
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Side-chain engineering of green color electrochromic polymer materials: toward adaptive camouflage application†
Hongtao
Yu‡
a,
Shan
Shao‡
a,
Lijia
Yan
b,
Hong
Meng
*ab,
Yaowu
He
a,
Chao
Yao
a,
Panpan
Xu
a,
Xiaotao
Zhang
cd,
Wenping
Hu
c and
Wei
Huang
*b
aSchool of Advanced Materials, Peking University Shenzhen Graduate School, Peking University, Shenzhen, 518055, China. E-mail: menghong@pkusz.edu.cn
bKey Lab for Flexible Electronics & Institute of Advanced Materials, Jiangsu National Synergistic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University, 30 South Puzhu Road, Nanjing, P. R. China. E-mail: iamwhuang@njtech.edu.cn
cBeijing National Laboratory for Molecular Sciences, Key Laboratory of Organic Solids, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China
dBroad (Beijing) Organic Optoelectronic Materials Research Engineering Co, Ltd, Beijing, China
First published on 16th February 2016
Abstract
The syntheses of adaptive camouflage devices based on novel side-chain engineered organic electrochromic materials have been demonstrated. Herein we report a molecule engineering approach for the tuning and syntheses of green-brown switchable electrochromic materials and also demonstrate their applications in chameleonic fabric devices. We have also successfully demonstrated the fabrication of chameleonic fabric devices.
Military adaptive camouflage technologies are being developed toward the development of lighter, more comfortable and more intelligent devices.1 Combining digital and lighting technologies, a proof of concept of a diffused lighting camouflage or counter-illumination camouflage has been proposed. The target is to make military armed forces more mobile and be better protected. Ideally, active camouflage could exhibit adaptive changes to the object surroundings by varying its patterns and colors, acting as perfect concealment from visual and infrared detections.
Electrochromic devices (ECDs) have been used in many applications such as anti-glare rear-view mirrors,2 smart windows,3 displays,4 data-storage devices,5 sunglasses6 and fabric electrochromics.7 Over the past few years, conjugated polymers have been used in electrochromic devices as active layers, because of their fast switching time,8 high optical contrasts,9 processability,10,11 and easy tuning of color with alternations in the structure.12 According to previous studies of conducting polymers, green-colored polymer materials in the neutral state are difficult to be attained for the realization of polymeric electrochromic display devices, because green color requires two absorption bands centered at the blue and red regions of the visible spectrum, and these absorption bands should be capable of being controlled simultaneously with the applied potentials. A green polymer PDDTP (poly(2,3-di(thien-3-yl)-5,7-di(thien-2-yl)thieno[3,4-b]-pyrazine)) was reported by Sonmez et al.13 Toppare et al. also reported a series of green polymer materials based on the molecule structure of PDPEQ (poly(2,3-diphenyl-5,8-(2,3-dihydrothieno[3,4-b][1,4]dioxin-7-yl)quinoxaline)).14–16 Previous studies have clearly demonstrated that the insertion of strong electron-donating alkoxy side chains into the polymer backbone not only enhances the ease of processing,17 but also modifies the electronic properties of the conjugated polymers,18 including the oxidation potential,19 bandgap,10,20 and stability of the oxidized state.21 Indeed, more interesting optical changes at longer wavelengths, extending from near infrared (NIR) to microwave regions were observed. While previous studies have focused on the fabrication of green color electrochromic materials for display applications, few efforts have been devoted to the study of the residual brown color in the oxidized form of these polymers and applying the polymer for camouflage applications.22–24 On the one hand, motivated by searching electrochromic materials with reversible color changes between vegetation green and soil-like brown color to endow the chameleonic devices with multiple camouflage abilities to fit the forest and sand-tone conditions; on the other hand, examining modified versions that might absorb higher energy to minimize radar or sonar detection; we hypothesized that these merits of the green-colored polymers in the neutral state and brown-colored in the oxidized state and their absorbing infrared longer wavelength range might meet the demands of chameleon materials in military applications. Inspired by the recent work in the design of conjugated polymers with branched groups for multichromophoric electrochromic polymers25 or high performance of organic thin film transistors (OTFTs),26 we applied this strategy in tuning organic conjugated materials through changing the alkoxy side chains of different spatial structure (short, straight and branched) to deliberately modify their electrochromic properties for the envisaged intelligent camouflage applications.
Three donor–acceptor-type compounds, namely 2,3-bis(3-methoxyphenyl)-5,8-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)quinoxaline (G1), 2,3-bis(3,4-bis(decyloxy)phenyl)-5,8-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)quinoxaline (G2), and 2,3-bis(3,4-bis((2-octyldodecyl)oxy)phenyl)-5,8-bis(2,3-dihydrothieno[3,4-b][1,4]dioxin-5-yl)quinoxaline (G3) were synthesized. Compared with G1 and G2, the polymer G3 presents an excellent solubility in common solvents, super-hydrophobicity, a more vegetable-green color in the neutral state and exceptional sand-tone color in the oxidized state at the lowest conversion voltage while switching absorption in NIR and IR range. Herein, the electrochromic fabric of the polymer G3 has also been fabricated using a spray-coating method and a novel device architecture was developed for the fabrication of ECD. The fabric can reversibly change from the fully neutral state (vegetable-green color) to the fully oxidized state (sand-tone color) at a low applied voltage. The high solubility, super-hydrophobic, multiple natural color and absorption in NIR and IR range of PG3 exactly meet the requirements as intelligent electrochromic chameleonic materials in military applications.
The monomer structures of G1, G2 and G3 are shown in Fig. 1a and the specific synthetic routes are exhibited in Fig. S1 (ESI†). Conceivably, the side chains of the polymer function primarily as solubilizing groups, and also have substantial impacts on polymer packing motifs, film morphologies and optical properties. We chose three specifically designed molecules to fine-tune the optical properties. The designed monomer G1 has a rigid-body model with short side chain methoxyl substitutes. The monomer G2 contains the straight long C12 alkoxyl side chain which is tilted relative to the quinoxaline unit backbone. The G3 monomer was constructed with the more bulky double-alkoxyl side chains which have large spaces between alkyl side chains. This may hinder the interchain π–π stacking twisted relative to the quinoxaline moiety of conjugated polymers. All the designed molecules are studied to fine-tune the molecule structures and hence optical properties. The appearance of the synthetic G1, G2 and G3 are shown in Fig. 1b–d, respectively. The state and color of the samples changes from solid to liquid and light-orange to brown, which suggests that the alkoxyl side chains of quinoxaline have a great effect on the interaction of monomer molecules. However, these monomers all have a high solubility in some common solvent (Table S1, ESI†), which is beneficial for further polymerization by chemical or electrochemical methods.
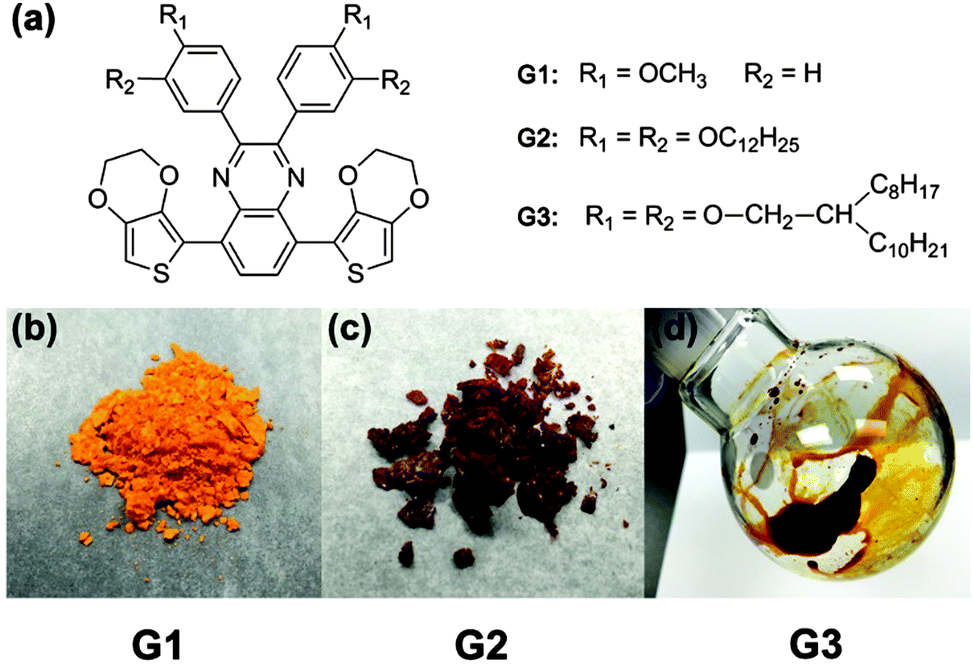 | ||
| Fig. 1 (a) The molecular structure of G1, G2 and G3, (b), (c) and (d) the appearance of G1, G2 and G3 monomers, respectively. | ||
In order to elucidate the polymer properties, electrochemical polymerization of monomers was applied by cyclic voltammetry on a platinum button electrode (area: 0.02 cm2) or indium tin oxide (ITO) coated glasses by oxidative electropolymerization in a dichloromethane (DCM) solution containing a 0.01 M monomer and 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6). The representative electrochemical growth processes revealing electroactivities of monomers G1, G2 and G3 and formation of corresponding polymers are given in Fig. S2 (ESI†). The oxidation of G1, G2 and G3 on a bare electrode starts at 0.74 V, 0.80 V and 0.85 V versus Ag wire pseudo-reference electrode in TBAPF6/DCM system, respectively. The increase of oxidation potential can be attributed to the improvement of acceptor capacity of the quinoxaline unit as the change of the side-chains. Redox couples for both monomers (G1 and G2) rapidly grow at relatively low potentials (at −0.25 V, −0.01 V for G1, and −0.01 V, 0.10 V for G2 vs. the same reference electrode), which indicates the formation of highly electroactive polymers except G3 (Fig. S2a–c, ESI†). It can be observed that polymer G1 (PG1) and G2 (PG2) could be deposited on a Pt or ITO glass slide, and the deposition rate of PG1 is faster than that of PG2. Moreover, some insoluble solids were formed near the electrodes in the process of electrochemical polymerization of G2, which suggests that PG1 and PG2 both have poor solubility in DCM and PG1 is more stable. To decrease the solubility of PG3 and increase the polymer forming performance of PG3, electrochemical polymerization of G3 was achieved potentiodynamically in a mixture of DCM and acetonitrile (ACN) (50/50, v/v) solution containing 0.01 M monomer and 0.1 M TBAPF6, because ACN was a more efficient medium to produce adhesive films.9 It should be noted that the redox couples rapidly grow at relatively high potentials (0.75 V, 0.65 V) compared to PG1 and PG2 (Fig. S2d, ESI†). It was attributed to a decrease in conductivity and poor hydrophilic character resulting from the vast alkyl-substituent, which prevents the polymerization process of G3 and provides a worse adherence to the electrodes. These results above imply that the presence of alkyl side chains not only allows an easier processing of electroactive polymers but also affects the electronic properties of the conjugated main chain.18 The alkoxyl side-chain greatly increased the solubility of corresponding polymers, leading to an excellent solubility of PG3 with the bulky double-alkoxyl in common solvents.
To further verify the properties of polymers, PG1, PG2 and PG3 were obtained by conventional chemical polymerization method as reported.27 As expected, the obtained PG1 and PG2 both are insoluble and PG3 has an excellent solubility in common solvents, such as DCM and TOL. As a polymeric electrochromic material, solution process of PG3 is convenient for the large-scale production of electrochromic devices using spin-coating or spray-coating methods.
To compare the electrochemical behaviors of different alkoxy-substituted polymers, the cyclic voltammetry studies were performed. Fig. 2a shows a stable cyclic voltammograms of PG1, PG2 and PG3 (obtained from electrochemical polymerization) films coated on a Pt electrode at scan rates of 100 mV s−1 in 0.1 M TBAPF6/ACN and very well-defined redox processes are observed, in consistency with the redox processes reported for the similar polymers with other alkyl substituted.14,15 It is surprising to note that, alkoxy side chain on pendant phenyl rings greatly affect the potentials for the oxidation of monomer and the redox couple of corresponding polymer. As observed for the redox potentials of different films, the value of E1/2 is increasing for PG1 with short methoxyl side chains, PG2 with straight long C12 alkoxyl side chains, to PG3 with more bulky double-alkoxyl side chains. This can be attributed to the fact that the substituent may distort the polymer backbone and decrease the degree of conjugation, leading to a lower conductivity. The charge injected/ejected during the CV was calculated by integrating the current density passing through the system and the value also decreased gradually, which suggests that the capacitive behavior of these materials decreases with an increase of alkyl-substituent side-chains. We can attribute this to a drop in electronic conductivity as interchain interactions decrease with the change of polymer side chains.18 The scan rate dependence of the anodic and cathodic peak currents is illustrated in Fig. 2b for PG3 and Fig. S3a and b (ESI†) for PG1 and PG2 films. A linear dependence demonstrates that these films are well adhered and the electrochemical processes are reversible and non-diffusion-controlled.18
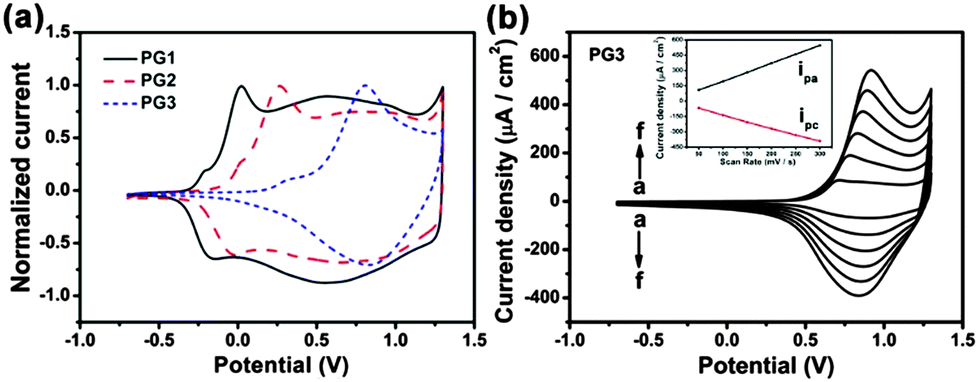 | ||
| Fig. 2 (a) Single scan cyclic voltammetry of different films at 100 mV s−1 in 0.1 M TBAPF6/ACN: PG1 (black, solid line), PG2 (red, dashed line), and PG3 (blue, short dashed line). (b) Scan rate dependence of the PG3 film in 0.1 M TBAPF6/ACN at (a) 50, (b) 100, (c) 150, (d) 200, (e) 250 and (f) 300 mV s−1 (inset is the plot of scan rate with anodic (ipa) and cathodic (ipc) peak currents). | ||
Spectroelectrochemistry experiments were performed to probe the optical changes at different applied potentials. Polymer films were deposited on ITO glass slides through electrochemical polymerization with a thickness of around 600 nm (Fig. S4, ESI†). The spectroscopic changes were investigated using a UV-Vis-NIR spectrophotometer in a 0.l M TBAPF6/ACN solution without monomers, as the increasing applied potentials. Fig. 3 reveals the spectroelectrochemistry and corresponding colors of PG1, PG2 and PG3. All polymers have two distinct absorption bands in the visible region (Fig. 3a–c), which is essential for neutral-state green conducting polymers. Although both of absorption bands are necessary, the values of maximum absorption wavelengths are critical for neutral-state green polymers. The absorption maxima of PG1, PG2 and PG3 are centered at 415, 420, 435 and 730, 714, 715 nm, which are essential values for a green color to be observed in the neutral state. It should be noted that, the potential of their fully neutral state (green color) greatly decreases as the increase of polymer alkoxyl side chains (−0.5 V for PG1, −0.3 V for PG2, and −0.1 V for PG3), leading to a more saturated green color. Meanwhile, their full oxidation potential (brown color) increases slowly (+0.8 V for PG1, +0.9 V for PG2, and +1.0 V for PG3) and gradually to produce a brown color analogous to the color of sands at the same applied potential of +0.8 V. The alkoxy groups affect the potentials not only for the oxidation of monomer and the redox couples of polymers, but also for their neutral and oxidized states. This behavior is attributed to the different electronic structures of the polymers, and to the different acceptor capacities of the quinoxaline derivatives, which leads to unique donor–acceptor matches with EDOT moieties. Compared to PG1 and PG2, PG3 exhibited unique vegetable green neutral and sand-tone oxidized color, and the corresponding (Y; x; y) values were shown in Fig. 3d. Representation of the hue and saturation for these three polymers is given in Fig. S5 (ESI†). Although an extremely transmissive colorless oxidized state is essential for the realization of polymer electrochromic-based display devices, PG3 is unique in the literature with its highly saturated green color in the neutral state and exceptional sand-tone color in the oxidized state (Fig. 3d). In addition, the absorption spectrum of all polymers in the NIR (Near Infrared Ray) and IR regions increases greatly during the bleaching of the visible absorption as the applied potential increases. It can act as an infrared electrochromic process for potential applications in modulation of thermal transmission and absorption. Regarding these superior properties of PG3, the green-colored in the neutral state and brown-colored in the oxidized state and its absorbing infrared exactly meet the demands of chameleonic materials in military applications.
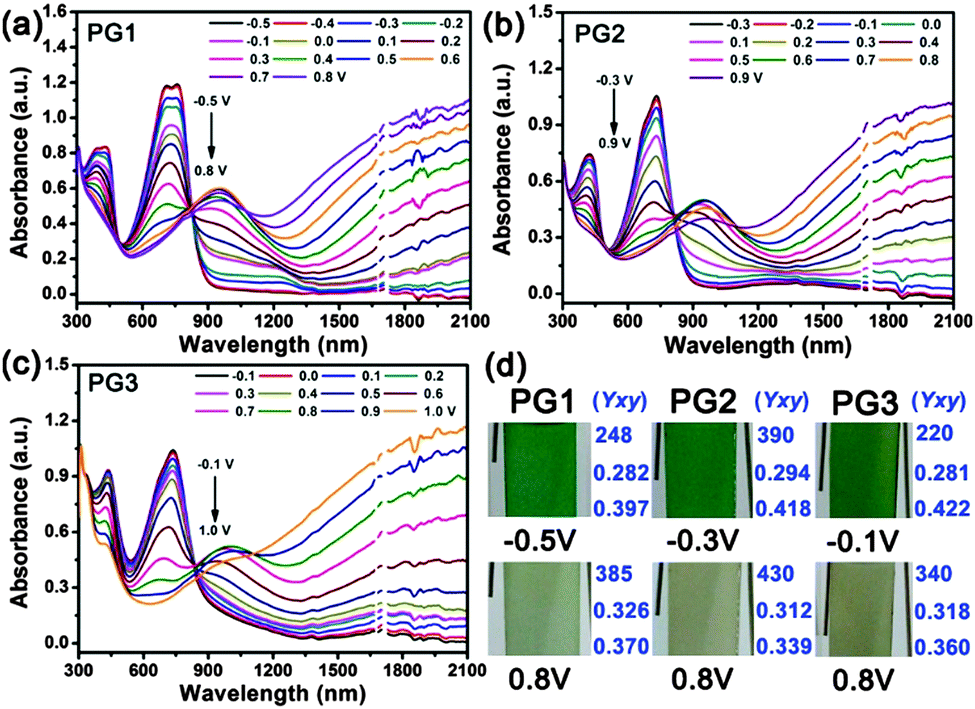 | ||
| Fig. 3 Spectroelectrochemistry of PG1, PG2 and PG3 film on an ITO-coated glass slide (0.5 cm width) in a monomer-free 0.1 M TBAPF6/ACN electrolyte–solvent couple at applied potentials: (a) −0.5 to 0.8 V, (b) −0.3 to 0.9 V, (c) −0.1 to 1.0 V. (d) Photographs and corresponding (Y; x; y) values of PG1, PG2 and PG3 film on an ITO-coated glass slide in the fully neutral and oxidized states. | ||
The electrochromic switching time and long-term stability of PG3 films are important for their potential applications in intelligent camouflage application. As illustrated in Fig. 4a, PG3 coated on a Pt electrode by spin-coating method was switched by stepping the potential between −0.1 V and +0.8 V with a switching interval of 5 s in a 0.1 M TBAPF6/ACN electrolyte–solvent system. PG3 showed a fast switching time of 1.0 s at fully neutral state and 1.5 s at fully oxidized state (defined as the time required to reach 95% of the full response). To investigate their stability, PG3 films were cycled between their fully neutral state (−0.1 V) and fully oxidized state (0.8 V) 5000 times in 0.1 M TBAPF6/ACN electrolyte–solvent system. PG3 showed a decrease by lower than 5% for the anodic peak current (ipa) and a decrease by lower than 4% for the cathodic (ipc) one (Fig. 4b), which highlights the robustness of the polymers upon switching between the neutral and oxidized states. Additionally, PG3 exhibited a super-hydrophobic property due to the bulky and long alkoxyl side chains (Fig. 4c and Movie S1, ESI†). As contact angles portrayed in Fig. 4c, the hydrophlic ITO (nearly 0°) and rough paper have been turned into a hydrophobic surface with much larger contact angles (about 120° and 135°).
 | ||
| Fig. 4 (a) Chronoamperometry for PG3 with a 5 s delay for each potential (−0.1 V and +0.8 V). (b) Stability of a 600 nm thick PG3 film cycled 5000 times with a scan rate of 100 mV s−1 in 0.1 M TBAPF6/ACN: the change in the anodic (□) and cathodic (○) peak currents as a function of number of cycles. (c) Photographs of the water droplet volume of 10 μL on the surface of ITO, PG3/ITO and PG3/rough paper, respectively. | ||
These findings indicate that PG3 is an excellent candidate for electrochromic devices with a low driving voltage and may find important applications in future camouflage devices.
To elucidate the effect of PG3 films in the ECDs, a simple sandwich-type PG3 EC cell was fabricated based on the reported method with modification,28 the letters “PKU” formed by PG3 films were prepared on an ITO-coated glass slide by spray-coating the 5 mg mL−1 solution of PG3/toluene on a mask. The appearance of PG3 and the corresponding solution (such as DCM or TOL) for spray-coating were shown in Fig. 5a. Switching tests were done in air using a two-electrode potentiostat (reference and counter electrode shorted together). Fig. 5b and Movie S2 (ESI†) shows the reversible changing process of letters between green color in the fully neutral state by applying a potential of −1.5 V and sand-color in the fully oxidized state by applying a potential of +1.5 V.
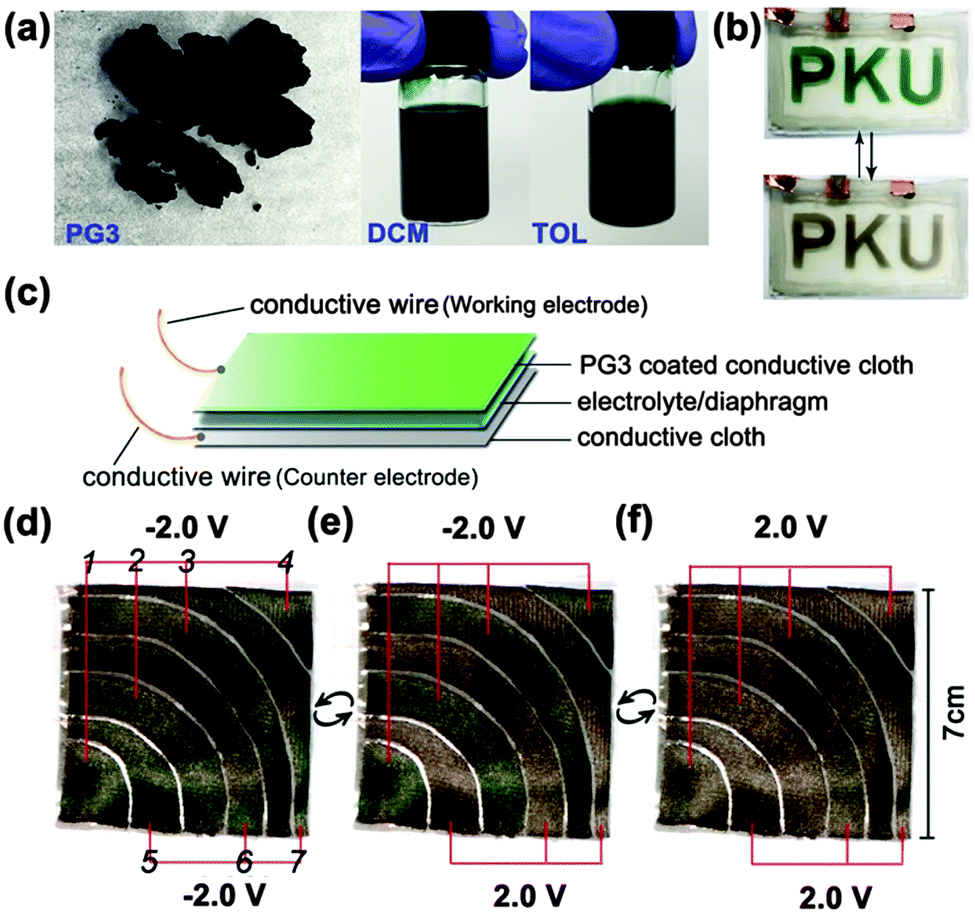 | ||
| Fig. 5 (a) The appearance of PG3, and in a DCM or TOL solution. Images of the PG3 camouflage fabric with space between each piece, (b) example of the patterned device in neutral and oxidized states, (c) fabric chameleonic electrochromic device cross-section for a typical device used in this study, (d) all pieces 1–7 as the working electrode in their neutral, (e) 1, 2, 3, 4 in neutral and 5, 6, 7 in oxidized, (f) in oxidized states. | ||
Aiming to fabricate the camouflage clothing, a chameleonic fabric device was built by spray-coating the 5 mg mL−1 solution of PG3/toluene onto the surface of a conductive fabric as the working electrode. Another conductive fabric without treatment was used as the counter electrode. A fiberglass separator soaked with a gel electrolyte (weight ratio of ACN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC:PMMA:LiClO4 was 70:20:7:3) was added to provide ions and prevent short circuit. The structure of the chameleonic fabric device is shown in Fig. 5c.
:PC:PMMA:LiClO4 was 70:20:7:3) was added to provide ions and prevent short circuit. The structure of the chameleonic fabric device is shown in Fig. 5c.
As demonstrated in Fig. 5d–f, Movies S3 and S4 in the (ESI†), the whole fabric device was composed by one or more piece working electrodes and a whole piece counter electrode on the other side of diaphragm. The potential was applied on the interval piece working electrodes (1, 2, 3, 4 or 5, 6, 7) simultaneously (Fig. 5d). The whole device can reversibly change between green color and sand-tone color at an applied potential just like a camouflage clothing. Moreover, the chameleonic ECDs could maintain a given redox state (green or brown color) when taken to open circuit. If desired, the color can be refreshed just by a short potential pulse without a continuous supply of electrical energy to operate.
Conclusions
Three donor–acceptor-type polymers have been synthesized and can be switched between green (neutral state) and brown (oxidized state) colors. Compared with other alkoxy side chain polymers PG1 and PG2, the novel PG3 with bulky double-alkoxyl side chains revealed a highly vegetable-green color at a lower applied voltage and a soil-brown color in oxidized-state, strong absorption in the NIR and IR regions, excellent solubility in common organic solvents, super-hydrophobic, very fast switching times, and high stability. Chameleonic fabric has also been fabricated, and the ability to reversibly change between green and sand colors is essential for adaptive camouflage to the conditions of forest and sands.Acknowledgements
This work was supported by Shenzhen Key Laboratory of Organic Optoelectromagnetic Functional Materials of Shenzhen Science and Technology Plan (ZDSYS20140509094114164), Guangdong Talents Project and NSFC (51373075). We also thank Prof. Liang, Y. Y. (the South University of Science and Technology of China) for UV measurements.References
- http://https://en.wikipedia.org/wiki/Camouflage .
- C. G. Granqvist, A. Azens, J. Isidorsson, M. Kharrazi, L. Kullman, T. Lindström, G. Niklasson, C.-G. Ribbing, D. Rönnow and M. S. Mattsson, J. Non-Cryst. Solids, 1997, 218, 273 CrossRef CAS.
- (a) M.-H. Yeh, L. Lin, P.-K. Yang and Z. L. Wang, ACS Nano, 2015, 9, 4757 CrossRef CAS PubMed; (b) A. L. Dyer, R. H. Bulloch, Y. Zhou, B. Kippelen, J. R. Reynolds and F. Zhang, Adv. Mater., 2014, 26, 4895 CrossRef CAS PubMed.
- (a) J. Remmele, D. E. Shen, T. Mustonen and N. Fruehauf, ACS Appl. Mater. Interfaces, 2015, 7, 12001 CrossRef CAS PubMed; (b) H. C. Moon, T. P. Lodge and C. D. Frisbie, Chem. Mater., 2015, 27, 1420 CrossRef CAS.
- G. Sonmez and H. B. Sonmez, J. Mater. Chem., 2006, 16, 2473 RSC.
- A. M. Österholm, D. E. Shen, J. A. Kerszulis, R. H. Bulloch, M. Kuepfert, A. L. Dyer and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2015, 7, 1413 Search PubMed.
- M. A. Invernale, Y. Ding and G. A. Sotzing, ACS Appl. Mater. Interfaces, 2010, 2, 296 CAS.
- (a) S. A. Sapp, G. A. Sotzing and J. R. Reynolds, Chem. Mater., 1998, 10, 2101 CrossRef CAS; (b) A. Kumar, D. M. Welsh, M. C. Morvant, F. Piroux, K. A. Abboud and J. R. Reynolds, Chem. Mater., 1998, 10, 896 CrossRef CAS; (c) J. Jensen, M. Hösel, I. Kim, J.-S. Yu, J. Jo and F. C. Krebs, Adv. Funct. Mater., 2014, 24, 1228 CrossRef CAS.
- L. Groenendaal, G. Zotti, P. H. Aubert, S. M. Waybright and J. R. Reynolds, Adv. Mater., 2003, 15, 855 CrossRef CAS.
- G. Sonmez, H. B. Sonmez, C. K. F. Shen, R. W. Jost, Y. Rubin and F. Wudl, Macromolecules, 2005, 38, 669 CrossRef CAS.
- (a) J. Padilla, A. M. Österholm, A. L. Dyer and J. R. Reynolds, Sol. Energy Mater. Sol. Cells, 2015, 140, 54 CrossRef CAS; (b) Z. Xu, X. Chen, S. Mi, J. Zheng and C. Xu, Org. Electron., 2015, 26, 129 CrossRef CAS; (c) A. Kumar, M. T. Otley, F. A. Alamar, Y. Zhu, B. G. Arden and G. A. Sotzing, J. Mater. Chem. C, 2014, 2, 2510 RSC; (d) J. Jensen, M. Hösel, A. L. Dyer and F. C. Krebs, Adv. Funct. Mater., 2015, 25, 2073 CrossRef CAS.
- J. A. Kerszulis, C. M. Amb, A. L. Dyer and J. R. Reynolds, Macromolecules, 2014, 47, 5462 CrossRef CAS.
- G. Sonmez, C. K. F. Shen, Y. Rubin and F. Wudl, Angew. Chem., Int. Ed., 2004, 43, 1498 CrossRef CAS PubMed.
- G. E. Gunbas, A. Durmus and L. Toppare, Adv. Funct. Mater., 2008, 18, 2026 CrossRef CAS.
- G. E. Gunbas, A. Durmus and L. Toppare, Adv. Mater., 2008, 20, 691 CrossRef CAS.
- A. Durmus, G. E. Gunbas and L. Toppare, Chem. Mater., 2007, 19, 6247 CrossRef CAS.
- K. Yoshino, P. Love, M. Onoda and R.-i. Sugimoto, Jpn. J. Appl. Phys., 1988, 27, L2388 CrossRef CAS.
- G. Sönmez, I. Schwendeman, P. Schottland, K. Zong and J. R. Reynolds, Macromolecules, 2003, 36, 639 CrossRef.
- B. Karabay, L. C. Pekel and A. Cihaner, Macromolecules, 2015, 48, 1352 CrossRef CAS.
- T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190 CrossRef CAS PubMed.
- R. Hanna and M. Leclerc, Chem. Mater., 1996, 8, 1512 CrossRef CAS.
- G. Sonmez, Chem. Commun., 2005, 5251 RSC.
- S. Beaupre, A. C. Breton, J. Dumas and M. Leclere, Chem. Mater., 2009, 21, 1504 CrossRef CAS.
- P. M. Beaujuge and R. R. John, Chem. Rev., 2010, 110, 268 CrossRef CAS PubMed.
- L. Beverina, G. A. Pagani and M. Sassi, Chem. Commun., 2014, 50, 5413 RSC.
- (a) H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478 CrossRef CAS PubMed; (b) J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130 CrossRef CAS PubMed; (c) H. Meng, J. Zheng, A. J. Lovinger, B.-C. Wang, P. G. Van Patten and Z. Bao, Chem. Mater., 2003, 15, 1778 CrossRef CAS.
- H. Meng and F. Wudl, Macromolecules, 2001, 34, 1810 CrossRef CAS.
- G. Sonmez, H. Meng and F. Wudl, Chem. Mater., 2004, 16, 574 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tc00197a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |