
Enhancing the sensing specificity of a MoS2 nanosheet-based FRET aptasensor using a surface blocking strategy
Alisha
Geldert†
a,
Kenry†
 abc,
Xiao
Zhang
de,
Hua
Zhang
d and
Chwee Teck
Lim
*abcf
abc,
Xiao
Zhang
de,
Hua
Zhang
d and
Chwee Teck
Lim
*abcf
aDepartment of Biomedical Engineering, National University of Singapore, Singapore 117576. E-mail: ctlim@nus.edu.sg
bNUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, Singapore 117456
cCentre for Advanced 2D Materials and Graphene Research Centre, National University of Singapore, Singapore 117543
dCenter for Programmable Materials, School of Materials Science and Engineering, Nanyang Technological University, Singapore 639798
eEnergy Research Institute @NTU (ERI@N), Interdisciplinary Graduate School, Nanyang Technological University, Singapore 637553
fMechanobiology Institute, National University of Singapore, Singapore 117411
First published on 17th May 2017
Abstract
Aptamer-based biosensing, which uses short, single-stranded nucleic acid segments to bind to a target, can be advantageous over antibody-based diagnostics due to the ease of synthesis and high stability of aptamers. However, the development of most aptamer-based sensors (aptasensors) is still in its initial stages and many factors affecting their performance have not been studied in great detail. Here, we enhance the sensing specificity of a fluorescence resonance energy transfer (FRET)-based MoS2 nanosheet aptasensor in detecting the malarial biomarker Plasmodium lactate dehydrogenase (pLDH). In this sensing scheme, the presence of target is signaled by an increase in fluorescence when fluorescently-labeled aptamers bind to pLDH and release from a quenching material. Interestingly, unlike most of the reported literature on aptasensors, we observe that non-target proteins also cause a considerable increase in the detected fluorescence. This may be due to the nonspecific adsorption of proteins onto the fluorescence quencher, leading to the displacement of aptamers from the quencher surface. To reduce this nonspecific association and to enhance the sensor specificity, we propose the application of a surface blocking agent to the quenching material. Importantly, we demonstrate that the sensing specificity of the MoS2 nanosheet-based aptasensor towards target pLDH biomolecules can be significantly enhanced through surface passivation, thus contributing to the development of highly selective and robust point-of-care malaria diagnostics.
Introduction
Point-of-care medical testing benefits greatly from simple biosensors which can accurately and specifically detect and signal the presence of disease biomarkers to enable rapid and accessible disease diagnosis. Conventionally, most rapid diagnostic tests rely on antibodies to bind to biomarkers in a clinical sample. However, aptamers – short, single-stranded nucleic acid segments which can be developed to bind specifically to a range of biomolecules – have gained growing interest as an alternative tool for biosensing in recent years. Aptamers can be selected for essentially any biomolecule through a process known as systematic evolution of ligands by exponential enrichment (SELEX), and many reported aptamers have similar affinity to their targets as antibodies do.1 As compared to antibody production, aptamer synthesis is affordable and more highly reproducible. Aptamers are also more easily modified with functionalities such as fluorescent labels, biotin tags, or chemical species to render them more nuclease-resistant.2 They tend to be smaller in size than antibodies, enabling aptamers to be more densely packed on the surface of a sensor for improved sensitivity. While antibodies denature irreversibly at high temperatures, nucleic acid denaturation is reversible, facilitating the long-term storage, transport, and use of aptamer-based diagnostics, especially at high temperatures and in other harsh conditions. Thus, aptamers are potentially cheaper, simpler, and more robust affinity reagents for biosensing applications.1,3,4One simple scheme commonly employed in aptamer-based biosensing (i.e., aptasensing) is fluorescence resonance energy transfer (FRET). In FRET, fluorescently-labeled aptamers are typically combined with a fluorescence quenching material, to which aptamers will spontaneously adsorb and their fluorescence will be quenched. Interestingly, adsorption to the quenching material also protects the aptamers from nuclease degradation due to steric hindrance.5 Upon the addition of an analyte containing the aptamer target, the aptamer will change its conformation to bind to its target, causing the aptamer to be released from the quencher and resulting in a measurable fluorescence increase. Two-dimensional (2D) nanomaterials, such as graphene-based and solution-processed transition metal chalcogenide nanosheets, may be ideal fluorescence quenching materials. These nanomaterials have been actively explored for bioapplications6–15 and are increasingly being used as fluorescence quenchers due to their large specific surface area, high hydrophilicity, and exceptional fluorescence quenching abilities.16–18
In our previous contribution, we demonstrated the development of a FRET-based aptasensor using MoS2 nanosheets for the detection of a malarial biomarker, Plasmodium lactate dehydrogenase (pLDH).13 pLDH is a soluble metabolic enzyme produced by all species of blood-stage Plasmodium parasites, and the pLDH concentration in blood has been found to be directly correlated to the level of parasitemia.19 The aptamer-based detection of pLDH is highly attractive, as the current antibody-based pLDH-targeted diagnostics typically suffer from poor performance in hot and humid conditions, such as in tropical climates in which malaria is prevalent.20,21 The pLDH concentration in the blood samples of Plasmodium vivax-infected patients has been found to vary widely, with a mean of 3.9 ± 6.1 μg mL−1 (112 ± 175 nM). Importantly, over 24% of patients had pLDH levels of less than 50 ng mL−1 (1.43 nM), but the three evaluated commercial antibody-based rapid diagnostic tests had limits of detection between 25 and 50 ng mL−1, indicating insufficient sensitivity of these sensors.22 As a proof of concept, using pLDH-spiked solutions, we showed that our aptamer-based sensor possesses a limit of detection of 19.2 ng mL−1 (550 pM) pLDH, suggesting its promise as an alternative malaria diagnostic tool.13 However, the sensitivity and specificity of the sensor will need to be further improved for the handling of clinical blood samples.
Our FRET aptasensor is simple and attractive as it operates based on spontaneous adsorption and desorption without the need for complex synthesis or assay procedures. However, the simplicity of FRET-based aptasensors may also limit their sensing performance. In fact, fluorescence quenchers such as graphene oxide (GO), molybdenum disulfide (MoS2), and other 2D nanomaterials have been found to possess moderate affinities not only to aptamers but also to a range of proteins.23–25 Importantly, the nonspecific adsorption of both target and non-target biomolecules onto these fluorescence quenchers may affect the sensor performance. The adsorption of target protein molecules onto the quencher surface reduces the specific signal by decreasing the number of target molecules available to bind to and release the aptamers from the quencher surface to recover fluorescence, as was reported for a GO-based aptasensor for ochratoxin A.26 Furthermore, non-target proteins found in the analyte may adsorb to the quencher surface and interfere with fluorescence recovery or induce nonspecific recovery, eventually reducing the sensor specificity. Also, it may be challenging to determine the aptamer-to-quencher ratio which maximizes the signal-to-noise ratio because of nonspecific adsorption on the quencher surface. Currently, there has been little investigation into the details involved in FRET-based aptasensing or various factors affecting sensor performance.
As such, in this contribution, we aim to examine the existence and origin of nonspecific fluorescence recovery in our pLDH aptasensor and propose a facile method to improve the sensor specificity. We first investigate the interactions between pLDH aptamer and its target as well as nonspecific proteins typically found in a blood sample. In light of our experimental data, we propose that the nonspecific fluorescence recovery may be caused by proteins adsorbing nonspecifically onto the fluorescence quencher and displacing aptamers from its surface. To mitigate this nonspecific protein adsorption, the quencher surface is blocked in order to improve the sensor specificity significantly. Although quenching materials in FRET-based aptasensing schemes have occasionally been coated with blocking agents, the underlying motivation as well as the effect of this blocking have not been clearly demonstrated.26–28 Consequently, through this work, we aim to elucidate the mechanism of nonspecific fluorescence recovery in FRET-based aptasensors and demonstrate the application of surface blocking to enhance sensor specificity.
Results and discussion
An aptamer which has been demonstrated to bind to pLDH,29 with the fluorescent dye 6-carboxyfluorescein (FAM) conjugated to the 3′ end, was employed throughout this study. We first characterized the optical properties of the aptamer using absorbance, fluorescence, and circular dichroism (CD) spectroscopy. The aptamer possessed an absorbance peak at around 505 nm and a fluorescence peak at around 525 nm (Fig. 1a), which is characteristic of the FAM dye. This showed that the free aptamer can produce a detectable fluorescent signal, which is crucial to FRET-based sensing. Proper aptamer folding is also required to form the specific stem, loop, or quadruplex structures necessary for target recognition and binding. Because aptamer folding is dependent on temperature, ionic concentration of the solvent, and many other factors,30,31 we aimed to ensure that the pLDH aptamer could fold into its proper conformation in the prepared buffer solution, even with its fluorescent label. To do this, the CD spectrum of the aptamer was characterized and we observed that the spectrum exhibited a characteristic positive peak near 280 nm and negative peak near 240 nm (Fig. 1b), matching closely with that reported in literature. Altogether, these results suggested that the pLDH aptamer used here had proper fluorescence and secondary structure to bind to its target and produce a detectable signal to indicate the presence of its target biomolecule.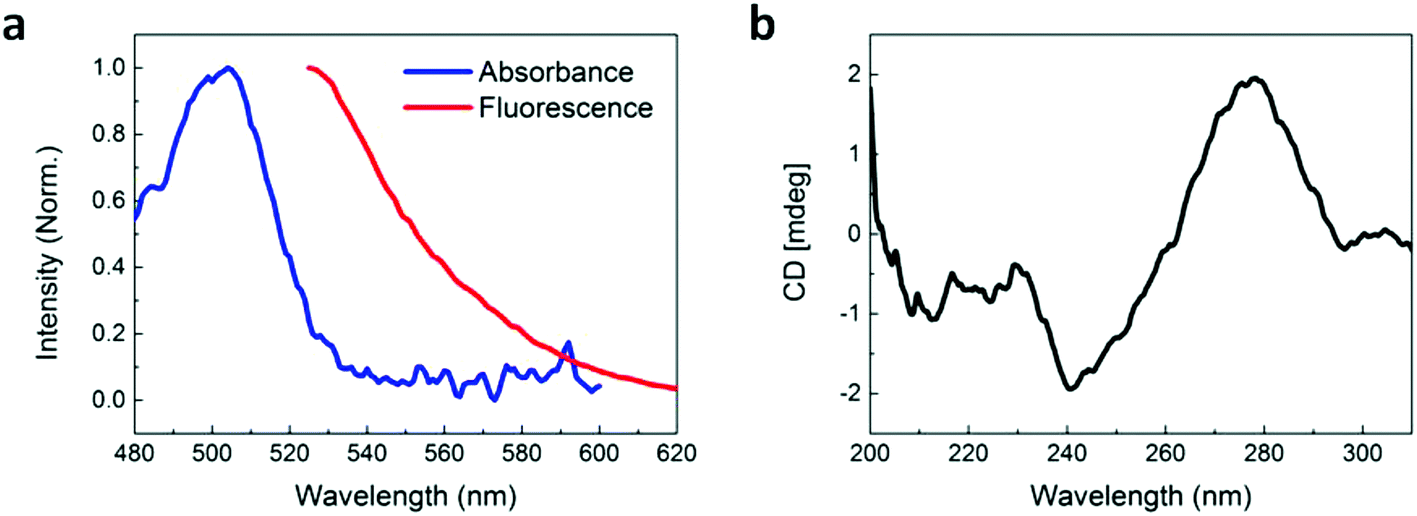 | ||
| Fig. 1 Characterization of the fluorescently-labeled pLDH aptamer. (a) Normalized absorbance and fluorescence of the pLDH aptamer. Fluorescence measurements were taken at an excitation wavelength of 495 nm, corresponding to the absorbance maximum of the FAM dye label. (b) CD spectrum of the pLDH aptamer, with negative and positive peaks around 240 nm and 280 nm, respectively. | ||
In order to have value as a diagnostic tool, the aptamer must bind specifically to its target, pLDH protein, and not to other biomolecules, so as to minimize false positive test results. As such, we used CD spectroscopy to probe the interactions between pLDH aptamer and both pLDH protein (Fig. 2a) and common plasma proteins present in a blood sample (i.e., fibrinogen (Fig. 2b), γ-globulin (Fig. 2c), and albumin (Fig. 2d)). The CD spectra of aptamer–protein mixtures were measured at several timepoints: immediately and after 30 and 60 minutes of incubation. In the far-UV range (i.e., <250 nm), the protein CD spectra exhibited negative peaks associated with the alpha helix and beta sheet secondary structures, while the aptamer CD spectrum remained near zero. Thus, differences in the 200–250 nm range of the CD spectra of aptamer–protein mixtures may be attributed to changes in protein structure, as the aptamer contributes negligibly to the CD spectrum at this wavelength range. Similarly, at around 280 nm, the CD spectrum of the pLDH aptamer displayed a peak (albeit smaller due to the lesser chirality of single-stranded DNA), while the CD spectra of all tested proteins were near zero. Thus, changes in the 280 nm peak in the CD spectra of aptamer–protein mixtures represent changes in the aptamer structure.
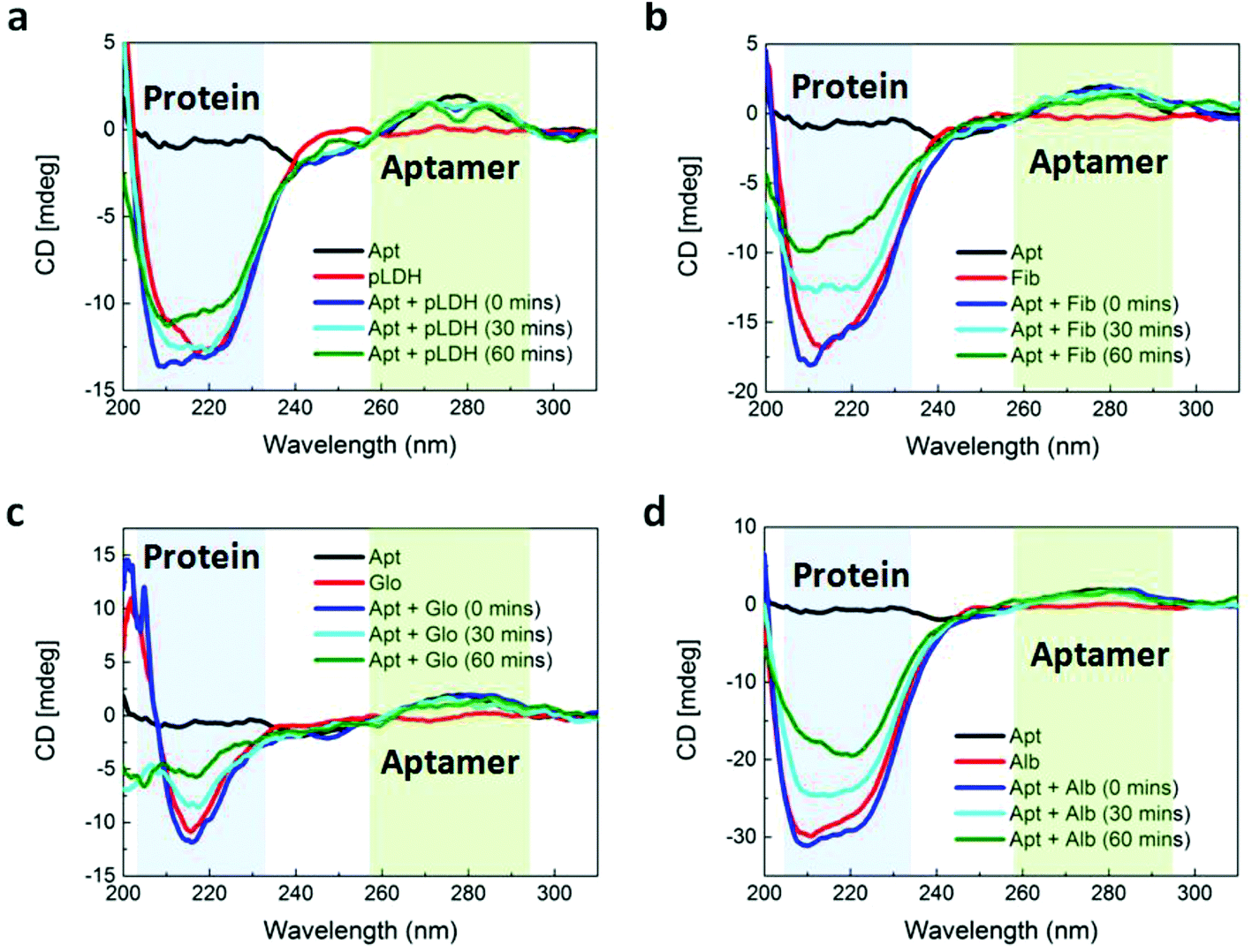 | ||
| Fig. 2 CD characterization of the structures of aptamer, proteins, and aptamer–protein mixtures. The CD spectra of 5 μM pure pLDH aptamer (Apt), 2.5 μM of pure: (a) pLDH, (b) fibrinogen (Fib), (c) γ-globulin (Glo), and (d) albumin (Alb) proteins, and 5 μM pLDH aptamer incubated with 2.5 μM proteins for varying durations. The shaded blue and green regions indicate the wavelength ranges containing the characteristic CD peaks of protein and aptamer structures, respectively. | ||
Aptamer–ligand binding is typically described by an induced-fit model, in which a ligand induces an aptamer to fold to precisely fit the ligand. When the two molecules bind, the aptamer undergoes a significant conformational change, while the target typically does not change its conformation.32,33 According to this model, we would anticipate the CD peak corresponding to the aptamer to be altered when the aptamer is incubated with its target, reflecting the change in aptamer structure induced by its target ligand. Previous studies have reported changes in the aptamer peak of the CD spectrum when aptamers bound to targets such as L-tyrosinamide34 and thrombin.35 Similarly, we observed a distinct change in the shape of the aptamer peak at 280 nm when pLDH aptamer was incubated with its target, pLDH protein (Fig. 2a). As the aptamer and its target were incubated for longer durations, the signal intensity near 280 nm decreased, forming a spectrum with two smaller peaks on either side of 280 nm. When the aptamer was incubated with nonspecific proteins, however, the 280 nm peak retained its original shape (Fig. 2b–d). This indicated that the structure of the pLDH aptamer changed more significantly when it was incubated with pLDH protein than with nonspecific proteins, which suggested that the aptamer bound specifically to its target.
We also aimed to use CD spectroscopy to assess the conformation of proteins when they were incubated with pLDH aptamer. To the best of our knowledge, previous studies have only used CD spectroscopy to assess conformational changes of aptamers, not proteins, when the two are incubated together. Based on the induced-fit model, we anticipated that the conformations of all proteins would remain unaffected during incubation with the aptamer. However, the far-UV region of the obtained CD spectra, corresponding to the protein secondary structure, yielded ambiguous results. When pLDH aptamer was incubated with its target, pLDH protein, a small gradual change in the magnitude of the negative peak of the CD spectra could be observed over time. In contrast, as pLDH aptamer was incubated with nonspecific plasma proteins for the same amount of time, the magnitude of their negative peaks varied more considerably. This may suggest that the conformation of pLDH protein might not change significantly upon aptamer binding, which would agree with the induced-fit model. However, there was also variability in the shapes of the protein and aptamer–protein CD spectra, which would complicate the analysis. As such, further studies using crystallography or other more advanced techniques would be beneficial in elucidating the effect of aptamers on the conformations of target and nonspecific proteins.
To further assess and confirm the specificity of the aptamer, we performed a binding assay in which fluorescently-labeled pLDH aptamer was incubated on top of a protein-coated surface for 10 minutes and then rinsed to wash away any unbound aptamer. Areas which fluoresced after the rinsing process indicated the occurrence of aptamer–protein binding. Here, we observed more fluorescent regions on the pLDH-coated surface (Fig. 3a) as compared to the fibrinogen-, γ-globulin-, and albumin-coated surfaces (Fig. 3b–d), supporting our earlier finding that pLDH aptamer binds specifically to its target. The results were further quantified by measuring the fluorescent area from several random locations in the images of each protein surface (Fig. 3e). Though the fluorescent area observed in the aptamer-pLDH incubation varied between images, the specific pairing exhibited a much higher total amount of aptamer–protein binding than any of the nonspecific incubations, which all had a negligible total fluorescent area.
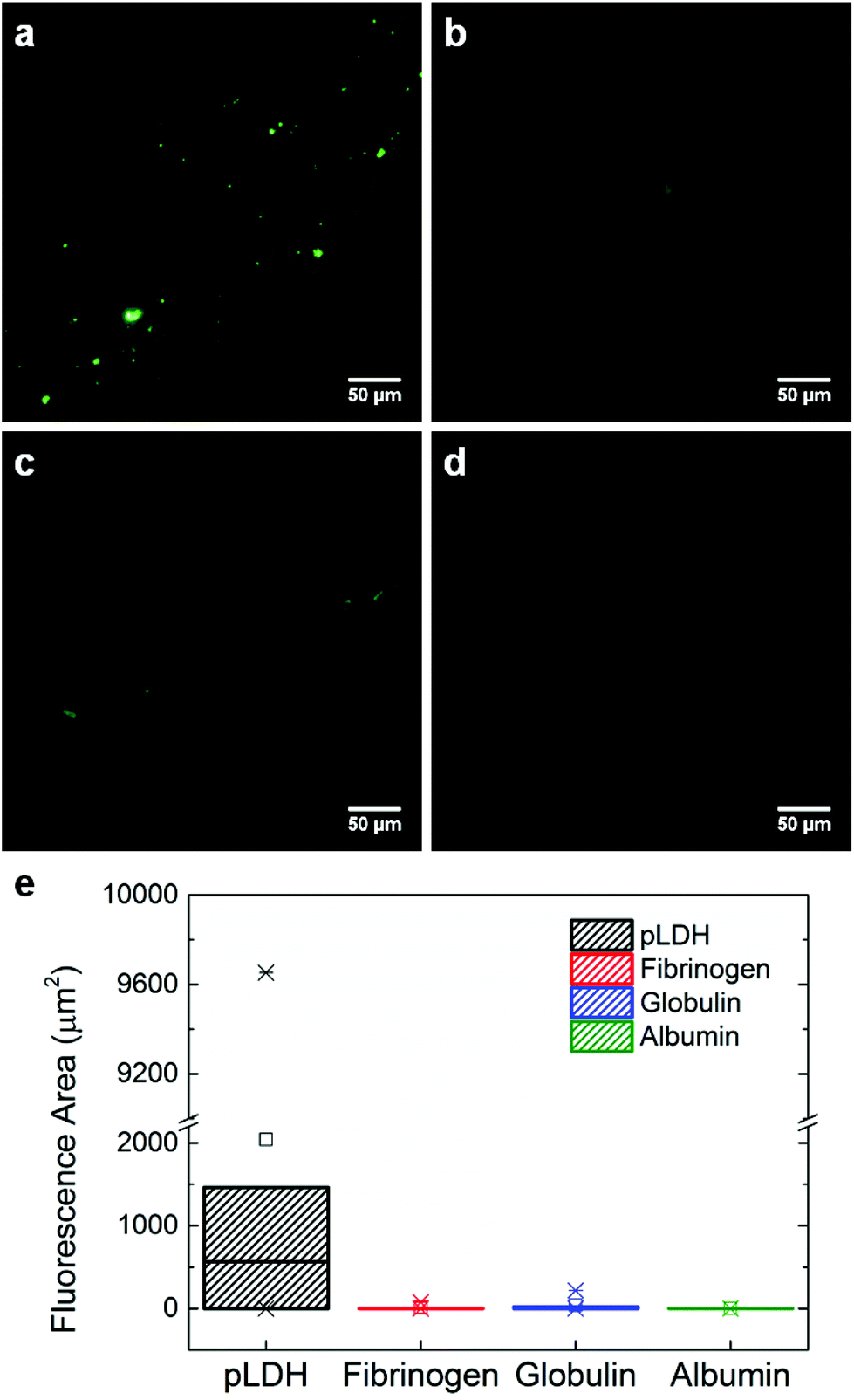 | ||
| Fig. 3 Aptamer–protein binding assay. Fluorescent areas indicate the binding between pLDH aptamer and (a) pLDH, (b) fibrinogen, (c) γ-globulin, or (d) albumin protein. 7.16 μM of protein was adsorbed onto a mica surface, on top of which 10 μM fluorescently-labeled pLDH aptamer was incubated and then rinsed. (e) Box plot quantifying the fluorescent areas measured from images at six randomly chosen locations for each aptamer–protein incubation. The higher number of fluorescent areas observed in the pLDH aptamer-pLDH protein incubation shows that pLDH aptamer binds specifically to its target. | ||
After investigating the aptamer–protein interactions and verifying the proper functionality of pLDH aptamer, we assessed the specific and nonspecific fluorescence recovery of our FRET aptasensor. Fluorescently-labeled pLDH aptamer was incubated with single-layer MoS2 nanosheets (as synthesized and characterized in our previous work)36 for 10 minutes to allow the aptamer to adsorb to and be quenched by MoS2 nanosheets. Subsequently, an analyte solution containing pLDH or a nonspecific protein was added to the aptamer-MoS2 incubation, and fluorescence readings were taken after 30 minutes to measure the amount of fluorescence recovery induced by the protein. Although our experimental results on the interactions between pLDH aptamer and a wide range of proteins suggested that pLDH binds specifically to its target, intriguingly, the plasma proteins fibrinogen, globulin, and albumin all induced considerable fluorescence recovery of almost half the magnitude of that of the specific recovery due to pLDH (Fig. 4a, unblocked bars).
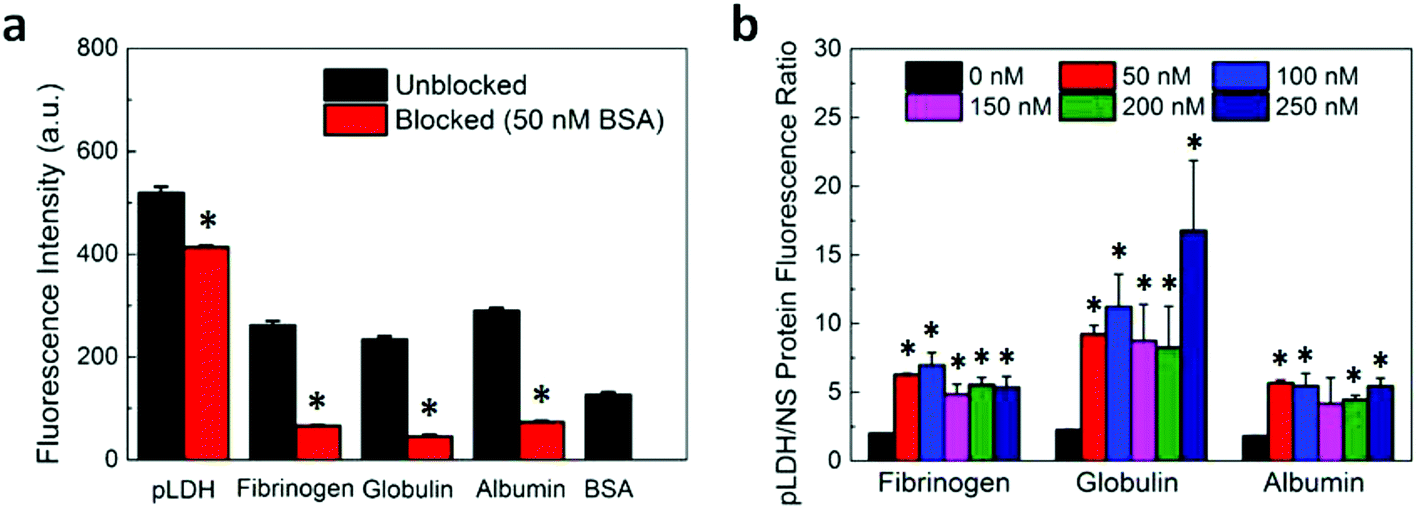 | ||
| Fig. 4 Effect of surface blocking on the sensing specificity of the pLDH aptasensor. (a) Fluorescence intensity after the addition of 250 nM protein to either an aptasensor which was unblocked or blocked with 50 nM BSA. The fluorescence recovery induced by the blocking agent (i.e., 50 nM BSA) is also shown. (b) Ratio of the fluorescence measured after the addition of 250 nM pLDH protein to the fluorescence measured after the addition of 250 nM of a nonspecific protein when the aptasensor was blocked with varying concentrations of BSA. All fluorescence measurements were taken with an excitation wavelength of 495 nm and an emission wavelength of 527 nm. The * indicates that the fluorescence ratios of the blocked samples are significantly different than that of the unblocked sample (i.e., p < 0.05). | ||
As our characterization data (Fig. 2 and 3) indicate that the aptamer binds specifically to pLDH, we hypothesized that the observed nonspecific fluorescence recovery was not primarily caused by nonspecific proteins binding to aptamers and releasing them from the MoS2 nanosheet surface. Instead, it is possible that the nonspecific proteins adsorbed to the MoS2 nanosheet and displaced the aptamers from the nanosheet surface, causing the aptamer fluorescence to be restored. In this case, preventing nonspecific protein adsorption onto the nanosheet surface should reduce the amount of nonspecific fluorescence recovery (Scheme 1). In fact, nonspecific protein adsorption has been known to pose challenges to the development of various biomedical devices, notably medical implants. One common strategy to combat nonspecific protein adsorption on implants is to immobilize blocking molecules on the surface of the biomaterial, as Kenausis et al. demonstrated by coating metal oxide surfaces with a blocking agent to reduce the adsorption of fibrinogen, albumin, and other serum proteins.37 Motivated by this, we implemented a similar blocking strategy, using bovine serum albumin (BSA) as a blocking agent to coat the nanosheet surface and prevent nonspecific protein adsorption. BSA, in general, possesses a surface passivating effect and has been actively utilized as an inexpensive surface blocking agent to mitigate nonspecific adsorption on photoresists, polystyrene, and other materials.38–40 However, BSA has rarely been explored as a blocking agent for 2D nanomaterials.
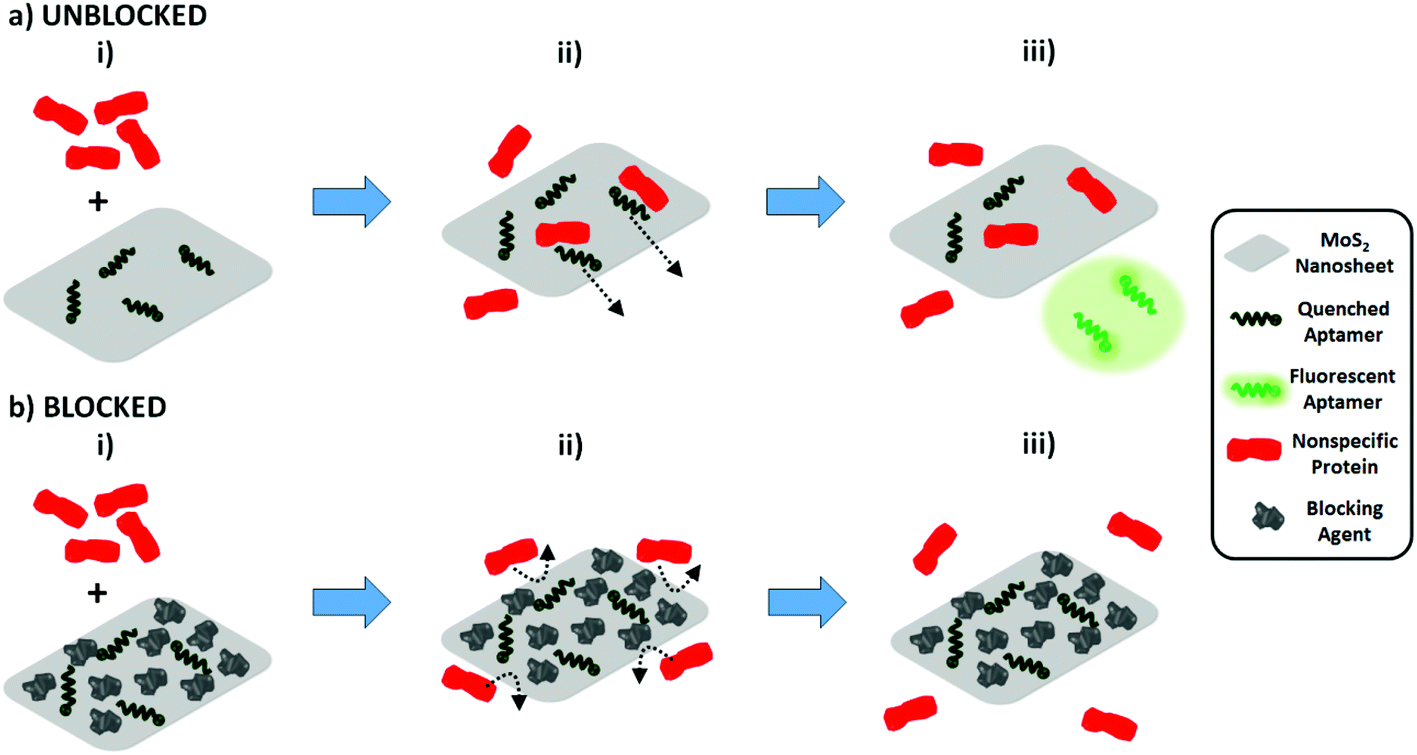 | ||
| Scheme 1 Effect of MoS2 nanosheet surface blocking on nonspecific fluorescence recovery. (a) Unblocked MoS2 nanosheets. (i) When nonspecific proteins are added to a solution of unblocked MoS2 nanosheets with aptamers adsorbed on the nanosheet surface, (ii) nonspecific proteins may adsorb onto the nanosheet and displace aptamers from the surface, (iii) restoring the aptamers’ fluorescence. (b) Surface-blocked MoS2 nanosheets. (i) In contrast, when the MoS2 nanosheet surface (with aptamers adsorbed) is blocked prior to protein addition, the amount of available space on the nanosheet will be reduced. (ii) This will prevent the adsorption of nonspecific proteins onto the nanosheet, (iii) thus mitigating aptamer displacement from the quencher surface and reducing the amount of nonspecific fluorescence recovery significantly. | ||
For blocked samples, BSA was incubated with the aptamer-nanosheet solution for 10 minutes to allow it to adsorb to available spaces on the nanosheet surface, prior to analyte addition. The fluorescence recovery due to BSA alone was subtracted from fluorescence recovery of blocked samples after analyte addition to account for any aptamer displacement caused by BSA. Interestingly, we observed that although the recovery due to pLDH was slightly lower in the blocked case, the recovery due to nonspecific proteins decreased to a large extent when blocking was applied (Fig. 4a). As a signal-to-noise metric, we calculated the ratio of the fluorescence recovery induced by pLDH to the recovery induced by nonspecific proteins. This signal-to-noise ratio was significantly higher when BSA blocking was implemented prior to analyte addition as compared to when the analyte was added directly to the unblocked aptamer-MoS2 nanosheet incubation (Fig. 4b). Particularly, the pLDH fluorescence recovery was only about twice as high as nonspecific protein recovery in unblocked samples, while in blocked samples, the specific pLDH fluorescence recovery was at least four times as high as the nonspecific recovery. Also, the effect of BSA blocking was not concentration-dependent, as a wide range of BSA concentrations generated similar signal-to-noise ratios. Ultimately, in all cases, BSA blocking resulted in higher ratios of pLDH-induced to nonspecific protein-induced fluorescence recovery, demonstrating that surface blocking of the fluorescence quencher can reduce the background signal. As it requires only one additional incubation step, blocking serves as an attractive strategy to significantly improve sensor specificity while maintaining the simplicity of the FRET-based aptasensing technique.
Conclusions
We investigated the interactions of an aptamer for a malarial biomarker, pLDH, with both its specific target and nonspecific plasma proteins commonly present in a blood sample to identify the existence and origin of the nonspecific fluorescence recovery in a MoS2 nanosheet-based FRET aptasensor. We then adopted a blocking strategy to improve the sensing specificity of the aptasensor for the diagnosis of malaria, in which the surface of the fluorescence quenching nanosheets was passivated with BSA prior to analyte detection. This significantly reduced the fluorescence recovery induced by nonspecific proteins, most likely by preventing them from binding to the quencher and displacing the aptamers from the quencher surface. We believe that this study has provided useful insights into various factors affecting the performance of FRET-based aptasensors. This study has also demonstrated the successful combination of a FRET aptasensing technique with a blocking strategy for the detection of the malarial biomarker pLDH with high specificity against other proteins commonly found in the blood, supporting the development of a simple and stable rapid diagnostic test for malaria.Methods
Materials
MoS2 nanosheets were synthesized using the electrochemical lithium intercalation/exfoliation method reported by our group.17,36 Fluorescently-labeled pLDH aptamer with the sequence 5′-GTT CGA TTG GAT TGT GCC GGA AGT GCT GGC TCG AAC-FAM-3′ was ordered from Integrated DNA Technologies. The aptamer was dissolved in a buffer of 20 mM Tris-HCl, 50 mM NaCl, 5 mM KCl, and 5 mM MgCl2 (pH 8). Recombinant falciparum pLDH protein was purchased from Sino Biological and dissolved in water, as per the manufacturer's instructions. All other proteins from human sources were purchased from Sigma-Aldrich, and used directly without further purification. These protein powders were dissolved in 1× PBS before use.Circular dichroism
CD spectra were measured with a Jasco J-810 spectropolarimeter with a scan speed of 500 nm min−1, using a cuvette with a 0.1 cm path length. All reported spectra are an average of five scans. The final concentrations of aptamer and protein were 5 μM and 2.5 μM, respectively, for all measurements. An aptamer concentration of 5 μM had to be used to obtain a detectable CD spectrum, due to the lesser chirality of nucleic acids. The protein concentration of 2.5 μM was chosen because it was sufficiently high to match the order of magnitude of the aptamer concentration, yet low enough to keep the absorbance of the sample within reasonable bounds to improve the signal to noise ratio.41 The protein and aptamer concentrations used for our CD experiments match closely those utilized in previous work using CD spectroscopy to study aptamer–thrombin interactions.35Microscopy binding assay
7.16 μM of either pLDH or a nonspecific protein was incubated on top of freshly cleaved mica for 10 minutes to allow the protein to adsorb to the surface. A high concentration of protein was used to ensure that proteins coated as much mica surface as possible. Maximizing the protein coating on the mica surface would minimize the nonspecific binding of aptamer to the mica surface. Subsequently, 10 μM of fluorescently-labeled pLDH aptamer was incubated on top of the protein-coated surface for 10 minutes. The mica surface was then rinsed three times with water and left to dry for 5 minutes. Images of six randomly-chosen regions on the mica were taken with a fluorescence microscope. All images were taken at 20× with 1 second exposure.Image processing
To quantify the fluorescent area in each set of images, all images were background-subtracted and converted to 8-bit. A threshold range of 18–255 was applied to distinguish between fluorescent and non-fluorescent areas. Total fluorescent areas were measured using the Analyze Particles function in ImageJ. All fluorescent areas larger than 0.025 μm2 were considered.Absorbance and fluorescence measurements
All absorbance measurements were taken with a Nanodrop 2000 UV-vis spectrophotometer, and all fluorescence measurements were taken with a Tecan Infinite M200 microplate reader. The reported absorbance and fluorescence spectra of the aptamer are an average of three independent readings and were normalized using the formula: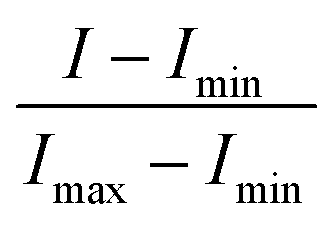 | (1) |
For fluorescence recovery experiments, 100 nM pLDH aptamer was incubated with 20 μg mL−1 MoS2 for 10 minutes to allow for the aptamer to adsorb to and be quenched by the MoS2 surface. For unblocked samples, 250 nM of either pLDH or a nonspecific protein was then added to the aptamer-MoS2 solution. For blocked samples, BSA (at 50–250 nM, depending on the experiment) was added to the aptamer-MoS2 solution and incubated for an additional 10 minutes prior to 250 nM protein addition. In one blocked sample, PBS was added instead of a protein solution analyte, to assess the amount of fluorescence recovery caused solely by the blocking agent. The fluorescence of this sample (block + PBS) was subtracted from the fluorescence of other blocked samples (block + 250 nM protein) to account for the additional recovery caused by the blocking agent itself. All concentrations are reported as the final concentrations in each sample, and all samples had a total volume of 200 μL. Fluorescence intensity was measured 30 minutes after protein (or PBS) was added to the solution. Each reported value is the average of measurements from three independent samples.
Acknowledgements
Kenry and A. Geldert would like to acknowledge the NUS Graduate School for Integrative Sciences and Engineering Scholarship and Whitaker International Program Fellowship, respectively. This research was supported by the National Research Foundation, Prime Minister's Office, Singapore under its medium-sized centre programme, Centre for Advanced 2D Materials and its Research Centre of Excellence, Mechanobiology Institute, as well as the MechanoBioEngineering Laboratory of the Department of Biomedical Engineering of the National University of Singapore. This work was also supported by MOE under AcRF Tier 2 (ARC 26/13, no. MOE2013-T2-1-034; ARC 19/15, no. MOE2014-T2-2-093; MOE2015-T2-2-057) and AcRF Tier 1 (RG5/13), NTU under Start-Up Grant (M4081296.070.500000) and iFood Research Grant (M4081458.070.500000), and Singapore Millennium Foundation.References
- S. D. Jayasena, Clin. Chem., 1999, 45, 1628 CAS.
- T. Mairal, V. Cengiz Özalp, P. Lozano Sánchez, M. Mir, I. Katakis and C. K. O'Sullivan, Anal. Bioanal. Chem., 2008, 390, 989–1007 CrossRef CAS PubMed.
- K. Groff, J. Brown and A. J. Clippinger, Biotechnol. Adv., 2015, 33, 1787–1798 CrossRef CAS PubMed.
- W. Zhou, P.-J. Jimmy Huang, J. Ding and J. Liu, Analyst, 2014, 139, 2627–2640 RSC.
- C.-H. Lu, J. Li, M.-H. Lin, Y.-W. Wang, H.-H. Yang, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2010, 49, 8454–8457 CrossRef CAS PubMed.
- W. C. Lee, C. H. Lim, Kenry, C. Su, K. P. Loh and C. T. Lim, Small, 2015, 11, 963–969 CrossRef CAS PubMed.
- A. M. H. Ng, Kenry, C. Teck Lim, H. Y. Low and K. P. Loh, Biosens. Bioelectron., 2015, 65, 265–273 CrossRef CAS PubMed.
- Kenry, K. P. Loh and C. T. Lim, Small, 2015, 11, 5105–5117 CrossRef CAS PubMed.
- Kenry, K. P. Loh and C. T. Lim, Nanoscale, 2016, 8, 9425–9441 RSC.
- Kenry, P. K. Chaudhuri, K. P. Loh and C. T. Lim, ACS Nano, 2016, 10, 3424–3434 CrossRef CAS PubMed.
- Kenry, K. P. Loh and C. T. Lim, RSC Adv., 2016, 6, 46558–46566 RSC.
- Kenry, J. C. Yeo, J. Yu, M. Shang, K. P. Loh and C. T. Lim, Small, 2016, 12, 1593–1604 CrossRef CAS PubMed.
- Kenry, A. Geldert, X. Zhang, H. Zhang and C. T. Lim, ACS Sens., 2016, 1, 1315–1321 CrossRef CAS.
- Kenry, A. Geldert, Z. Lai, Y. Huang, P. Yu, C. Tan, Z. Liu, H. Zhang and C. T. Lim, Small, 2017, 13, 1601925 CrossRef PubMed.
- Kenry and C. T. Lim, ChemNanoMat, 2017, 3, 5–16 CrossRef CAS.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS PubMed.
- C. Zhu, Z. Zeng, H. Li, F. Li, C. Fan and H. Zhang, J. Am. Chem. Soc., 2013, 135, 5998–6001 CrossRef CAS PubMed.
- X. Zhang, Z. Lai, C. Tan and H. Zhang, Angew. Chem., Int. Ed., 2016, 55, 8816–8838 CrossRef CAS PubMed.
- M. T. Makler and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 205–210 CrossRef CAS PubMed.
- M. L. McMorrow, M. Aidoo and S. P. Kachur, Clin. Microbiol. Infect., 2011, 17, 1624–1631 CrossRef CAS PubMed.
- C. Wongsrichanalai, M. J. Barcus, S. Muth, A. Sutamihardja and W. H. Wernsdorfer, Am. J. Trop. Med. Hyg., 2007, 77, 119–127 Search PubMed.
- J. W. Jang, C. H. Cho, E. T. Han, S. S. A. An and C. S. Lim, Malar. J., 2013, 12, 181 CrossRef CAS PubMed.
- J. Lee, P. Dak, Y. Lee, H. Park, W. Choi, M. A. Alam and S. Kim, Sci. Rep., 2014, 4, 7352 CrossRef CAS PubMed.
- J. Zhang, F. Zhang, H. Yang, X. Huang, H. Liu, J. Zhang and S. Guo, Langmuir, 2010, 26, 6083–6085 CrossRef CAS PubMed.
- G. Zuo, X. Zhou, Q. Huang, H. Fang and R. Zhou, J. Phys. Chem. C, 2011, 115, 23323–23328 CAS.
- L. Sheng, J. Ren, Y. Miao, J. Wang and E. Wang, Biosens. Bioelectron., 2011, 26, 3494–3499 CrossRef CAS PubMed.
- L. Peng, Z. Zhu, Y. Chen, D. Han and W. Tan, Biosens. Bioelectron., 2012, 35, 475–478 CrossRef CAS PubMed.
- Y. Pu, Z. Zhu, D. Han, H. Liu, J. Liu, J. Liao, K. Zhang and W. Tan, Analyst, 2011, 136, 4138–4140 RSC.
- S. Lee, K.-M. Song, W. Jeon, H. Jo, Y.-B. Shim and C. Ban, Biosens. Bioelectron., 2012, 35, 291–296 CrossRef CAS PubMed.
- E. Baldrich, A. Restrepo and C. K. O'Sullivan, Anal. Chem., 2004, 76, 7053–7063 CrossRef CAS PubMed.
- B. I. Kankia and L. A. Marky, J. Am. Chem. Soc., 2001, 123, 10799–10804 CrossRef CAS PubMed.
- T. Hermann and D. J. Patel, Science, 2000, 287, 820 CrossRef CAS PubMed.
- A. D. Gelinas, D. R. Davies and N. Janjic, Curr. Opin. Struct. Biol., 2016, 36, 122–132 CrossRef CAS PubMed.
- P.-H. Lin, S.-L. Yen, M.-S. Lin, Y. Chang, S. R. Louis, A. Higuchi and W.-Y. Chen, J. Phys. Chem. B, 2008, 112, 6665–6673 CrossRef CAS PubMed.
- P.-H. Lin, R.-H. Chen, C.-H. Lee, Y. Chang, C.-S. Chen and W.-Y. Chen, Colloids Surf., B, 2011, 88, 552–558 CrossRef CAS PubMed.
- Z. Zeng, Z. Yin, X. Huang, H. Li, Q. He, G. Lu, F. Boey and H. Zhang, Angew. Chem., Int. Ed., 2011, 50, 11093–11097 CrossRef CAS PubMed.
- G. L. Kenausis, V. Voros, D. L. Elbert, N. Huang, R. Hofer, L. Ruiz-Taylor, M. Textor, J. A. Hubbell and N. D. Spencer, J. Phys. Chem. B, 2000, 104, 3298–3309 CrossRef CAS.
- L. Convert, V. Chabot, P.-J. Zermatten, R. Hamel Jr., J.-P. Cloarec, R. Lecomte, V. Aimez and P. G. Charette, Sens. Actuators, B, 2012, 173, 447–454 CrossRef CAS.
- Y. L. Jeyachandran, J. A. Mielczarski, E. Mielczarski and B. Rai, J. Colloid Interface Sci., 2010, 341, 136–142 CrossRef CAS PubMed.
- K. Reimhult, K. Petersson and A. Krozer, Langmuir, 2008, 24, 8695–8700 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |