also very close to 10 ± 2 MPa that was obtained for PCL/keratin by Edwards et al.168 In another study by Zhao et al.203 the mechanical properties and biocompatibility of the PCL/keratin (7:3) were improved by the incorporation of hydroxyapatite particles into the mixture and a maximum Young's modulus of 25.92 MPa was achieved, with the authors suggesting the fibre for bone tissue engineering. Despite the significant improvement in the mechanical properties, some important fibre properties such as specific surface area, porosity and the processability of the HA incorporated PCL/keratin still require further research. It is important to consider that HA particles can adversely affect the processability of the fibre and probably reduce the fibre porosity. The bone extracellular matrix has a 3D structure that supports cells as a scaffold, therefore composite scaffolds with a 3D structure are more preferred in tissue engineering compared to one-layered film composites. However, there are fabrication technical difficulties. Three-dimensional (3D) ultrafine fibrous keratin/PLA was electrospun by Xu et al. (Fig. 14).208 The authors concluded that the mesenchymal stem cells proliferated and differentiated better in 3D scaffolds compared to the traditional 2D structures, i.e. keratin hydrogels.
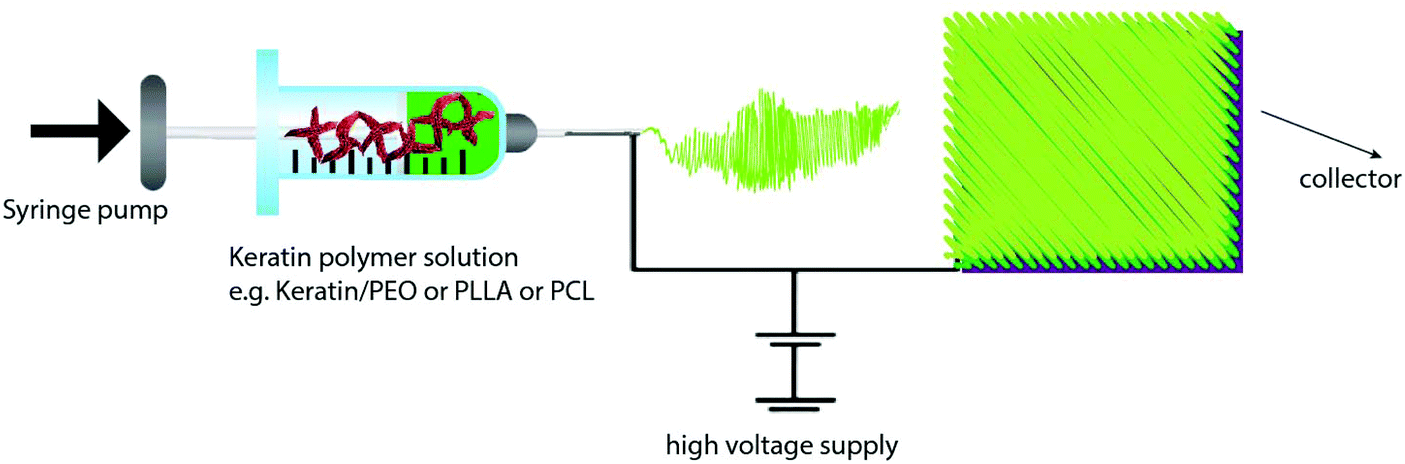 | ||
| Fig. 14 The schematic diagram of electrospinning of the keratin solution into a three-dimensional fibrous scaffolds. | ||
| Polymer | Keratin extraction method and concentration (wt%) | Total solution concentration (wt%) | Polymer/keratin ratios | Results | ES conditions (volume, distance, voltage, flow rate, thickness, time) | Ref. |
|---|---|---|---|---|---|---|
| PEO | Sulfitolysis, 6% | 7% | 30:70, 40:60, 50:50, 60:40, 70:30, 80:20, 90:10 |
Maximum keratin concentrations for the formation of bead-free nanofibers were 70 wt% in the blends | 5 ml, 15 cm, 10 to 30 kV, 0.5 ml min−1 | 272 |
| PEO | Sulfitolysis, 1–7% | 7% | 90/10, 70/30, 50/50, 30/70, 10/90 | Keratin/PEO 70/30 | 3 ml, 20 cm, 20 kV, 0.01 ml min−1, 12–30 μm, 20 min | 204 |
| Max. Young's modulus 31 MPa and stress 6 MPa obtained for (30/70) | ||||||
| Max. strain at break was 117.5 MPa for (10/90) | ||||||
| PEO | Sulfitolysis, 2.5, 3.5, and 5.0 | 5, 7, and 10 wt% | 50/50 | 50:50 keratin/PEO with 7 and 10 wt% polymer concentrations |
50 ml, 20 cm, 30 kV, 0.03 ml min−1 | 205 |
| PEO | Sulfitolysis | 7% | 90/10, 70/30, 50/50, 30/70, 10/90 | 50:50 keratin |
20 kV, 0.6 ml h−1, 20 cm | 151 |
| PVA | Sulfitolysis | 10% by adding PVA into keratin solution | 15 kV, 15 cm, 10% glyoxal at pH 2–3 | 270 | ||
| PCL 10% | Peracetic acid, 10% | — | PCL/keratin ratios of 90:10, 80:20, 70:30, and 60:40. |
PCL/keratin ratios from 100/00 to 70/30 showed good uniformity in fibre morphology and suitable mechanical properties | 10 ml, 23 cm, 25–27 kV, 100–160 | 168 |
| PCL | Sulfitolysis | 10% | PCL keratin weight ratio of 7:3, in HFIP solvent. HA/PCL 1:10 |
20 kV, 14 cm, 1 ml h−1 | 270 | |
| PLA 7% | NaOH, cysteine, 25% | — | — | Keratin and PLA fibres generated separately | PLA: 18 kV, 1 ml h−1, 15 cm, 1200 rpm, | 208 |
| keratin: 45 kV, 25 cm, needle negative charge |
3.3 Keratin hydrogels for drug release/delivery
Natural and synthetic materials are widely used to deliver therapeutic agents to the target tissue. A good drug release system can carry antibiotics to prevent infection or special drugs for healing and support a controlled release over a specified time period. In some instances, in order to be effective and to avoid the possible side effects, the drug only needs to be delivered to the targeted tissue. A number of different parameters, such as the route of drug administration, stability of the carrier to prevent the leakage and the desired drug release pattern, determine the efficiency of the delivery system. On the other hand, the delivery device can play a protective role as well, since the encapsulation of the bioactive compound may prevent the body immune system to neutralize the compound before the therapeutic effect to take place.Materials devised for drug delivery should be biocompatible, biodegradable, and of course commercially interesting. Drug delivery materials can be in the form of a gel, film, scaffold or microparticles. However, in any form, it is important to minimize the amount of solvent for the fabrication of the drug carrier to minimize any possible side effects. Also the release profile of the carrier construct should be adjustable to meet the specific requirements for the release concentration of specific drugs. Various synthetic polymers such as polyorthoesters, polyphosphazene and polyanhydrides209 have been used for drug delivery. Alginate, chitosan, collagen and keratin have also been investigated for this purpose,210,211 representing the natural polymers. Natural polymers have the advantages of being biocompatible, biodegradable and have similar biological properties due to the presence of hydrophilic groups such as hydroxyl, carboxyl, and amines. Due to this hydrophilic capability, the natural polymer constructs can interact with biological molecules. In addition to their low toxicity, safety, and high abundance, proteins are gaining wide interest for drug delivery due to their technical unique properties. Protein has emulsifying, gelling and good water holding ability that may protect them from the immune system through an aqueous steric barrier.212,213
Keratin has been investigated for drug delivery in different forms of films and hydrogels (Table 10). A simple way of fabrication of a keratin drug carrier is through dissolving keratin into keratin solution. It is important to consider the possible effect of this dissolution process on the physiological and chemical properties of the drug in order to prevent unwanted changes to the drug structure through this processing technique. The keratin films for this purpose have been discussed above; in this part of the review important keratin hydrogels that have been used for drug delivery will be the main focus. Keratose, as the water soluble fraction of keratin, has been widely used for the preparation of a keratin hydrogel due to its easy to prepare method which only uses water as a solvent.142,214 Ciprofloxacin has been incorporated into a keratin hydrogel through electrostatic interaction, and it has been observed that 60% of the loaded drug was released during 10 days to prevent the growth of the bacteria.24 The keratin hydrogel can also play an important role in the prevention of postoperative adhesion. The adhesion after some surgeries like abdominal or peritoneal occurs in more than 90% of the patients which can cause severe consequences such as chronic pain, bowel obstruction or even death, necessitating a second operation to relieve the symptoms.215 Peyton et al.215 devised a physicochemical adhesion inhibitor by using halofuginone (HF) as the drug and the keratose hydrogel as the physical barrier. The HF–keratin hydrogel was able to reduce the quantity and density of the adhesion in the rodent cecal abrasion model. It was suggested that the hydrogel can also carry the drug to the target site and function as a multipurpose hydrogel.215 Despite the ease of processing techniques and the promising results reported in the above studies, several issues remain to be solved. For example, the high degradation rate of the keratose (usually within 2 weeks) practically limits its versatile drug release application. Secondly, the drug incorporation into the keratin matrix is through weak electrostatic interactions (van der Waals, hydrophobic or hydrogen) and its stability depends on the physical stability of the carrier that in this case is a highly degradable keratose gel. Therefore, the drug release and the gel stability depend on the physiological environment of the target tissue that makes it difficult to apply the gel as a controlled release matrix. Covalent bonding, cross-linking175 and using nanoparticle drug carriers have been suggested to overcome this problem. However, these strategies might not be possible when the target material should be in the form of a hydrogel. Covalent binding provides a stable attachment of the therapeutic agent to the keratin, which results in a longer controlled release time of the therapeutic agent compared to the electrostatic interaction.216 For example, lysozyme was immobilised in the keratin sponge through disulfide and thioester bonds. Using this method, lysozyme remained in the structure for two months using thioester bonds and more than 3 weeks using disulfide bonding.217 Proteins like keratin have different functional groups which allow modification of the protein's physicochemical properties and fine tuning of the protein interaction with various active agents, cell membranes and receptors. Therefore, some studies modified the amino acid constituents of the keratin to improve its drug carrier ability and stability.213 For example, the tertiary and quaternary structures of the protein can be strengthened by the addition of disulfide bonds and possibly use this functionality when a longer controlled release duration is needed. The concentration of glutathione (GSH) is significantly higher in the cellular membrane of the cells with metastatic activity in comparison to healthy cells; therefore, an ideal chemotherapeutic drug carrier should have a GSH responsive activity to release its anti-cancer drug payload in the cellular membrane of the unhealthy cells.218 Keratin was coupled with polyethylene glycol-40 stearate as a hydrophobic block through radical grafting to produce an amphiphilic, GSH responsive polymer with the ability to load both hydrophobic and hydrophilic drugs (curcumin and methotrexate respectively).213 In another study, a pH-sensitive keratin hydrogel was prepared by grafting copolymerization using methacrylic acid (MAA) as a functional monomer and two different drugs with small molecules (rhodamine B) and macromolecules (bovine serum albumin) were loaded. The authors observed that 97% of the small molecules were released in 24 h at pH 8.4 while they experienced a better control over the release of the macromolecules (89% at pH 7.4) and suggested that the large molecule release behaviour can be controlled better by pH.
| Product | Conditions | Results | Application | Ref. |
|---|---|---|---|---|
| Keratin gel | Rabbit liver lethal injury, 20 wt% keratin gel | Keratin was better than commercial wound patches (QuickClot, HemCon) | Haemostatic dressing gel | 221 |
| Keratin-based scaffold | 15% keratin gel | Improved electrophysiological recovery, compared with empty conduits and sensory nerve autografts | Peripheral nerve regeneration | 144 |
| Keratin wound dressing | Porcine lethal extremity haemorrhage model | KeraStat and Nanosan increased survival | Haemostatic | 223 |
| Keratin gel | Lethal liver injury model 12% keratin solution | — | Haemostasis tissue engineering, regenerative medicine, drug and cell delivery, and trauma | 225 |
| Keratin gel filler | Rat 15 mm sciatic nerve defect | — | Peripheral nerve repair | 145 |
| Hydrophobically modified keratin | — | — | Nanocarriers for chemotherapeutic agents | 213 |
| Keratin gel | pH-Sensitive feather-keratin-based polymer hydrogel | The cumulative release rate was 97% for 24 h at pH 8.4 | Drug release | 148 |
| Keratin gel | — | Tuneable hydrogel erosion and drug delivery in tissue | Provide a matrix for cell attachment and proliferation | 138 |
| Injectable hydrogels | — | — | For cardiac tissue repair | 222 |
| Keratin hydrogel | 180 mg ml−1 | — | Cell substrate with drug releasing ability | 137 |
| Keratin hydrogel | 15% (w/v) hydrogel | — | Early cellular response to sciatic nerve injury in a rat model | 273 |
| Keratin hydrogel | Keratin + PVA | Hydrogels prepared by electron beam irradiation | 9 | |
| Keratin hydrogels | 5% PVA | Wound healing process in vivo | 274 | |
| 5% keratin | ||||
| Keratin hydrogels | Adhesions in a rodent cecal abrasion model | Halofuginone (HF) is a type-1 collagen synthesis inhibitor + keratin | Adhesions in a rodent cecal abrasion model | 215 |
| Keratin hydrogels | 9% gel | — | Skin regeneration after burns | 275 |
| Keratin hydrogel | — | — | Haemostatic agents on coagulation | 224 |
| Keratin hydrogel | — | — | Release of bioactive ciprofloxacin | 24 |
| Keratin hydrogel | 3% glycerol to make a 20% (w/v) solution | Its cytocompatibility was statistically equivalent to the collagen hydrogel | Pulp-tissue engineering. enhanced odontoblast cell behaviour | 276 |
| Keratin hydrogel | 15% gel | Rapid regeneration of peripheral nerves | 135 | |
| Injectable keratin | — | — | Delivery of rhBMP-2 in a porcine mandible defect | |
| Keratin hydrogels | — | — | Culturing fibroblasts | 277 |
Keratose is the keratin extracted via the oxidation process, and its thiol groups cannot establish covalent disulfide cross-linking due to sulfonic acid groups that capped the thiol groups, therefore, the hydrogel produced from keratose has a physical binding that is formed by chain entanglement rather than covalent binding. On the other hand, keratin extracted during the reduction method (kerateine) has free thiol groups that have covalent binding ability as well as chain entanglement capability. Therefore, considering that the drug release rate of the keratin hydrogel is related to its rate of erosion, hydrogels with different degradation and drug release abilities can be fabricated using various combinations of these two keratins.82,219 Han et al.138 modified the thiol groups on the kerateine in order to modulate the erosion of the hydrogel and consequently tune the drug release rate. To achieve this, the thiol groups were alkylated (capped) using iodoacetamide as the alkylating agent. Although the authors concluded that this process does not have any toxic effect on the tested cells, possible interactions of the alkylation and the alkylating agent on the loaded drugs and the release of any possible residual alkylating agent are not clear and so it might be only safe for some specific drugs and experimental conditions evaluated by the authors. In another study, the free cysteine residue of the reduced keratin was subjected to acetamidation, carboxymethylation or aminomethylation,137 and the result showed that the hydrogel produced from the acetamidated keratin was able to sustain and release the drug (salicylic acid, p-acetamidophenol, and aminopyrine) in a 3 day-period while the drug was released within only one day from the two other modified forms. A keratin graft polyethylene glycol copolymer was synthesized by Li et al.220 The copolymer was reported as an effective carrier for the doxorubicin hydrochloride salt with a high loading capacity (18.1% w/w) which can be used for the intercellular delivery of drugs for cancer treatment. In a similar study, polyethyleneglycol-40 stearate coupled to keratin through radical grafting and the synthesized polymerosome was loaded with two different drugs namely, hydrophilic methotrexate and hydrophobic curcumin. The authors reported that the redox-responsive vesicles can be used for drug delivery for cancer therapy.213 Once different drugs are used in different studies, it is not realistic to provide a fair comparison between the controlled release properties of the modified keratin carriers in this study and the alkylated keratin reported earlier.
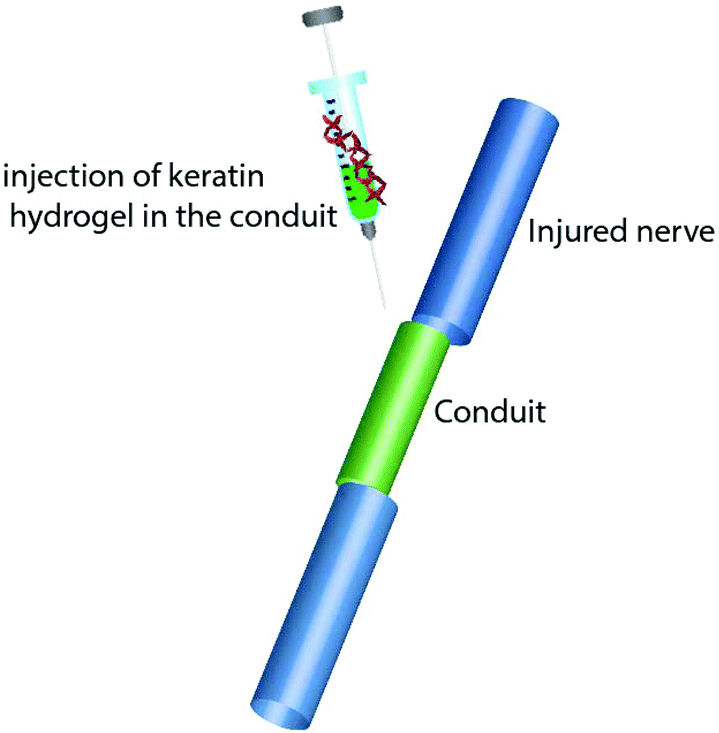 | ||
| Fig. 15 Nerve regeneration via keratin hydrogel injection into the conduits placed in the defective nerve segment. | ||
According to Apel et al.144 the keratin hydrogel can facilitate nerve regeneration in three major ways:
1. The hydrogel provides a biocompatible scaffold matrix that Schwann cells and macrophages can infiltrate, with time dependent biodegradation compatible with the axonal ingrowth;227
2. The keratin hydrogel with fibronectin-like cell binding domains facilitates cell adhesion;135
3. It has biological activities that facilitate the proliferation of the Schwann cells.
4 Concluding remarks and future challenges
Wool is very resistant to weak alkalis, acids and organic solvents due to its high content of disulfite bonds. Chemical hydrolysis of wool to extract keratin often brings serious pollution to the environment. Chemical methods using thiols, such as 2-mercaptoethanol, are the benchmark for good yield and undamaged keratin. This method is based on the breakage of disulfite bonds through reduction and so conversion of cystine to cysteine but environmental harm and high-cost mean that they are not industrially viable. In addition, it is hard to remove chemicals such as mercaptoethanol and the method is time-consuming. On the other hand, the higher cost of enzymes, with a long production cycle, has thus far limited the development of industrial processes using an enzymatic method. Methods used to oxidise the material and convert disulfite into sulfate groups such as cysteic acid residues are well established. Using these methods different fractions of keratin (α, β and γ) can be separated. However, the process is time-consuming and requires large amounts of oxidising agents. Steam flash explosion and microwave are thermal methods, which mainly employ water and heat treatment. However, the process has not been able to reach high yields without the addition of chemicals to the reaction, and a large amount of cysteine as a semi-essential amino acid is lost during the reaction. The nature of the solubilized keratin widely depends on the method used for the solubilisation. Therefore, the right selection of the appropriate method largely depends on the scale and the final targeted product(s). There is, thus, an opportunity for research avenues on the improvement of methods for the sustainable isolation of keratin, including new methodologies as is the case of ionic liquids as potent solvents or, more recently, deep eutectic solvents,228,229 similar in principal to ionic liquids (although being a mixture, behave thermally as a pure substance), but where ionic interactions are substituted by hydrogen-bonds by adequate blending of the solvents.Moreover, the main target of the studies addressing the isolation of keratin is the abundant by-products resulting from processing livestock, as wool, horns and feathers. Nevertheless, less conventional sources can also play an important role in the scientific and industrial/market arenas, particularly when considering high added value applications, such as the ones directed for cosmetics and biomedical sectors, where the value is on the (bio)technological innovation and not on the availability of huge amounts of raw-materials. In this regard, the marine environment can arise, given the recent developments in technology allowing the exploration of farther (open) and deeper waters. In fact, keratin can also be found in marine organisms, not only on baleen230 of a sub-order of whales, but also in the rays of fish as sturgeon and bichir,231 or in hagfish slime threads.232 These marine originated keratins, including those from marine birds and reptiles, have been recently reviewed by Hermann Ehrlich,233 and deserve more attention given the growth of marine materials for biomedical applications.234
Various keratin-based biomaterials have been developed over the last decade for biomedical applications such as sponges, hydrogels, wound patches, films or fibres. Despite the promising results reported for the applications of these biomaterials, only a few of these progressed to clinical trials. This review discussed the diversity of keratin biomaterials and highlighted the limitations and strengths of these biomaterials. Keratin biomaterials have some unique pluses such as matchless chemistry and the chemical structure that make it possible to generate a variety of specifically designed biomaterials by fine-tuning and modification of the structure. Keratin biomaterials are also biocompatible, biodegradable and have positive cell interaction, and importantly keratin sources are cheap materials such as wool and hair. However, keratin biomaterials only have a small share of the big market of biomaterials compared to the major players. In order to make keratin a mainstream biomaterial, there are certain issues that need to be addressed in future:
• The keratin interaction with cells and its role in supporting the cells need to be understood better. Consequently, wound healing, nerve, bone and skin regeneration processes using keratin biomaterials will experience a significant advancement.
• The mechanical–physical properties of keratin-based materials such as films, composites and sponges need to be improved and the keratin interactions with other natural or synthetic polymers have to be elucidated at the molecular level in order to optimize the structure and function of the biomaterial.
• Further investigations are required to find simple, cost effective and yet efficient methodologies such as chemical or enzymatic assisted methods, using new classes of green solvents such as ILs or DES, or deeply exploring the supercritical fluid technology, for the better extraction of a different fraction of keratin from hair and wool, or from less conventional sources.
When these properties of keratin are achieved, it is expected that keratin biomaterials will turn into a mainstream biomaterial for clinical trials.
References
- Y. Zhang, R. Yang and W. Zhao, J. Agric. Food Chem., 2014, 62, 2745–2751 CrossRef CAS PubMed.
- J. R. Barone, W. F. Schmidt and C. F. E. Liebner, J. Appl. Polym. Sci., 2005, 97, 1644–1651 CrossRef CAS.
- FAOSTAT, FAO statistical databases, agricultural data, Rome, Italy, 2013 Search PubMed.
- E. Jin, N. Reddy, Z. Zhu and Y. Yang, J. Agric. Food Chem., 2011, 59, 1729–1738 CrossRef CAS PubMed.
- Y. Dou, X. Huang, B. Zhang, M. He, G. Yin and Y. Cui, RSC Adv., 2015, 5, 27168–27174 RSC.
- N. M. Neves and R. L. Reis, Biomaterials from Nature for Advanced Devices and Therapies, Wiley, 2016 Search PubMed.
- S. Isarankura Na Ayutthaya, S. Tanpichai and J. Wootthikanokkhan, J. Polym. Environ., 2015, 23, 506–516 CrossRef CAS.
- A. Aluigi, C. Tonetti, F. Rombaldoni, D. Puglia, E. Fortunati, I. Armentano, C. Santulli, L. Torre and J. M. Kenny, J. Mater. Sci., 2014, 49, 6257–6269 CrossRef CAS.
- M. Park, B.-S. Kim, H. K. Shin, S.-J. Park and H.-Y. Kim, Mater. Sci. Eng., C, 2013, 33, 5051–5057 CrossRef CAS PubMed.
- A. Poole, R. Lyons and J. Church, J. Polym. Environ., 2011, 19, 995–1004 CrossRef CAS.
- T. Korniłłowicz-Kowalska and J. Bohacz, Waste Manage., 2011, 31, 1689–1701 CrossRef PubMed.
- C. Tonin, A. Aluigi, A. Varesano and C. Vineis, Keratin-based Nanofibres, 2010 Search PubMed.
- R. D. B. Fraser, T. P. MacRae and G. E. Rogers, Keratins: Their Composition, Structure, and Biosynthesis, Thomas, 1972 Search PubMed.
- K. Yamauchi, A. Yamauchi, T. Kusunoki, A. Kohda and Y. Konishi, J. Biomed. Mater. Res., 1996, 31, 439–444 CrossRef CAS PubMed.
- J. H. Buchanan, Biochem. J., 1977, 167, 489–491 CrossRef CAS PubMed.
- M. Zoccola, A. Aluigi, A. Patrucco, C. Vineis, F. Forlini, P. Locatelli, M. C. Sacchi and C. Tonin, Text. Res. J., 2012, 82, 2006–2018 CrossRef.
- Y. Tsuda and Y. Nomura, Anim. Sci. J., 2014, 85, 180–185 CrossRef CAS PubMed.
- C. Tonin, M. Zoccola, A. Aluigi, A. Varesano, A. Montarsolo, C. Vineis and F. Zimbardi, Biomacromolecules, 2006, 7, 3499–3504 CrossRef CAS PubMed.
- K. Katoh, T. Tanabe and K. Yamauchi, Biomaterials, 2004, 25, 4255–4262 CrossRef CAS PubMed.
- H. Xie, S. Li and S. Zhang, Green Chem., 2005, 7, 606–608 RSC.
- A. Ghosh, S. Clerens, S. Deb-Choudhury and J. M. Dyer, Polym. Degrad. Stab., 2014, 108, 108–115 CrossRef CAS.
- C. Earland and C. S. Knight, Biochim. Biophys. Acta, 1955, 17, 457–461 CrossRef CAS.
- J. A. Maclaren, Text. Res. J., 1987, 57, 87–92 CrossRef CAS.
- J. M. Saul, M. D. Ellenburg, R. C. de Guzman and M. Van Dyke, J. Biomed. Mater. Res., Part A, 2011, 98, 544–553 CrossRef PubMed.
- D. R. Goddard and L. Michaelis, J. Biol. Chem., 1935, 112, 361–371 CAS.
- W. I. Patterson, W. B. Geiger, L. R. Mizell and H. Milton, J. Res. Natl. Bur. Stand., 1951, 27, 89–103 CrossRef.
- W. E. Savige, Text. Res. J., 1960, 30, 1–10 CrossRef.
- H. Zahn, F. W. Kunitz and D. Hildebrand, J. Text. Inst., Trans., 1960, 51, T740–T755 CrossRef.
- D. H. Baker, J. Nutr., 2006, 136, 1670s–1675s CAS.
- M. Friedman, J. Agric. Food Chem., 1999, 47, 1295–1319 CrossRef CAS PubMed.
- E. Thompson and I. O'donnell, Aust. J. Biol. Sci., 1962, 15, 757–768 CrossRef CAS.
- H. Xu, Z. Ma and Y. Yang, J. Mater. Sci., 2014, 49, 7513–7521 CrossRef CAS.
- P. M. M. Schrooyen, P. J. Dijkstra, R. C. Oberthür, A. Bantjes and J. Feijen, J. Colloid Interface Sci., 2001, 240, 30–39 CrossRef CAS PubMed.
- T. Kitahara and H. Ogawa, J. Dermatol. Sci., 1991, 2, 402–406 CrossRef CAS PubMed.
- A. Nakamura, M. Arimoto, K. Takeuchi and T. Fujii, Biol. Pharm. Bull., 2002, 25, 569–572 CAS.
- T. H. Barrows, Biomaterials made from human hair, US9072818 B2, 2012 Search PubMed.
- S. Blackburn and G. R. Lee, Biochim. Biophys. Acta, 1956, 19, 505–512 CrossRef CAS.
- Y. Zhang, W. Zhao and R. Yang, ACS Sustainable Chem. Eng., 2015, 3, 2036–2042 CrossRef CAS.
- R. L. Evans, Regenerated keratin, US2434688 A, 1948 Search PubMed.
- M. Harris, J. Res. Natl. Bur. Stand., 1935, 15, 63–71 CrossRef CAS.
- K. M. Arai, R. Takahashi, Y. Yokote and K. Akahane, Eur. J. Biochem., 1983, 132, 501–507 CrossRef CAS PubMed.
- C. J. B. Thor and R. A. Gortner, J. Biol. Chem., 1932, 99, 383–403 Search PubMed.
- Y. Nagai and T. Nishikawa, Agric. Biol. Chem., 1970, 34, 16–22 Search PubMed.
- Y. Nagai and T. Nishikawa, Agric. Biol. Chem., 1970, 34, 575–584 CAS.
- X. U. Jiang-tao, Z. H. U. Ping, Z. Lin, S. U. I. Shu-ying, D. Zhao-hong and Z. U. O. Yi, Wool Text. J., 2015, 43, 38–42 Search PubMed.
- J. A. Maclaren and B. Milligan, Wool Science: The Chemical Reactivity of the Wool Fibre, Science Press, 1981 Search PubMed.
- W. S. Simpson, G. H. Crawshaw and T. Institute, Wool: Science and Technology, CRC Press, 2002 Search PubMed.
- F. Happey and R. L. Wormell, J. Text. Inst., Trans., 1949, 40, T855–T869 CrossRef CAS.
- I. J. O'Donnell, Aust. J. Biol. Sci., 1973, 26, 401–413 CrossRef.
- L. T. Zhou, G. Yang, X. X. Yang, Z. J. Cao and M. H. Zhou, Environ. Sci. Pollut. Res. Int., 2014, 21, 5730–5736 CrossRef CAS PubMed.
- W. Leitner, Pure Appl. Chem., 2004, 76, 635–644 CrossRef CAS.
- J. F. Fernández, D. Waterkamp and J. Thöming, Desalination, 2008, 224, 52–56 CrossRef.
- T. Ueki, Polym. J., 2014, 46, 646–655 CrossRef CAS.
- H. Wang, G. Gurau and R. Rogers, in Structures and Interactions of Ionic Liquids, ed. S. Zhang, J. Wang, X. Lu and Q. Zhou, Springer, Berlin Heidelberg, 2014, vol. 151, ch. 3, pp. 79–105 Search PubMed.
- J. Dupont, R. F. de Souza and P. A. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS PubMed.
- C. F. Poole and S. K. Poole, J. Chromatogr. A, 2010, 1217, 2268–2286 CrossRef CAS PubMed.
- M. A. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976–4983 CrossRef CAS PubMed.
- H. Hagiwara, Y. Sugawara, K. Isobe, T. Hoshi and T. Suzuki, Org. Lett., 2004, 6, 2325–2328 CrossRef CAS PubMed.
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- T. Torimoto, T. Tsuda, K.-i. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef CAS PubMed.
- R. Li and D. Wang, J. Appl. Polym. Sci., 2013, 127, 2648–2653 CrossRef CAS.
- Y. Ji, J. Chen, J. Lv, Z. Li, L. Xing and S. Ding, Sep. Purif. Technol., 2014, 132, 577–583 CrossRef CAS.
- W. Zhao, R. Yang, Y. Zhang and L. Wu, Green Chem., 2012, 14, 3352–3360 RSC.
- P. Sun, Z.-T. Liu and Z.-W. Liu, J. Hazard. Mater., 2009, 170, 786–790 CrossRef CAS PubMed.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- L. Feng and Z.-l. Chen, J. Mol. Liq., 2008, 142, 1–5 CrossRef.
- Y.-X. Wang and X.-J. Cao, Process Biochem., 2012, 47, 896–899 CrossRef CAS.
- A. Idris, R. Vijayaraghavan, U. A. Rana, D. Fredericks, A. F. Patti and D. R. MacFarlane, Green Chem., 2013, 15, 525–534 RSC.
- J. M. Gillespie and F. G. Lennox, Biochim. Biophys. Acta, 1953, 12, 481–482 CrossRef CAS.
- C. Tonin, A. Aluigi, M. Bianchetto Songia, C. D'Arrigo, M. Mormino and C. Vineis, J. Therm. Anal. Calorim., 2004, 77, 987–996 CrossRef CAS.
- A. Idris, R. Vijayaraghavan, U. A. Rana, A. F. Patti and D. R. MacFarlane, Green Chem., 2014, 16, 2857–2864 RSC.
- N. Hameed and Q. Guo, Cellulose, 2010, 17, 803–813 CrossRef CAS.
- C. D. Tran and T. M. Mututuvari, Langmuir, 2015, 1516–1526 CrossRef CAS PubMed.
- D. M. Phillips, L. F. Drummy, D. G. Conrady, D. M. Fox, R. R. Naik, M. O. Stone, P. C. Trulove, H. C. De Long and R. A. Mantz, J. Am. Chem. Soc., 2004, 126, 14350–14351 CrossRef CAS PubMed.
- H. J. Jin, S. V. Fridrikh, G. C. Rutledge and D. L. Kaplan, Biomacromolecules, 2002, 3, 1233–1239 CrossRef CAS PubMed.
- G. J. Weston, Biochim. Biophys. Acta, 1955, 17, 462–464 CrossRef CAS.
- A. Strasheim and K. Buijs, Biochim. Biophys. Acta, 1961, 47, 538–541 CrossRef CAS.
- H. H. Stein and J. Guarnaccio, Text. Res. J., 1959, 29, 492–496 CrossRef CAS.
- C. Robbins, Text. Res. J., 1967, 37, 811–813 CrossRef CAS.
- M. C. Corfield, A. Robson and B. Skinner, Biochem. J., 1958, 68, 348–352 CrossRef CAS PubMed.
- C. R. Robbins, Chemical and Physical Behavior of Human Hair, Springer, Berlin Heidelberg, 2012 Search PubMed.
- R. C. de Guzman, M. R. Merrill, J. R. Richter, R. I. Hamzi, O. K. Greengauz-Roberts and M. E. Van Dyke, Biomaterials, 2011, 32, 8205–8217 CrossRef CAS PubMed.
- G. Toennies and R. P. Homiller, J. Am. Chem. Soc., 1942, 64, 3054–3056 CrossRef CAS.
- D. Simmonds, Aust. J. Biol. Sci., 1955, 8, 114–121 CAS.
- C. H. W. Hirs, W. H. Stein and S. Moore, J. Biol. Chem., 1954, 941–950 CAS.
- E. l. Smith and A. Stockell, J. Biol. Chem., 1953, 207, 501–514 Search PubMed.
- C. R. Blanchard, S. F. Timmons and R. A. Smith, Keratin-based hydrogel for biomedical applications and method of production, US5932552 A, 1999 Search PubMed.
- M. A. Aslaksen, O. H. Romarheim, T. Storebakken and A. Skrede, Anim. Feed Sci. Technol., 2006, 128, 320–330 CrossRef CAS.
- Z. Yu, B. Zhang, F. Yu, G. Xu and A. Song, Bioresour. Technol., 2012, 121, 335–341 CrossRef CAS PubMed.
- A. T. W. M. Hendriks and G. Zeeman, Bioresour. Technol., 2009, 100, 10–18 CrossRef CAS PubMed.
- M. N. Anglès, F. Ferrando, X. Farriol and J. Salvadó, Biomass Bioenergy, 2001, 21, 211–224 CrossRef.
- H. M. Mason, Apparatus for and process of explosion fibration of lignocellulose material, US1655618 A, http://www.google.com/patents/US1655618 Search PubMed.
- Ó. J. Sánchez and C. A. Cardona, Bioresour. Technol., 2008, 99, 5270–5295 CrossRef PubMed.
- X. F. Sun, F. Xu, R. C. Sun, Z. C. Geng, P. Fowler and M. S. Baird, Carbohydr. Polym., 2005, 60, 15–26 CrossRef CAS.
- L.-H. Zhang, D. Li, L.-J. Wang, T.-P. Wang, L. Zhang, X. D. Chen and Z.-H. Mao, Bioresour. Technol., 2008, 99, 8512–8515 CrossRef CAS PubMed.
- R. A. Young and M. Akhtar, Environmentally friendly technologies for the pulp and paper industry, J. Wiley, 1998 Search PubMed.
- P. McKendry, Bioresour. Technol., 2002, 83, 47–54 CrossRef CAS PubMed.
- T. Miyamoito, T. Amiya and H. Inagaki, Kobunshi Ronbunshu, 1982, 39, 679–685 CrossRef.
- W. Xu, G. Ke, J. Wu and X. Wang, Eur. Polym. J., 2006, 42, 2168–2173 CrossRef CAS.
- F. Bertini, M. Canetti, A. Patrucco and M. Zoccola, Polym. Degrad. Stab., 2013, 98, 980–987 CrossRef CAS.
- L. Shen, X. Wang, Z. Wang, Y. Wu and J. Chen, Food Chem., 2008, 107, 929–938 CrossRef CAS.
- J. Yin, S. Rastogi, A. E. Terry and C. Popescu, Biomacromolecules, 2007, 8, 800–806 CrossRef CAS PubMed.
- J. Chen, S. Ding, Y. Ji, J. Ding, X. Yang, M. Zou and Z. Li, Chem. Eng. Process., 2015, 87, 104–109 CrossRef CAS.
- G. S. J. Sturm, M. D. Verweij, A. I. Stankiewicz and G. D. Stefanidis, Chem. Eng. J., 2014, 243, 147–158 CrossRef CAS.
- S. Sanchez and A. L. Demain, Org. Process Res. Dev., 2011, 15, 224–230 CrossRef CAS.
- T. W. Johannes and H. Zhao, Curr. Opin. Microbiol., 2006, 9, 261–267 CrossRef CAS PubMed.
- D. J. Daroit and A. Brandelli, Crit. Rev. Biotechnol., 2014, 34, 372–384 CrossRef CAS PubMed.
- A. Brandelli, Food Bioprocess Technol., 2008, 1, 105–116 CrossRef.
- A. Brandelli, L. Sala and S. J. Kalil, Food Res. Int., 2015, 73, 3–12 CrossRef CAS.
- M. Saleem, A. Rehman, R. Yasmin and B. Munir, Mol. Biol. Rep., 2012, 39, 6399–6408 CrossRef CAS PubMed.
- V. Chaturvedi, K. Bhange, R. Bhatt and P. Verma, Biocatal. Agric. Biotechnol., 2014, 3, 167–174 Search PubMed.
- R. Gupta and P. Ramnani, Appl. Microbiol. Biotechnol., 2006, 70, 21–33 CrossRef CAS PubMed.
- A. A. Onifade, N. A. Al-Sane, A. A. Al-Musallam and S. Al-Zarban, Bioresour. Technol., 1998, 66, 1–11 CrossRef CAS.
- S. E. Tork, Y. E. Shahein, A. E. El-Hakim, A. M. Abdel-Aty and M. M. Aly, Int. J. Biol. Macromol., 2013, 55, 169–175 CrossRef CAS PubMed.
- D. J. Daroit, A. P. F. Corrêa and A. Brandelli, Int. Biodeterior. Biodegrad., 2009, 63, 358–363 CrossRef CAS.
- E. H. Burtt and M. I. Jann, Auk, 1999, 116, 364–372 CrossRef.
- R. Gupta, R. Sharma and Q. K. Beg, Crit. Rev. Biotechnol., 2013, 33, 216–228 CrossRef CAS PubMed.
- S. C. B. Gopinath, P. Anbu, T. Lakshmipriya, T.-H. Tang, Y. Chen, U. Hashim, A. R. Ruslinda and M. K. M. Arshad, BioMed Res. Int., 2015, 2015, 10 Search PubMed.
- J. M. Kim, W. J. Lim and H. J. Suh, Process Biochem., 2001, 37, 287–291 CrossRef CAS.
- J. Kunert, in Revista Iberoamericana de Micologia Volume 17, Revista Iberoamericana de Micología, Bilbao, 2000, pp. 77–85 Search PubMed.
- I. N. Dozie, C. N. Okeke and N. C. Unaeze, World J. Microbiol. Biotechnol., 1994, 10, 563–567 CrossRef CAS PubMed.
- J. J. Noval and W. J. Nickerson, J. Bacteriol., 1959, 77, 251–263 CAS.
- W. J. Nickerson, J. J. Noval and R. S. Robison, Biochim. Biophys. Acta, 1963, 77, 73–86 CrossRef CAS.
- T. Korniłłowicz-Kowalska, Acta Mycol., 1997, 32, 65–78 Search PubMed.
- J. Kunert, Z. Allg. Mikrobiol., 1976, 16, 97–105 CrossRef CAS PubMed.
- T. Matsui, Y. Yamada, H. Mitsuya, Y. Shigeri, Y. Yoshida, Y. Saito, H. Matsui and K. Watanabe, Appl. Microbiol. Biotechnol., 2009, 82, 941–950 CrossRef CAS PubMed.
- G. W. Nam, D. W. Lee, H. S. Lee, N. J. Lee, B. C. Kim, E. A. Choe, J. K. Hwang, M. T. Suhartono and Y. R. Pyun, Arch. Microbiol., 2002, 178, 538–547 CrossRef CAS PubMed.
- X. Lin, C. G. Lee, E. S. Casale and J. C. Shih, Appl. Environ. Microbiol., 1992, 58, 3271–3275 CAS.
- N. Eslahi, F. Dadashian and N. H. Nejad, Prep. Biochem. Biotechnol., 2013, 43, 624–648 CrossRef CAS PubMed.
- A. Bertsch and N. Coello, Bioresour. Technol., 2005, 96, 1703–1708 CrossRef CAS PubMed.
- J. Kunert, Experientia, 1972, 28, 1025–1026 CrossRef CAS PubMed.
- B. Léchenne, U. Reichard, C. Zaugg, M. Fratti, J. Kunert, O. Boulat and M. Monod, Microbiology, 2007, 153, 905–913 CrossRef PubMed.
- M. Monod, Mycopathologia, 2008, 166, 285–294 CrossRef PubMed.
- P. Ruffin, S. Andrieu, G. Biserte and J. Biguet, Sabouraudia, 1976, 14, 181–184 CrossRef CAS PubMed.
- P. Sierpinski, J. Garrett, J. Ma, P. Apel, D. Klorig, T. Smith, L. A. Koman, A. Atala and M. Van Dyke, Biomaterials, 2008, 29, 118–128 CrossRef CAS PubMed.
- F. Bordeleau, J. Bessard, Y. Sheng and N. Marceau, Biochem. Cell Biol., 2008, 86, 352–359 CrossRef CAS PubMed.
- R. Nakata, Y. Osumi, S. Miyagawa, A. Tachibana and T. Tanabe, J. Biosci. Bioeng., 2015, 120, 111–116 CrossRef CAS PubMed.
- S. Han, T. R. Ham, S. Haque, J. L. Sparks and J. M. Saul, Acta Biomater., 2015, 23, 201–213 CrossRef CAS PubMed.
- J.-S. Li, Y. Li, X. Liu, J. Zhang and Y. Zhang, J. Mater. Chem. B, 2013, 1, 432–437 RSC.
- A. Tachibana, Y. Nishikawa, M. Nishino, S. Kaneko, T. Tanabe and K. Yamauchi, J. Biosci. Bioeng., 2006, 102, 425–429 CrossRef CAS PubMed.
- S. Reichl, M. Borrelli and G. Geerling, Biomaterials, 2015, 42, 112 CrossRef PubMed.
- T. R. Ham, R. T. Lee, S. Han, S. Haque, Y. Vodovotz, J. Gu, L. R. Burnett, S. Tomblyn and J. M. Saul, Biomacromolecules, 2016, 17, 225–236 CrossRef CAS PubMed.
- R. Nakata, A. Tachibana and T. Tanabe, Mater. Sci. Eng., C, 2014, 41, 59–64 CrossRef CAS PubMed.
- P. J. Apel, J. P. Garrett, P. Sierpinski, J. Ma, A. Atala, T. L. Smith, L. A. Koman and M. E. Van Dyke, J. Hand Surg. Am., 2008, 33, 1541–1547 CrossRef PubMed.
- Y. C. Lin, M. Ramadan, M. Van Dyke, L. E. Kokai, B. J. Philips, J. P. Rubin and K. G. Marra, Plast. Reconstr. Surg., 2012, 129, 67–78 CrossRef CAS PubMed.
- S. Xu, L. Sang, Y. Zhang, X. Wang and X. Li, Mater. Sci. Eng., C, 2013, 33, 648–655 CrossRef CAS PubMed.
- X.-C. Yin, F.-Y. Li, Y.-F. He, Y. Wang and R.-M. Wang, Biomater. Sci., 2013, 1, 528–536 RSC.
- J. Guo, S. Pan, X. Yin, Y.-F. He, T. Li and R.-M. Wang, J. Appl. Polym. Sci., 2015, 132, 41572 Search PubMed.
- C. Tonin, A. Aluigi, C. Vineis, A. Varesano, A. Montarsolo and F. Ferrero, J. Therm. Anal. Calorim., 2007, 89, 601–608 CrossRef CAS.
- J. Li, X. Liu, J. Zhang, Y. Zhang, Y. Han, J. Hu and Y. Li, J. Biomed. Mater. Res., Part B, 2012, 100, 896–902 CrossRef PubMed.
- A. Varesano, A. Aluigi, C. Vineis and C. Tonin, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 1193–1201 CrossRef CAS.
- A. Aluigi, G. Sotgiu, A. Torreggiani, A. Guerrini, V. T. Orlandi, F. Corticelli and G. Varchi, ACS Appl. Mater. Interfaces, 2015, 7, 17416–17424 CAS.
- K. Katoh, M. Shibayama, T. Tanabe and K. Yamauchi, Biomaterials, 2004, 25, 2265–2272 CrossRef CAS PubMed.
- X. Yang, H. Zhang, X. Yuan and S. Cui, J. Colloid Interface Sci., 2009, 336, 756–760 CrossRef CAS PubMed.
- S. Reichl, Biomaterials, 2009, 30, 6854–6866 CrossRef CAS PubMed.
- S. Reichl, M. Borrelli and G. Geerling, Biomaterials, 2011, 32, 3375–3386 CrossRef CAS PubMed.
- Y. Feng, M. Borrelli, T. Meyer-ter-Vehn, S. Reichl, S. Schrader and G. Geerling, Curr. Eye Res., 2014, 1 Search PubMed.
- Lusiana, S. Reichl and C. C. Müller-Goymann, Eur. J. Pharm. Biopharm., 2011, 78, 432–440 CrossRef CAS PubMed.
- F. Selmin, F. Cilurzo, A. Aluigi, S. Franzè and P. Minghetti, Results Pharma Sci., 2012, 2, 72–78 CrossRef PubMed.
- T. Fujii, D. Ogiwara and M. Arimoto, Biol. Pharm. Bull., 2004, 27, 89–93 CAS.
- A. Vasconcelos, A. P. Pêgo, L. Henriques, M. Lamghari and A. Cavaco-Paulo, Biomacromolecules, 2010, 11, 2213–2220 CrossRef CAS PubMed.
- R. C. de Guzman, J. M. Saul, M. D. Ellenburg, M. R. Merrill, H. B. Coan, T. L. Smith and M. E. Van Dyke, Biomaterials, 2013, 34, 1644–1656 CrossRef CAS PubMed.
- H. Uludag, W. Friess, D. Williams, T. Porter, G. Timony, D. D'Augusta, C. Blake, R. Palmer, B. Biron and J. Wozney, Ann. N. Y. Acad. Sci., 1999, 875, 369–378 CrossRef CAS PubMed.
- A. Kavitha, K. Boopalan, G. Radhakrishnan, S. Sankaran, B. N. Das and T. P. Sastry, J. Macromol. Sci., Part A: Pure Appl. Chem., 2005, 42, 1703–1713 CrossRef.
- T. Tanabe, N. Okitsu and K. Yamauchi, Mater. Sci. Eng., C, 2004, 24, 441–446 CrossRef.
- T. Tanabe, N. Okitsu, A. Tachibana and K. Yamauchi, Biomaterials, 2002, 23, 817–825 CrossRef CAS PubMed.
- J. Fan and W. D. Yu, Waste Manag. Res., 2010, 28, 44–50 CrossRef CAS PubMed.
- A. Edwards, D. Jarvis, T. Hopkins, S. Pixley and N. Bhattarai, J. Biomed. Mater. Res., Part B, 2015, 103, 21–30 CrossRef PubMed.
- A. Vasconcelos and A. Cavaco-Paulo, Curr. Drug Targets, 2013, 14, 612–619 CrossRef CAS PubMed.
- M. George Dan, G. Alexandru Mihai and C. Mariana Carmen, Curr. Drug Targets, 2014, 15, 518–530 CrossRef.
- J. G. Rouse and M. E. Van Dyke, Materials, 2010, 3, 999 CrossRef.
- J. E. Gough, C. A. Scotchford and S. Downes, J. Biomed. Mater. Res., 2002, 61, 121–130 CrossRef CAS PubMed.
- B. Piotrowska, K. Sztuka, I. Kołodziejska and E. Dobrosielska, Food Hydrocolloids, 2008, 22, 1362–1371 CrossRef CAS.
- C. Larre, C. Desserme, J. Barbot and J. Gueguen, J. Agric. Food Chem., 2000, 48, 5444–5449 CrossRef CAS PubMed.
- L. Cui, J. Gong, X. Fan, P. Wang, Q. Wang and Y. Qiu, Eng. Life Sci., 2012, 149–155 CAS.
- G. Rocha Plácido Moore, S. Maria Martelli, C. Gandolfo, P. José do Amaral Sobral and J. Borges Laurindo, Food Hydrocolloids, 2006, 20, 975–982 CrossRef.
- S. Maria Martelli, G. Moore, S. Silva Paes, C. Gandolfo and J. B. Laurindo, LWT – Food Sci. Technol., 2006, 39, 292–301 CrossRef.
- S. M. Martelli and J. B. Laurindo, Int. J. Polym. Mater. Polym. Biomater., 2012, 61, 17–29 CrossRef CAS.
- R. Bazargan-Lari, M. E. Bahrololoom and A. Nemati, World Appl. Sci. J., 2009, 7, 763–768 CAS.
- K. Y. Lee, Fibers Polym., 2001, 2, 71–74 CrossRef CAS.
- K. Y. Lee, S. J. Kong, W. H. Park, W. S. Ha and I. C. Kwon, J. Biomater. Sci., Polym. Ed., 1998, 9, 905–914 CrossRef CAS PubMed.
- A. Vasconcelos, G. Freddi and A. Cavaco-Paulo, Biomacromolecules, 2008, 9, 1299–1305 CrossRef CAS PubMed.
- M. Zoccola, A. Montarsolo, A. Aluigi, A. Varesano, C. Vineis and C. Tonin, e-polym., 2007, 7, 1204 Search PubMed.
- J. Borges and J. F. Mano, Chem. Rev., 2014, 114, 8883–8942 CrossRef CAS PubMed.
- X. Yang, H. Zhang, X. Yuan and S. Cui, J. Colloid Interface Sci., 2009, 336, 756–760 CrossRef CAS PubMed.
- H. Zhang, Y. Yu and S. Cui, Colloids Surf., A, 2011, 384, 501–506 CrossRef CAS.
- G. Jin, L.-M. Jiang, D.-M. Yi, H.-Z. Sun and H.-C. Sun, ChemPhysChem, 2015, 16, 3687–3694 CrossRef CAS PubMed.
- A. Tachibana, Y. Furuta, H. Takeshima, T. Tanabe and K. Yamauchi, J. Biotechnol., 2002, 93, 165–170 CrossRef CAS PubMed.
- A. Tachibana, S. Kaneko, T. Tanabe and K. Yamauchi, Biomaterials, 2005, 26, 297–302 CrossRef CAS PubMed.
- P. V. Peplow and G. J. Dias, J. Mater. Sci.: Mater. Med., 2004, 15, 1217–1220 CrossRef CAS PubMed.
- G. J. Dias, P. Mahoney, M. Swain, R. J. Kelly, R. A. Smith and M. A. Ali, J. Biomed. Mater. Res., Part A, 2010, 95, 1084–1095 CrossRef PubMed.
- A. Belcarz, G. Ginalska, J. Zalewska, W. Rzeski, A. Slosarczyk, D. Kowalczuk, P. Godlewski and J. Niedzwiadek, J. Biomed. Mater. Res., Part B, 2009, 89, 102–113 CrossRef PubMed.
- P. Kakkar, S. Verma, I. Manjubala and B. Madhan, Mater. Sci. Eng., C, 2014, 45, 343–347 CrossRef CAS PubMed.
- D. Wawro and L. Pighinelli, Int. J. Mol. Sci., 2011, 12, 7286–7300 CrossRef CAS PubMed.
- S. Hirano, in Biotechnol. Annu. Rev, ed. M. R. El-Gewely, Elsevier, 1996, vol. 2, pp. 237–258 Search PubMed.
- S. Singaravelu, G. Ramanathan, M. D. Raja, S. Barge and U. T. Sivagnanam, Mater. Lett., 2015, 152, 90–93 CrossRef CAS.
- S. Balaji, R. Kumar, R. Sripriya, P. Kakkar, D. V. Ramesh, P. N. K. Reddy and P. K. Sehgal, Mater. Sci. Eng., C, 2012, 32, 975–982 CrossRef CAS.
- P. Kakkar, S. Verma, I. Manjubala and B. Madhan, Mater. Sci. Eng., C, 2014, 45, 343–347 CrossRef CAS PubMed.
- C. Flores-Hernández, A. Colín-Cruz, C. Velasco-Santos, V. Castaño, J. Rivera-Armenta, A. Almendarez-Camarillo, P. García-Casillas and A. Martínez-Hernández, Polymers, 2014, 6, 686 CrossRef.
- K. K. Nayak and P. Gupta, Int. J. Biol. Macromol., 2015, 81, 1–10 CrossRef CAS PubMed.
- S. Hamasaki, A. Tachibana, D. Tada, K. Yamauchi and T. Tanabe, Mater. Sci. Eng., C, 2008, 28, 1250–1254 CrossRef CAS.
- A. A. El-Kheir, C. Popescu, S. Mowafi, M. Salama and H. El-Sayed, Fibers Polym., 2015, 16, 537–542 CrossRef CAS.
- X. Zhao, Y. S. Lui, C. K. C. Choo, W. T. Sow, C. L. Huang, K. W. Ng, L. P. Tan and J. S. C. Loo, Mater. Sci. Eng., C, 2015, 49, 746–753 CrossRef CAS PubMed.
- A. Aluigi, C. Vineis, A. Varesano, G. Mazzuchetti, F. Ferrero and C. Tonin, Eur. Polym. J., 2008, 44, 2465–2475 CrossRef CAS.
- A. Aluigi, A. Varesano, A. Montarsolo, C. Vineis, F. Ferrero, G. Mazzuchetti and C. Tonin, J. Appl. Polym. Sci., 2007, 104, 863–870 CrossRef CAS.
- J. Yuan, Z.-C. Xing, S.-W. Park, J. Geng, I.-K. Kang, J. Shen, W. Meng, K.-J. Shim, I.-S. Han and J.-C. Kim, Macromol. Res., 2009, 17, 850–855 CrossRef CAS.
- X. Li, Q. Feng and F. Cui, Mater. Sci. Eng., C, 2006, 26, 716–720 CrossRef CAS.
- H. Xu, S. Cai, L. Xu and Y. Yang, Langmuir, 2014, 30, 8461–8470 CrossRef CAS PubMed.
- L. S. Nair and C. T. Laurencin, in Tissue Engineering I, ed. K. Lee and D. Kaplan, Springer, Berlin, Heidelberg, 2006, pp. 47–90, DOI:10.1007/b137240.
- J. F. Mano, G. A. Silva, H. S. Azevedo, P. B. Malafaya, R. A. Sousa, S. S. Silva, L. F. Boesel, J. M. Oliveira, T. C. Santos, A. P. Marques, N. M. Neves and R. L. Reis, J. R. Soc., Interface, 2007, 4, 999–1030 CrossRef CAS PubMed.
- P. B. Malafaya, G. A. Silva and R. L. Reis, Adv. Drug Delivery Rev., 2007, 59, 207–233 CrossRef CAS PubMed.
- A. O. Elzoghby, W. S. El-Fotoh and N. A. Elgindy, J. Controlled Release, 2011, 153, 206–216 CrossRef CAS PubMed.
- M. Curcio, B. Blanco-Fernandez, L. Diaz-Gomez, A. Concheiro and C. Alvarez-Lorenzo, Bioconjugate Chem., 2015, 26, 1900–1907 CrossRef CAS PubMed.
- S. Tomblyn, E. L. Pettit Kneller, S. J. Walker, M. D. Ellenburg, C. J. Kowalczewski, M. Van Dyke, L. Burnett and J. M. Saul, J. Biomed. Mater. Res., Part B, 2016, 104(5), 864–879 CrossRef CAS PubMed.
- C. C. Peyton, T. Keys, S. Tomblyn, D. Burmeister, J. H. Beumer, J. L. Holleran, J. Sirintrapun, S. Washburn and S. J. Hodges, J. Surg. Res., 2012, 178, 545–552 CrossRef CAS PubMed.
- M. Biondi, F. Ungaro, F. Quaglia and P. A. Netti, Adv. Drug Delivery Rev., 2008, 60, 229–242 CrossRef CAS PubMed.
- A. Kurimoto, T. Tanabe, A. Tachibana and K. Yamauchi, J. Biosci. Bioeng., 2003, 96, 307–309 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS PubMed.
- P. Hill, H. Brantley and M. Van Dyke, Biomaterials, 2010, 31, 585–593 CrossRef CAS PubMed.
- Q. Li, L. Zhu, R. Liu, D. Huang, X. Jin, N. Che, Z. Li, X. Qu, H. Kang and Y. Huang, J. Mater. Chem., 2012, 22, 19964–19973 RSC.
- T. Aboushwareb, D. Eberli, C. Ward, C. Broda, J. Holcomb, A. Atala and M. Van Dyke, J. Biomed. Mater. Res., Part B, 2009, 90, 45–54 Search PubMed.
- A. Hasan, A. Khattab, M. A. Islam, K. A. Hweij, J. Zeitouny, R. Waters, M. Sayegh, M. M. Hossain and A. Paul, Adv. Sci., 2015, 2, 1500122 CrossRef PubMed.
- L. R. Burnett, J. G. Richter, M. B. Rahmany, R. Soler, J. A. Steen, G. Orlando, T. Abouswareb and M. E. Van Dyke, J. Biomater. Appl., 2014, 28, 869–879 CrossRef PubMed.
- M. B. Rahmany, R. R. Hantgan and M. Van Dyke, Biomaterials, 2013, 34, 2492–2500 CrossRef CAS PubMed.
- L. R. Burnett, M. B. Rahmany, J. R. Richter, T. A. Aboushwareb, D. Eberli, C. L. Ward, G. Orlando, R. R. Hantgan and M. E. Van Dyke, Biomaterials, 2013, 34, 2632–2640 CrossRef CAS PubMed.
- V. Vipin, V. Poonam, R. Pratima and R. R. Alok, Biomed. Mater., 2008, 3, 025007 CrossRef PubMed.
- R. O. Labrador, M. Butí and X. Navarro, Exp. Neurol., 1998, 149, 243–252 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- I. M. Aroso, J. C. Silva, F. Mano, A. S. D. Ferreira, M. Dionísio, I. Sá-Nogueira, S. Barreiros, R. L. Reis, A. Paiva and A. R. C. Duarte, Eur. J. Pharm. Biopharm., 2016, 98, 57–66 CrossRef CAS PubMed.
- L. J. Szewciw, D. G. de Kerckhove, G. W. Grime and D. S. Fudge, Proc. R. Soc. London, Ser. B, 2010, 277, 2597–2605 CrossRef CAS PubMed.
- M. Schaffeld, M. Haberkamp, S. Schätzlein, S. Neumann and C. Hunzinger, Front. Zool., 2007, 4, 16 CrossRef PubMed.
- D. S. Fudge and J. M. Gosline, Proc. R. Soc. London, Ser. B, 2004, 271, 291–299 CrossRef CAS PubMed.
- H. Ehrlich, Biological Materials of Marine Origin: Vertebrates, Springer, Netherlands, 2014 Search PubMed.
- T. H. Silva, A. Alves, B. M. Ferreira, J. M. Oliveira, L. L. Reys, R. J. F. Ferreira, R. A. Sousa, S. S. Silva, J. F. Mano and R. L. Reis, Int. Mater. Rev., 2012, 57, 276–306 CrossRef CAS.
- S. Singaravelu, G. Ramanathan, M. D. Raja, S. Barge and U. T. Sivagnanam, Mater. Lett., 2015, 152, 90–93 CrossRef CAS.
- B. S. Harrap and J. M. Gillespie, Aust. J. Biol. Sci., 1963, 16, 542–556 CrossRef CAS.
- J. M. Gillespie, Aust. J. Biol. Sci., 1964, 17, 282–300 CAS.
- I. J. O'Donnell and E. O. P. Thompson, Aust. J. Biol. Sci., 1964, 19, 737–989 Search PubMed.
- P. M. M. Schrooyen, P. J. Dijkstra, R. G. Oberthü, A. Bantjes and J. Feijen, J. Agric. Food Chem., 2000, 48, 4326–4334 CrossRef CAS PubMed.
- C. H. Zeng and Q. Lu, Adv. Mater. Res., 2014, 881–883, 551–555 CrossRef.
- M. Canetti, A. Cacciamani and F. Bertini, J. Polym. Res., 2013, 20, 1–8 CrossRef CAS.
- C. Wu, J. Li, D. Wicks, S. Morgan and R. Smith, Hydratable keratin compositions, EP1888099 B1, 2008 Search PubMed.
- J. D. Leeder and R. C. Marshall, Text. Res. J., 1982, 52, 245–249 CrossRef CAS.
- S. J. Hong, G. S. Park, B. K. Jung, A. R. Khan, Y. J. Park, C. H. Lee and J. H. Shin, J. Appl. Biol. Chem., 2015, 58, 247–251 CrossRef.
- A. Riffel, F. Lucas, P. Heeb and A. Brandelli, Arch. Microbiol., 2003, 179, 258–265 CrossRef CAS PubMed.
- S. Sangali and A. Brandelli, J. Appl. Microbiol., 2000, 89, 735–743 CrossRef CAS PubMed.
- A. Grazziotin, F. A. Pimentel, S. Sangali, E. V. de Jong and A. Brandelli, Bioresour. Technol., 2007, 98, 3172–3175 CrossRef CAS PubMed.
- J. D. Allpress, G. Mountain and P. C. Gowland, Lett. Appl. Microbiol., 2002, 34, 337–342 CrossRef CAS PubMed.
- Z. Fang, J. Zhang, B. Liu, G. Du and J. Chen, Int. Biodeterior. Biodegrad., 2013, 82, 166–172 CrossRef CAS.
- J. H. Jeong, O. M. Lee, Y. D. Jeon, J. D. Kim, N. R. Lee, C. Y. Lee and H. J. Son, Process Biochem., 2010, 45, 1738–1745 CrossRef CAS.
- Z. J. Cao, Q. Zhang, D. K. Wei, L. Chen, J. Wang, X. Q. Zhang and M. H. Zhou, J. Ind. Microbiol. Biotechnol., 2009, 36, 181–188 CrossRef CAS PubMed.
- W.-H. Lo, J.-R. Too and J.-Y. Wu, J. Biosci. Bioeng., 2012, 114, 640–647 CrossRef CAS PubMed.
- S. Mousavi, M. Salouti, R. Shapoury and Z. Heidari, Jundishapur J. Microbiol., 2013, 6, e7160 Search PubMed.
- S. Rahayu, D. Syah and M. Thenawidjaja Suhartono, Biocatal. Agric. Biotechnol., 2012, 1, 152–158 CAS.
- T. I. Zaghloul, A. M. Embaby and A. R. Elmahdy, Bioresour. Technol., 2011, 102, 2387–2393 CrossRef CAS PubMed.
- L. Vidal, P. Christen and M. N. Coello, World J. Microbiol. Biotechnol., 2000, 16, 551–554 CrossRef.
- A. G. Kumar, S. Swarnalatha, S. Gayathri, N. Nagesh and G. Sekaran, J. Appl. Microbiol., 2008, 104, 411–419 CAS.
- A. Lateef, I. A. Adelere and E. B. Gueguim-Kana, Biotechnol. Biotechnol. Equip., 2015, 29, 54–63 CrossRef CAS PubMed.
- A. Bose, S. Pathan, K. Pathak and H. Keharia, Waste Biomass Valorization, 2014, 5, 595–605 CrossRef CAS.
- S. Kaul and G. Sumbali, Mycopathologia, 1997, 139, 137–140 CrossRef CAS PubMed.
- G. Mitola, F. Escalona, R. Salas, E. García and A. Ledesma, Mycopathologia, 2003, 156, 163–169 CrossRef CAS.
- P. Anbu, S. C. Gopinath, A. Hilda, N. Mathivanan and G. Annadurai, Can. J. Microbiol., 2006, 52, 1060–1069 CrossRef CAS PubMed.
- N. Rodrigues Marcondes, C. Ledesma Taira, D. Cirena Vandresen, T. I. Estivalet Svidzinski, M. K. Kadowaki and R. M. Peralta, Microb. Ecol., 2008, 56, 13–17 CrossRef PubMed.
- H. Gradišar, S. Kern and J. Friedrich, Appl. Microbiol. Biotechnol., 2000, 53, 196–200 CrossRef.
- R. M. D. B. Santos, A. A. P. Firmino, C. M. De Sá and C. R. Felix, Curr. Microbiol., 1996, 33, 364–370 CrossRef CAS PubMed.
- A. M. Mazotto, S. Couri, M. C. T. Damaso and A. B. Vermelho, Int. Biodeterior. Biodegrad., 2013, 85, 189–195 CrossRef CAS.
- L. Cao, H. Tan, Y. Liu, X. Xue and S. Zhou, Lett. Appl. Microbiol., 2008, 46, 389–394 CrossRef CAS PubMed.
- P. Sierpinski, J. Garrett, J. Ma, P. Apel, D. Klorig, T. Smith, L. A. Koman, A. Atala and M. Van Dyke, Biomaterials, 2008, 29, 118–128 CrossRef CAS PubMed.
- C. E. Tanase and I. Spiridon, Mater. Sci. Eng., C, 2014, 40, 242–247 CrossRef CAS PubMed.
- J. Choi, G. Panthi, Y. Liu, J. Kim, S.-H. Chae, C. Lee, M. Park and H.-Y. Kim, Polymer, 2015, 58, 146–152 CrossRef CAS.
- M. Park, B. S. Kim, H. K. Shin, S. J. Park and H. Y. Kim, Mater. Sci. Eng., C, 2013, 33, 5051–5057 CrossRef CAS PubMed.
- L. Yong, L. Jia, F. Jie and W. Meng, Matéria, 2014, 19, 382–388 Search PubMed.
- L. A. Pace, J. F. Plate, T. L. Smith and M. E. Van Dyke, Biomaterials, 2013, 34, 5907–5914 CrossRef CAS PubMed.
- M. Park, H. K. Shin, B.-S. Kim, M. J. Kim, I.-S. Kim, B.-Y. Park and H.-Y. Kim, Mater. Sci. Eng., C, 2015, 55, 88–94 CrossRef CAS PubMed.
- D. Poranki, W. Whitener, S. Howse, T. Mesen, E. Howse, J. Burnell, O. Greengauz-Roberts, J. Molnar and M. Van Dyke, J. Biomater. Appl., 2013, 29, 26–35 CrossRef PubMed.
- L. Ajay Sharma, M. A. Ali, R. M. Love, M. J. Wilson and G. J. Dias, Int. Endod. J., 2016, 49(5), 471–482 CrossRef CAS PubMed.
- S. Wang, Z. Wang, S. E. M. Foo, N. S. Tan, Y. Yuan, W. Lin, Z. Zhang and K. W. Ng, ACS Appl. Mater. Interfaces, 2015, 7, 5187–5198 CAS.
- E. Senoz, R. P. Wool, C. W. J. McChalicher and C. K. Hong, Polym. Degrad. Stab., 2012, 97, 297–307 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |