
A high throughput microfluidic platform for size-selective enrichment of cell populations in tissue and blood samples†
Nivedita
Nivedita‡
 a,
Neha
Garg‡
bc,
Abraham P.
Lee§
bcd and
Ian
Papautsky§
*ce
a,
Neha
Garg‡
bc,
Abraham P.
Lee§
bcd and
Ian
Papautsky§
*ce
aDepartment of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27514, USA
bBioMiNT Lab, Biomedical Engineering Department, University of California, Irvine, CA 92697, USA
cNSF Center for Advanced Design and Manufacturing of Integrated Microfluidics (CADMIM), E-mail: papauts@uic.edu
dMechanical & Aerospace Engineering Department, University of California, Irvine, CA 92697, USA
eDepartment of Bioengineering, University of Illinois at Chicago, Chicago, IL 60607, USA
First published on 21st June 2017
Abstract
Numerous applications in biology and medicine require the efficient and reliable separation of cells for disease diagnosis, genetic analysis, drug screening, and therapeutics. In this work, we demonstrate a novel technology that integrates a passive and an active device to separate, enrich and release cells on-demand from a complex blood sample, or cancer cells derived from a tissue biopsy. We exploit the high throughput (>1 mL min−1), size-based sorting capability of the passive spiral inertial microfluidic (iMF) device to focus particles/cells towards an active lateral cavity acoustic transducer (LCAT) device for size-selective enrichment. We demonstrate that this platform is capable of efficiently (>90%) removing smaller cells, such as RBCs in a blood sample or smaller cancer cells in a heterogeneous cell line, and providing 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× enrichment from the remaining sample within 5 min of device operation. Finally, we use this platform for two applications: selective enrichment of the side-population of DU-145 cells from tissue biopsy and isolation of larger monocytes from blood. Our platform integrates the high throughput (processing rate) capacity of spiral iMF with the high selectivity of LCAT, thereby offering a unique route for highly-selective, label-free particle/cell sorting, with potential application in lab-on-chip platforms for liquid biopsy and diagnostics applications.
000× enrichment from the remaining sample within 5 min of device operation. Finally, we use this platform for two applications: selective enrichment of the side-population of DU-145 cells from tissue biopsy and isolation of larger monocytes from blood. Our platform integrates the high throughput (processing rate) capacity of spiral iMF with the high selectivity of LCAT, thereby offering a unique route for highly-selective, label-free particle/cell sorting, with potential application in lab-on-chip platforms for liquid biopsy and diagnostics applications.
Introduction
The field of microfluidics has seen rapid development in the last decade, especially for applications involving the isolation and enrichment of cells or particles.1,2 The enrichment of cells is particularly critical to the sample preparation of complex biological fluids and heterogeneous cell samples, for a variety of applications.1–3 For example, sub-populations of cancer cells have been associated with stem cell like properties4–6 and may offer insights into cancer progression and metastasis,4,7–9 while the heterogeneity in peripheral blood monocytes has been linked to differentiation into mature tissue macrophages, osteoclasts and dendrite cells.10,11 The advantages of using microfluidic sorting and enrichment systems lie in the ease of use, small sample volume requirements, high efficiency, cost-effectiveness and disposable platforms, along with a wide range of separation modalities based on both the physical and chemical properties of cells or particles.2,3Recently, microfluidic devices have been reported for the separation of particles/cells based on a variety of properties such as magnetic properties, electrical polarizability, deformability and size.12,13 These approaches can be broadly classified into passive and active, based on the method of actuation and operation. Passive approaches such as inertial microfluidics (iMF), filtration and deterministic lateral displacement (DLD) rely on pressure driven flows, hydrodynamic forces and micro-channel geometry. On the other hand, active approaches like magnetophoresis, dielectrophoresis and acoustophoresis use an external force field to enable isolation. Passive methods based on DLD use pillars and posts for size and deformability based isolation. For example, Liu et al.14 demonstrated label-free cell isolation using a DLD microfluidic chip with high throughput (∼2 mL min−1), although the trade-off included high shear stress and fabrication complexity. In addition to DLD, McFaul et al.15 used microstructured constraints to filter lymphoma cells based on their size and deformability, but the filtration technique generally suffers from clogging and releasing of viable cells. Furthermore, Hur et al.16 and Sollier et al.17 used inertial migration in straight channels with rectangular reservoirs for size-based isolation, with enrichment ratios of 8× and 103× respectively. Additionally, Warkiani et al.18 used spiral inertial microfluidic devices with a trapezoidal cross section which combine cross sectional Dean flows with inertial migration to separate a variety of cancer cells spiked in blood with ∼80% recovery rate, while Sun et al.19 used a double spiral to demonstrate separation from diluted blood with 90% recovery rate. Although these devices present promising sorting systems with high throughput (1–2 mL min−1), they are limited to size-based selectivity and subsequent purity of the sample obtained.
Active approaches for particle/cell isolation provide the much needed high selectivity and enrichment. For example, the ApoStream chip from Gupta et al.20 used dielectrophoretic isolation and Ding et al.21 designed a device with tilted-angle standing surface acoustic waves to isolate and separate cancer cells from WBCs with different compressibility (viability of >97% and throughput of 18–25 μL min−1). While these devices demonstrate good separation efficiency, they add fabrication complexity and an extra step of blood pre-processing for RBC lysis. In addition to electrophoresis, a number of microfluidic devices use the differential effect of the magnetic field on cells labeled with magnetic beads or cells which are magnetically responsive.22–24 Although highly selective, magnetophoresis is limited either by sample contamination due to labeling or by the inherent magnetic properties of cells.25,26 Recently, size based separation dependent on acoustic actuation has gained importance due to higher cell viability, and the maintenance of cell function post acoustic actuation. Augustsson et al.27 used acoustophoresis for enrichment of prostate cancer cells and achieved a high recovery of 94–98% but the method suffers from erythrocyte concentration in whole blood (used RBC lysed blood). Overall, size based active microfluidic devices have suffered from trade-offs in selectivity, isolation efficiency and throughput. High throughput is particularly critical when processing of a large sample volume is required for target cell isolation such as rare cell isolation from a blood draw which may need dilution during sample preparation. Hence, for the isolation of particles/cells, especially where selective enrichment is important, sorting devices should be able to combine a high sample processing rate (1–2 mL min−1) with high enrichment ratios (>104 from RBCs in blood samples).
In this work, we present a novel integrated microfluidic platform that combines a passive sorting technique with an active enrichment method for high throughput, size-selective isolation of cells. The concept is illustrated in Fig. 1a. At the front end of the platform, a spiral inertial microfluidic (iMF) separator exploits inertial focusing and Dean flows to remove non-target cells at a high processing rate and directs the remaining sample to a lateral cavity acoustic transducer (LCAT) at the back end, for further selective enrichment of the target particles/cells (Fig. 1b). The integrated platform was first characterized using polystyrene particles and the limit of enrichment was tested by spiking the particles in blood. Following parametric optimization, the integrated platform was tested for two applications: enrichment of a larger sub-population of prostate cancer cells derived from brain metastasis (DU-145 cells)28,29 and enrichment of larger monocytes10,11 from human peripheral blood. The inherent mismatch between the operational conditions and design restrictions for each of the two systems presented a number of design challenges that needed to be addressed before integration. Thus, we first optimized each system individually keeping the channel height fixed in both systems for ease of fabrication. The payoff of this optimization and subsequent integration was the successful combination of two distinct sample preparation steps of extraction, isolation, and enrichment in a single platform with a sorting throughput of 50 million cells per min, >85% sorting efficiency and 44000× enrichment with respect to RBCs. The demonstrated work is particularly relevant for applications requiring selective enrichment of particles/cells from a complex mixture, such as biochemical assays, enrichment of a cell population from a tissue biopsy (such as a sub-population of cancer cells) or isolation of rare cells (such as larger monocytes) from blood.
 | ||
| Fig. 1 An integrated platform for the separation of particles from blood. (a) A spiral iMF device is integrated into the front end for RBC extraction, with an LCAT in the back end for target cell isolation and enrichment. (b) Schematic illustrating the size-based separation dynamics in the integrated platform. | ||
Results and discussion
Spiral iMF cell extractor
| FL = ρG2CLap4 | (1) |
In a curved rectangular microchannel, the net lift force interacts with the curvature induced Dean drag (due to Dean vortices) leading to a single equilibrium position per particle size.31,34,35 The Dean drag force (FD) can be defined as
| FD = 3πμUDap | (2) |
 | ||
| Fig. 2 Design and optimization of individual devices (spiral cell extractor and LCAT isolator). (a) Schematic of formation of cross-sectional Dean vortices in a curved rectangular channel and the cell/particle focusing regime. (b) Intensity plot across the channel width at the outer-most loop and before the expansion in the spiral sorter. Insets show the images of the collected sample at the inlet, outlet 1 and outlet 2. (c) Plot of the sorting efficiency of 25 and 7.32 μm diameter particles as a function of flow rate. Inset shows the fluorescent image of the focused 25 μm diameter particles eluting into outlet 2 and 7.32 μm diameter particles eluting into outlet 1. (d) Schematic of the operational regime of the LCAT device showing the entrapment of larger particles in the microvortices near the air/liquid interface. (e) Plot of the concentration of trapped particles as a function of bulk flow rate and two operational peak to peak voltages. (f) Trapped particles as a function of the concentration of spiked particles (input). Inset shows the bright field image showing the trapped larger (15 μm) particles and smaller (10 μm) particles following the bulk flow. | ||
At Re ∼ 160, the 25 μm diameter particles focused in a single stream near the inner channel wall and the 7.32 μm diameter particles focused in a band near the center of the channel. The intensity plot across the channel width at the end of the outermost loop of the spiral shows the peak of the focused stream of 25 μm diameter particles with the FWHM (full width at half maximum) of 25.11 μm, indicating a tightly focused stream (Fig. 2b). The second peak in the plot with the FWHM of 23.02 μm shows the band of 7.32 μm diameter particles focused towards the center of the channel (Fig. 2b). The insets in Fig. 2b show samples collected at the inlet and each of the outlets indicating the efficiency of separation. Sorting efficiency was then quantified as a function of flow rate and with respect to each of the particle sizes in the mixture (Fig. 2c). Sorting efficiency (SE) was calculated by counting the total number of particles/cells in each outlet as follows
 | (3) |
LCAT cell isolator
 | (4) |
The entrapment of larger particles in an LCAT device is dependent on the competition between the bulk flow velocity and the microstreaming velocity of the microvortices. The plot in Fig. 2e shows the trapping efficiency of the device as a function of flow rate for two different peak-to-peak voltages (2.75 V and 2.5 V). For flow rates up to 27.5 μL min−1, the observed trapping concentration was similar to the expected concentration, but as the flow rate further increased, there was a notable decrease in the concentration of the trapped particles. This decrease in the concentration of trapped particles can be attributed to the suppression of oscillation amplitude and the increase in dgap at higher flow rates. Thus, the diameter of trapped particles no longer remains greater than dgap and the particles then start to release from the microstreaming vortices. A similar trend was observed for both the applied voltages.
Once the operational parameters for the appropriate trapping efficiency were obtained, we determined the effect of sample concentration on the trapping efficiency to ascertain whether we would be able to enrich rare samples using LCAT. The standard curve in Fig. 2f illustrates the trapping efficiency as a function of input sample concentration (number of particles per mL). The device was operated at the bulk flow rate of 25 μL min−1 and a peak-to-peak voltage of 2.75Vpp. Voltage switching was used to remove the excess smaller particles (10 μm) from the outer streamlines, as described in the Methods section. The slope of the standard curve is 1.0623, suggesting that nearly all particles spiked into the sample were recovered experimentally, leading to the trapping efficiency of ∼99% even for spiked concentrations of <100 particles per mL. Following individual design and parametric optimization, both the iMF extractor and LCAT cell isolator were integrated to provide a novel platform for selective particle/cell isolation directly from a label-free, Ht = 0.5 blood sample and a sub-population isolation of the DU-145 cell line, as described in the following sections.
Integrated platform
 | ||
| Fig. 3 Integrated device design and parametric optimization. (a) Schematic of the integrated device with the spiral iMF and LCAT sections integrated into a single platform. (b) PIV scan and subsequent plots of the velocity magnitude as observed at (i) the spiral outlet channel, (ii) expansion channel and (iii) LCAT entry channel. (c) Intensity scans across the channel width of the spiral iMF section within the integrated device. The scans were taken at the innermost loop and the outermost loop, for the standard deviation z-stack. The inset shows the pseudo colored standard deviation z-stack of the focused 7.32 μm diameter particles and >18 μm diameter particles. (d) Bright field images of the 7.32 μm diameter particles eluting into outlet 1 and >18 μm diameter particles eluting into the LCAT, where they get trapped in the microvortices actuated at 6.5Vpp (second bright field image). | ||
Integration affected the flow and operational regime in both the components. Since the trapping efficiency in LCAT depends on the bulk flow velocity, the input flow rate at the spiral iMF inlet needed to be reduced to reach the optimum value. It was imperative to accomplish a reduction in volumetric flow rate, while maintaining the range of operational parameters and projected sorting and trapping efficiencies as determined in the individual optimization. For this purpose, the height of the entire platform was reduced to ∼75 μm. The aspect ratio of the spiral iMF was maintained (AR = 0.5) by reducing the width to 150 μm. The input Re was kept at ∼160 as determined in the previous sections.
To confirm the appropriate reduction in flow rate within the platform, we performed PIV (particle image velocimetry). The flow was seeded with fluorescently labeled polystyrene particles of diameter 2.1 μm (Fig. 3b). Vector plots were extracted for each of the critical sections in the design namely, (i) the outlet channel of the spiral, (ii) the expansion channel after the spiral extractor and (iii) the LCAT entry channel post the inter-connecting channel. Fig. 3b shows the velocity plots obtained for each of the aforementioned three sections. These plots illustrate the required reduction in the functional flow rate for LCAT to entrap the target particles, while keeping the net/input processing flow rate high.
We started with optimizing the integrated device parameters with polystyrene particles before testing the device with any biological sample. We mixed 7.32 μm and 18+ μm diameter particles (20 μm and 25 μm diameter particles) to simulate the opposite ends of the cell size spectrum in a blood sample. The 7.32 μm diameter particles at a concentration of 106 mL−1 represented non-target cells such as RBC/WBCs, while the 18+ μm diameter particles at a concentration of 10000 mL−1 represented larger target cells such as monocytes or side-populations from tumor biopsies. For the formation of air–liquid interfaces in the LCAT section of the integrated device, the sample solution was directly infused into the integrated device inlet. At Re ∼ 160, we observed that 7.32 μm particles eluted into outlet 1 and 18+ μm particles eluted into the interconnecting channel leading to LCAT. To illustrate the focusing regime, we plotted intensity scan across the iMF channel width at the inner-most loop and the outer-most loop (Fig. 3c). At the inner-loop, the entire channel had a uniform distribution of particles and as the flow progressed to the outer loop, 7.32 μm diameter particles focused in a band, thereby eluting into outlet 1. The 18+ μm diameter particles focused closer to the inner channel wall, thereby eluting into the interconnecting channel towards LCAT. The intensity scan was taken using the standard deviation z-stacking of the bright field images of the focused particles.
The bifurcation at the asymmetric outlet of the spiral iMF was designed to be blunt because sharp corners can often result in the formation of microvortices due to acoustic actuation, which in turn can interfere with inertial focusing. Hence, when PZT was turned on, we observed that the particles stay in the desired focusing regions irrespective of slight focusing fluctuations (Fig. 3d). After the actuation of the device at 6.5Vpp, the 18+ μm diameter particles were trapped in the microstreaming vortices as expected (Fig. 3d). Higher voltages were used for optimal trapping to compensate for the variation of bulk flow rate in the integrated device. Following the parametric determination, we tested the integrated device with particles spiked in a human blood sample.
000 particles per mL concentration (Fig. 4a). At Re ∼ 160, we observed that the particles focused closer to the inner channel wall and eluted into the interconnecting channel towards the LCAT. RBCs focused in a broad band near the center of the channel and eluted into outlet 1 (Fig. 4b and ESI, video 1†). To further optimize the sorting efficiency of the device, we tested blood samples with dilutions ranging from 20× to 200× and the resulting Ht of 2.4% to 0.24%.
 | ||
| Fig. 4 Integrated device operation with particles spiked in blood. (a) z-Stack of the bright field images of 25 μm diameter particles spiked in 20× diluted blood sample at the inlet of the integrated device. (b) z-Stack of the bright field images of focused 25 μm diameter particles eluting into the interconnecting channel towards the LCAT and RBCs eluting into outlet 1. (c) Sorting efficiency of the spiral iMF in the integrated device for 25 μm diameter particles and the purity of the sample that eluted into the LCAT. (d) Trapped 25 μm diameter particles in microvortices in the LCAT section of the integrated device at varying spiked concentrations. (e) Enrichment ratio evaluated at each of the spiked concentrations at the input of the integrated device, providing the enrichment factor for the target particles/cells. | ||
Since the dilution factor is dictated mainly by the inertial lift forces required for focusing, the sorting efficiency and purity of the 25 μm diameter particles were evaluated in the spiral iMF section of the integrated device. As expected, the sorting efficiency improved to 95% with higher dilution owing to the reduction in cell-to-cell interaction as well as more precise control over inertial lift forces (Fig. 4c). However, the ideal sample conditions dictate that there should be minimum sample dilution before running it through the device. Since we observed ∼85% sorting efficiency at 20× dilution in spiral iMF, we decided to operate the integrated device at 20× dilution or lower and evaluate the enrichment of 25 μm diameter particles in the trapped sample by the LCAT section of the integrated device.
After the majority of RBCs were separated using the spiral iMF, the 20× diluted sample with WBCs and larger particles (25 μm diameter) eluted into the LCAT section for further enrichment. The concentration of 25 μm diameter particles in the input blood sample was varied from 10000 particles per mL to 1 particle per ml to evaluate the enrichment limit of the integrated LCAT. A lower limit of 1 particle per mL was assigned to provide the baseline for extraction at rare concentrations and evaluate the sensitivity of entrapment to extreme requirements. The entrapment of the particles was observed at the PZT actuation of 6.5Vpp (Fig. 4d). In fact, the selectivity was high enough to allow entrapment even at the low concentration of 1 particle per mL. After the collection of the enriched sample by flowing buffer (PBS) through the device, we evaluated the enrichment ratio at each spiked concentration with respect to the remnant of RBCs and obtained enrichment ratios on the order of 104 (Fig. 4e). Additionally, we observed an increase in enrichment ratio with a decrease in spiked particle concentration, suggesting that the device is selective and sensitive enough to sort out even the most rare particle populations. The reduction in enrichment ratio for the concentration of 1 particle per mL (4.4 × 104) can be attributed to the entrapment of non-target particles in the empty microstreaming vortices over time. Hence, the integrated platform provides the highest enrichment (8.8 × 104) at the target concentration of 10 particles per mL. We also determined the recovery rate for enriched particles from blood to be 66.6%. These results suggest that the integrated device is capable of successfully extracting smaller non-target cells such as RBCs from the given heterogeneous sample at a high throughput and selectively trapping the desired particles/cells, thereby providing high enrichment of the target particles even at the lower concentrations. Following the optimization with particles spiked in blood, we tested the platform for two target applications: (1) sub-population isolation of DU-145 cells, and (2) enrichment of monocytes from human blood.
We used our integrated platform to significantly enrich (∼77×) a sub-population of larger DU-145 cells (>16 μm). The DU-145 cell line is derived from the prostate metastatic tumor in the brain.28,29 The cell size range of the DU-145 cell line was found to be very heterogeneous with cell sizes ranging from 7 μm to 28 μm.28,29 We prepared an input sample with an average concentration of 25000 cells per mL and infused the sample at a rate of ∼1.1 mL min−1. DU-145 cells are adherent cells; therefore, the cell concentration at the input sample was maintained at 25000 cells per mL to ensure that cells don't adhere to each other to form aggregates. We observed the entrapment of larger (>16 μm) cells in the microvortices at the optimized PZT actuation voltage of 6.5Vpp (Fig. 5a and ESI, video 2†). The imaging of the outlet sample revealed enriched larger DU-145 cells. We compared the inlet and the outlet samples using a box plot, which shows significant enrichment of larger cells in the outlet sample (Fig. 5b). The enrichment of DU-145 cells was achieved at a high processing rate of ∼28000 cells per mL and a high purity of ∼91.7%. Additionally, we were able to achieve a recovery rate of 67.5%, which is comparable to the recovery rate obtained for particles spiked in blood.
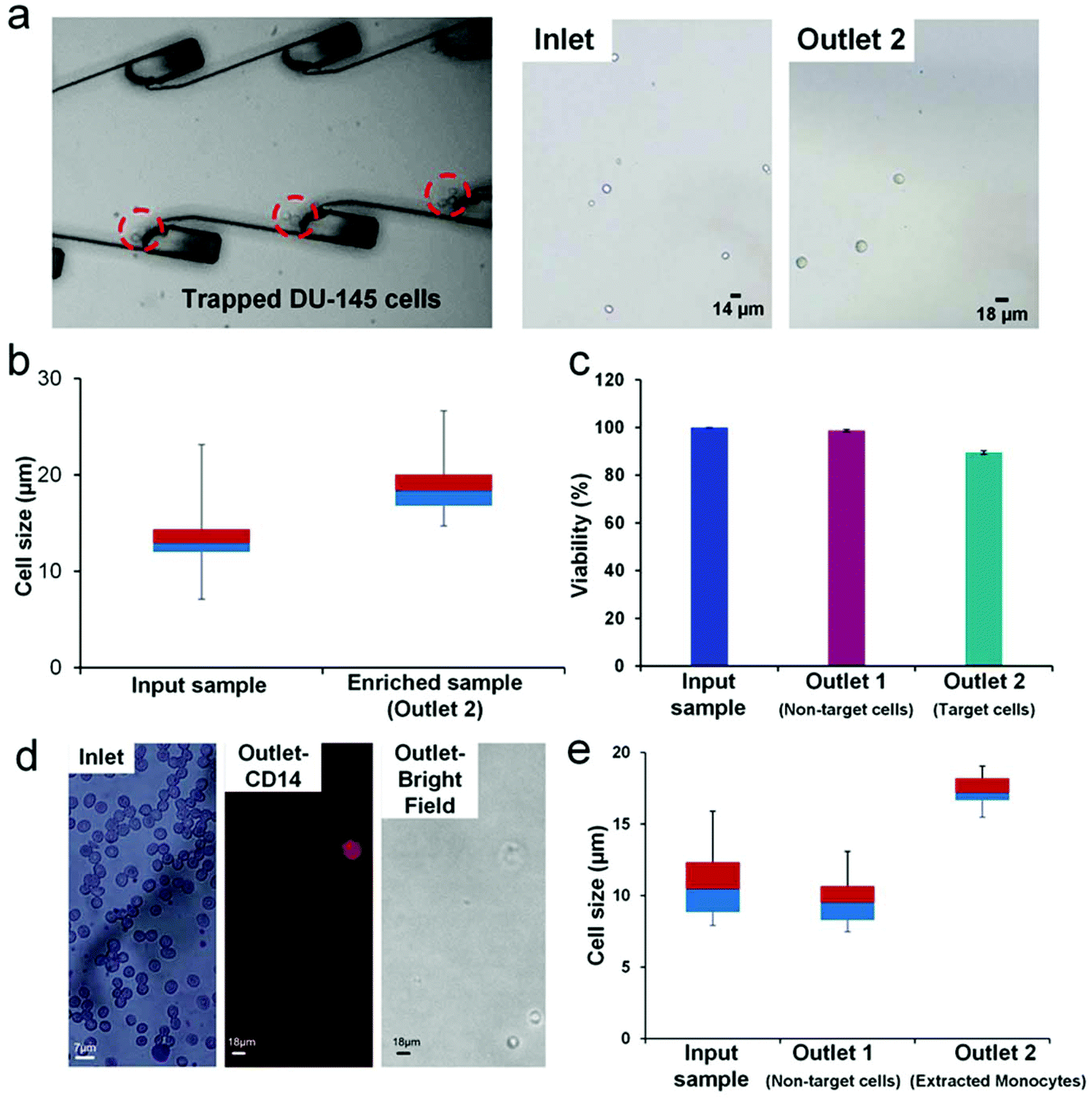 | ||
| Fig. 5 Integrated device used for two applications in cellular biology. (a) Bright field image of the larger DU-145 cells trapped in the micro-vortices of the integrated device (left), along with images showing the inlet sample and sample at the outlet (right). (b) Box plot showing the range of cell-sizes of both the DU-145 cell sample at the inlet and the sample obtained after the enrichment using the integrated platform. The plot shows the enrichment of larger DU-145 cells in the outlet sample. (c) Plot showing the viability of DU-145 cells. We obtained 90% viability of enriched target cells. (d) Images showing the use of the integrated device for the isolation of a sub-population of monocytes. The Wright–Geimsa stained inlet sample shows a majority of RBCs with a few WBCs (left). The outlet sample obtained contained only monocytes, the presence of which was confirmed using the CD14 fluorescent antibody (middle). (e) Box plot showing the size comparison of cells at the inlet (blood sample), outlet 1 (non-target cells) and extracted larger monocytes (target cells) from outlet 2. | ||
To assess the viability of cells post enrichment, we used Trypan Blue to determine the number of viable cells obtained after passing through the integrated platform (Fig. 5c). We obtained 98.7% viable cells in outlet 1 (non-target cells). These cells passed only through the first stage, spiral extractor of the integrated platform. Cells in outlet 2 (enriched target cells) exhibited 90% viability, having passed through the entire integrated platform (spiral extractor+ LCAT). The enrichment of a sub-population of cells that is morphologically different (size) from the rest of the cancer cell population provides a unique sample set for further investigation into cancer progression and metastasis. Post enrichment, these cells can be grown to the required confluency in a 3D culture and then specific assays can be performed to determine the migration and invasion ability, along with self-renewability of the enriched cells. This information will potentially provide vital insight into the development of tumors as well as regression in patients undergoing therapy.
In addition to cancer biology, size-based enrichment is particularly relevant to hematopoietic cells which are significantly heterogeneous in size. The monocyte heterogeneity has been linked to the macrophage lineage and differentiation into mature tissue osteoclasts and dendrite cells.10,11,48,49 The classification of monocytes into classical, intermediate and non-classical has been dependent on the expression of CD14 and CD 16.10,11 Recent work has shown that there may be sub-populations of monocytes that differ in size, granularity, functionality and even nuclear morphology.50 Furthermore, these sub-populations have not yet been fully characterized and the surface markers specific to each type have not yet been fully identified. For in depth investigation into how this differentiation is linked to the sub-populations of monocytes,11,48,49 it is significant to be able to sort these sub-populations without any labeling. The integrated platform proposed in this work can sort and enrich a rare sub-population of monocytes. Since this method is label free (size being the only marker), the collected cells can be further characterized for differentiation patterns, granularity and functionality. Here, we have used our platform to enrich the extremely rare sub-population of larger monocytes (18 μm+). We infused 10× diluted blood at the rate of 1.1 mL min−1 into the integrated device and were able to successfully obtain an extremely pure, enriched sample containing only larger monocytes (Fig. 5d). The presence of monocytes in the outlet sample was confirmed by CD14 staining. Owing to the extreme rarity of larger monocytes (∼2.2% of mononuclear cells),10,48,49 it was difficult to determine the exact number of cells in the inlet sample.
We estimated the enrichment of the target cells (large monocytes) with respect to non-target cells to be 987.6×. Fig. 5e shows the size comparison of the enriched sub-population of larger monocytes (compared to the input blood sample and the non-target cells (RBCs and smaller WBCs) collected at outlet 1). The average size of the enriched sub-population of monocytes was 17.3 μm. For this application, we used a human blood sample without any modifications to its cellular composition. Therefore, the exact recovery rate could not be calculated because the initial monocyte percentage varies from sample to sample; so the initial concentration can't be determined. However, we are able to report the recovery rate of DU-145 cells and particle spiked blood as 67%. Furthermore, the use of microstreaming for acoustic actuation instead of standing wave or cavitation preserves cell physiology and functionality for further analysis of enriched cells.51,52 The ability of the device to successfully enrich the sub-population of DU-145 and larger monocytes at the high processing rate along with high purity of the enriched sample shows the potential versatility and utility of the platform. Further improvement in enrichment ratio and throughput will ensure that the device is comparable to the current state of the art lab-on-chip enrichment devices.
Conclusions
In this work, we have successfully demonstrated an integrated iMF + LCAT device for the selective isolation and enrichment of cells from a heterogeneous mixture like cancer cell line and blood. Our integrated system combines passive and active microfluidic components for sample extraction and enrichment, despite the significant disparity in the operational flow rates of the two devices. Using this platform, we demonstrated successful removal (up to >90%) of smaller non-target cells (smaller DU-145 cells and RBCs and smaller WBCs) in spiral iMF at the front end for minimum interference in the isolation of larger target cells. LCAT or the back end of the platform was used to trap larger (18+) cells with ∼90% viability (larger DU-145 cells and large monocytes).The platform can enrich even the lowest of concentrations of spiked particles in blood (1 particle per mL) with high purity/enrichment ratio (44000×). The overall throughput is controlled by the amount of sample processed by the entire platform, which is ∼1 mL min−1 (for bulk non-target cell extraction). Additionally, the enrichment of target cells in 50 μL volume provides a highly concentrated sample of the target cell population. Using this platform, we were able to successfully enrich a larger side-population of DU-145 cells with >91% purity. Additionally, we were able to isolate rare larger monocytes from 10× diluted blood with an enrichment of 987×. In comparison with the current microfluidic devices that are limited by shear induced stress,14 low throughput53 or reliance on labeling,53,54 our integrated platform offers label free and size-selective sample sorting and enrichment in a single pass. In fact, our approach dispenses with the need for pre-processing of the input sample such as RBC lysing20,21,27 for pre-processing of a blood sample prior to enrichment, thereby maintaining the sample integrity.
Our integrated platform eliminates the trade-off between high purity and high throughput as experienced in previous size-based microfluidic devices. Furthermore, our approach offers a continuous, flow through, single step operation, unlike multi-step devices such as the vortex isolator12 that relies on a two-step process of trapping and release with different flow rates. In future, we will be working on fine-tuning the size-selection criteria of the platform for higher enrichment. Overall, this work demonstrates a highly promising approach to the selective isolation and enrichment of particles/cells, and ultimately could lead to the development of a single-step blood liquid-biopsy platform.
Methods
Device fabrication
The devices were fabricated using standard soft lithography. PDMS (polydimethyl siloxane) was cast on a mold fabricated using SU-8 (MicroChem) as a negative photoresist. After curing PDMS at 65 °C for 2 hours, it was peeled and bonded to a glass coverslip (0.17 mm thick) using a standard plasma bonder (Harrick). Bonding to the coverslip was carried out to ensure efficient actuation and induction of the acoustic field and the device was kept on a hot plate at 65 °C overnight to render hydrophobicity.Device operation
A syringe interfaced with clear Tygon tubing and FEP tubing (1/16′′ diameter) using a Luer connector was inserted in a syringe pump (New Era) to control the flow rate. For acoustic actuation, a function generator (Agilent Technologies) with a voltage amplifier (Krohn-Hite Corp.) was used to provide the desired amplitude (Vpp) square waveform. These square waves actuated a piezoelectric transducer (Steiner & Martins) at a resonating frequency of 49.8 kHz. For the individual spiral iMF and the integrated device particle optimization, a mixture of fluorescent polystyrene particles of diameter 7.32 μm (Bangs Lab, S.D. = 0.53 μm) (representing RBCs) and 25 μm (Polysciences Inc., S.D. = 2.5 μm) diameter particles (representing larger rare cells) was used. The distribution of particles across the channel was observed using an inverted fluorescence microscope (IX-71, Olympus). The images captured by the 12-bit CCD camera (Retiga EXi, QImaging) were z-stacked (∼10–100 images) using ImageJ software. The individual LCAT device was optimized using a mixture of 15 μm (Phosphorex Inc., S.D. = 0.140) and 10 μm (Phosphorex Inc., S.D. = 1.270) diameter particles. A 1′′ × 1′′ silicon wafer was glued (Krazy Glue) on the PZT to make the surface more reflective. PZT was mounted on a cover of a standard Petri dish using double sided tape. Ultrasound gel (Parker Labs Inc.) was used for improved conduction between the glass cover slip of the device and the silicon wafer attached to the PZT. Images were taken using a Phantom v310 (Vision Research Inc.) camera by means of an upright microscope (Nikon LV150). The flow was switched from the sample to PBS from a second syringe pump maintained at the original flow rate using a 2-way valve. PBS (Life Technologies) was used to release the trapped particles and collect the enriched sample from LCAT after the function generator was switched off. All cells/particles were counted by ImageJ using a Countess cell counting chamber slide (Life Technologies) under an inverted microscope (Olympus). De-identified male whole blood samples were obtained from Hoxworth Blood Center (University of Cincinnati) or from the Institute for Clinical and Translational Science (University of California, Irvine). The blood samples were diluted using 0.9% saline or PBS. For the spiral iMF section of the integrated device, bright field images were obtained with an exposure of 10 μs and ∼10–20 images were stacked to represent the focusing regime. Single images were obtained at an exposure of 300 μs for imaging trapped particles in microstreaming vortices formed in the LCAT section of the integrated device.Cell sample preparation and analysis
DU-145 cell culture and sample preparation: DU-145 cells were cultured as monolayers on a T-75 cell culture flask. ATCC-formulated Eagle's Minimum Essential Medium was used as the basal medium to which fetal bovine serum (FBS) was added (10% by volume). After the cells reached the desired confluency, the culture medium was discarded and the adherent cell layer was rinsed with 1× PBS. 2.5 mL of trypsin + EDTA was added to the flask to disassociate the adherent cells. Cells were observed to have disassociated in approximately 3 minutes. The cells were then transferred to a 10 mL Eppendorf tube and 7.5 mL of the growth medium with FBS was added. The cells were then centrifuged at 1000 rpm for 5 min and the pellet was re-suspended in 10 mL growth medium. The cell concentration in the resulting suspension was determined and appropriately diluted to 25000 cells per mL. This sample was then infused into the integrated device at the volumetric flow rate of 1.1 mL min−1 using a syringe pump. The PZT was actuated at 6.5Vpp for the entrapment of larger DU-145 cells in microvortices.
Viability assay
The viability of cells was tested by using the Trypan Blue viability test. In this test, the dye is taken up by dead cells which turn blue, whereas live cells stay clear. Cell samples (10 μL each) were taken from the inlet, outlet 1 (non-target cells) and outlet 2 (target cells). 10 μL of Trypan Blue was added to each of the samples. Live cells were counted by ImageJ using a Countess cell counting chamber slide (Life Technologies) to determine the viability of cells in each sample.Sample preparation for monocyte isolation
De-identified male blood sample was obtained from the Institute for Clinical and Translational Science, Irvine. The blood sample was diluted to 10× by adding 0.9% saline or 1× PBS. This diluted sample was then infused into the integrated platform using a single syringe pump at the volumetric flow rate of 1.1 mL min−1. The PZT was actuated at 6.5Vpp allowing larger monocytes to be trapped in the micro-vortices and subsequent collection at the outlet of the platform. On collection at the outlet, the collected monocytes were stained with CD14 antibody for confirmation. The protocol for CD14 antibody staining involved blocking the collected cell sample with FcR Blocking Reagent (Miltenyi Biotec) by the addition of 60 μL of staining buffer (1% BSA, 0.1% NaN3 in 1× PBS, pH 7.4) for the resuspension of cell pellets and addition of 20 μL FcR. The samples were incubated on ice for 10 min. 4 μL of CD14-PE/Dazzle 594 (BioLegend) antibodies were added to stain the samples for 30 min on ice. The stained sample was then washed twice with 1 mL of staining buffer and then centrifuged at 1000 rpm for 5 min. The cell pellet was then resuspended in fresh staining buffer and analyzed on a counter slide using a fluorescence microscope (TRITC filter).Acknowledgements
This work was supported by the NSF Center for Advanced Design and Manufacturing of Integrated Microfluidics (CADMIM) awards IIP-1362165 and IIP-1362048.References
- S. Haeberle and R. Zengerle, Lab Chip, 2007, 7, 1094–1110, 10.1039/B706364B.
- T. Konry, S. S. Bale, A. Bhushan, K. Shen, E. Seker, B. Polyak and M. Yarmush, Mikrochim. Acta, 2012, 176, 251–269, DOI:10.1007/s00604-011-0705-1.
- P. Sajeesh and A. K. Sen, Microfluid. Nanofluid., 2014, 17, 1–52, DOI:10.1007/s10404-013-1291-9.
- M. Boesch, A. G. Zeimet, H. Fiegl, B. Wolf, J. Huber, H. Klocker, G. Gastl, S. Sopper and D. Wolf, Oncoscience, 2016, 3, 85–87, DOI:10.18632/oncoscience.300.
- G. A. Challen and M. H. Little, Stem Cells, 2006, 24, 3–12, DOI:10.1634/stemcells.2005-0116.
- T. Chiba, K. Kita, Y. W. Zheng, O. Yokosuka, H. Saisho and A. Iwama, Hepatology, 2006, 44, 240–251, DOI:10.1002/hep.21227.
- M. Boesch, A. G. Zeimet, D. Reimer, S. Schmidt, G. Gastl, W. Parson, F. Spoeck, J. Hatina, D. Wolf and S. Sopper, Oncotarget, 2014, 5, 7027–7039 CrossRef PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572, DOI:10.1038/nrc865.
- L. Moserle, M. Ghisi, A. Amadori and S. Indraccolo, Cancer Lett., 2010, 288, 1–9, DOI:10.1016/j.canlet.2009.05.020.
- S. Gordon and P. R. Taylor, Nat. Rev. Immunol., 2005, 5, 953–964, DOI:10.1038/nri1733.
- B. K. Stansfield and D. A. Ingram, Clin. Transl. Med., 2014, 4, 5, DOI:10.1186/s40169-014-0040-3.
- Y. Chen, P. Li, P.-H. Huang, Y. Xie, J. D. Mai, L. Wang, N.-T. Nguyen and T. J. Huang, Lab Chip, 2014, 14, 626–645, 10.1039/c3lc90136j.
- P. Patil, Madhuprasad, T. Kumeria, D. Losic and M. Kurkuri, RSC Adv., 2015, 5, 89745–89762, 10.1039/C5RA16489C.
- Z. Liu, F. Huang, J. Du, W. Shu, H. Feng, X. Xu and Y. Chen, Biomicrofluidics, 2013, 7, 011801, DOI:10.1063/1.4774308.
- S. M. McFaul, B. K. Lin and H. Ma, Lab Chip, 2012, 12, 2369–2376, 10.1039/c2lc21045b.
- S. C. Hur, A. J. Mach and D. Di Carlo, Biomicrofluidics, 2011, 5, 22206, DOI:10.1063/1.3576780.
- E. Sollier, D. E. Go, J. Che, D. R. Gossett, S. O'Byrne, W. M. Weaver, N. Kummer, M. Rettig, J. Goldman, N. Nickols, S. McCloskey, R. P. Kulkarni and D. Di Carlo, Lab Chip, 2014, 14, 63–77, 10.1039/c3lc50689d.
- M. E. Warkiani, B. L. Khoo, D. S. Tan, A. A. S. Bhagat, W. Lim, Y. S. Yap, S. C. Lee, R. A. Soo, J. Han and C. T. Lim, Analyst, 2014, 139, 3245–3255, 10.1039/c4an00355a.
- J. Sun, M. Li, C. Liu, Y. Zhang, D. Liu, W. Liu, G. Hu and X. Jiang, Lab Chip, 2012, 12, 3952–3960 RSC.
- V. Gupta, I. Jafferji, M. Garza, V. O. Melnikova, D. K. Hasegawa, R. Pethig and D. W. Davis, Biomicrofluidics, 2012, 6, 024133, DOI:10.1063/1.4731647.
- X. Ding, Z. Peng, S. S. Lin, M. Geri, S. Li, P. Li, Y. Chen, M. Dao, S. Suresh and T. J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12992–12997, DOI:10.1073/pnas.1413325111.
- K. Han and A. B. Frazier, Lab Chip, 2006, 6, 265–273, 10.1039/B514539B.
- N. Pamme and A. Manz, Anal. Chem., 2004, 76, 7250–7256, DOI:10.1021/ac049183o.
- M. Zborowski and J. J. Chalmers, Anal. Chem., 2011, 83, 8050–8056, DOI:10.1021/ac200550d.
- S. Wyatt IV, C. D. Reyes and G. P. Lopez, Lab Chip, 2015, 15, 1230–1249, 10.1039/C4LC01246A.
- Y. Gao, W. Li and D. Pappas, Analyst, 2013, 138, 4714–4721, 10.1039/c3an00315a.
- P. Augustsson, C. Magnusson, M. Nordin, H. Lilja and T. Laurell, Anal. Chem., 2012, 84, 7954–7962, DOI:10.1021/ac301723s.
- K. R. Stone, D. D. Mickey, H. Wunderli, G. H. Mickey and D. F. Paulson, Int. J. Cancer, 1978, 21, 274–281 CrossRef CAS PubMed.
- A. van Bokhoven, M. Varella-Garcia, C. Korch, W. U. Johannes, E. E. Smith, H. L. Miller, S. K. Nordeen, G. J. Miller and M. S. Lucia, Prostate, 2003, 57, 205–225, DOI:10.1002/pros.10290.
- J. M. Martel and M. Toner, Annu. Rev. Biomed. Eng., 2014, 16, 371–396, DOI:10.1146/annurev-bioeng-121813-120704.
- D. Di Carlo, D. Irimia, R. G. Tompkins and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18892–18897, DOI:10.1073/pnas.0704958104.
- E. S. Asmolov, J. Fluid Mech., 1999, 381, 63–87, DOI:10.1017/S0022112098003474.
- A. A. S. Bhagat, S. S. Kuntaegowdanahalli and I. Papautsky, Phys. Fluids, 2008, 20, 101702, DOI:10.1063/1.2998844.
- S. S. Kuntaegowdanahalli, A. A. S. Bhagat, G. Kumar and I. Papautsky, Lab Chip, 2009, 9, 2973–2980, 10.1039/B908271A.
- N. Nivedita and I. Papautsky, Biomicrofluidics, 2013, 7, 054101, DOI:10.1063/1.4819275.
- G. Guan, L. Wu, A. A. Bhagat, Z. Li, P. C. Y. Chen, S. Chao, C. J. Ong and J. Han, Sci. Rep., 2013, 3, 1475 CrossRef PubMed.
- M. V. Patel, I. A. Nanayakkara, M. G. Simon and A. P. Lee, Lab Chip, 2014, 14, 3860–3872, 10.1039/C4LC00447G.
- C. Wang, S. V. Jalikop and S. Hilgenfeldt, Biomicrofluidics, 2012, 6, 12801–1280111, DOI:10.1063/1.3654949.
- A. R. Tovar, M. V. Patel and A. P. Lee, Microfluid. Nanofluid., 2011, 10, 1269–1278, DOI:10.1007/s10404-010-0758-1.
- R. H. Liu, J. Yang, M. Z. Pindera, M. Athavale and P. Grodzinski, Lab Chip, 2002, 2, 151–157, 10.1039/b201952c.
- A. R. Tovar and A. P. Lee, Lab Chip, 2009, 9, 41–43, 10.1039/b812435c.
- Y. Okabe and A. P. Lee, J. Lab. Autom., 2014, 19, 163–170, DOI:10.1177/2211068213495546.
- M. V. Patel, I. A. Nanayakkara, M. G. Simon and A. P. Lee, Lab Chip, 2014, 14, 3860–3872, 10.1039/c4lc00447g.
- C. Wang, S. V. Jalikop and S. Hilgenfeldt, Appl. Phys. Lett., 2011, 99, 034101, DOI:10.1063/1.3610940.
- M. V. Patel, A. R. Tovar and A. P. Lee, Lab Chip, 2012, 12, 139–145, 10.1039/c1lc20626e.
- W. Qi, C. Zhao, L. Zhao, N. Liu, X. Li, W. Yu and L. Wei, Cancer Cell Int., 2014, 14, 3, DOI:10.1186/1475-2867-14-3.
- M. Xavier, R. O. C. Oreffo and H. Morgan, Biotechnol. Adv., 2016, 34, 908–923, DOI:10.1016/j.biotechadv.2016.05.008.
- S. Y. Wang, K. L. Mak, L. Y. Chen, M. P. Chou and C. K. Ho, Immunology, 1992, 77, 298–303 CAS.
- B. Passlick, D. Flieger and H. W. Ziegler-Heitbrock, Blood, 1989, 74, 2527–2534 CAS.
- D. Strauss-Ayali, S. M. Conrad and D. M. Mosser, J. Leukocyte Biol., 2007, 82, 244–252, DOI:10.1189/jlb.0307191.
- M. Wiklund, Lab Chip, 2012, 12, 2018–2028, 10.1039/c2lc40201g.
- M. A. Burguillos, C. Magnusson, M. Nordin, A. Lenshof, P. Augustsson, M. J. Hansson, E. Elmér, H. Lilja, P. Brundin, T. Laurell and T. Deierborg, PLoS One, 2013, 8, e64233, DOI:10.1371/journal.pone.0064233.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS PubMed.
- S. L. Stott, C.-H. Hsu, D. I. Tsukrov, M. Yu, D. T. Miyamoto, B. A. Waltman, S. Michael Rothenberg, A. M. Shah, M. E. Smas, G. K. Korir, F. P. Floyd Jr., A. J. Gilman, J. B. Lord, D. Winokur, S. Springer, D. Irimia, S. Nagrath, L. V. Sequist, R. J. Lee, K. J. Isselbacher, S. Maheswaran, D. A. Haber and M. Toner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18392–18397, DOI:10.1073/pnas.1012539107.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an00290d |
| ‡ Co-first authors, equally contributed to the work presented. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |