
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halogen bonding of N-bromosuccinimide by grinding†
Juraj
Mavračić
,
Dominik
Cinčić
* and
Branko
Kaitner
Department of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, HR-10000 Zagreb, Croatia. E-mail: dominik@chem.pmf.hr; Fax: 00385 14606341; Tel: 00385 14606362
First published on 20th April 2016
Abstract
Two halogen bonded cocrystals of N-bromosuccinimide and 4,4′-bipyridine, with stoichiometric ratios 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1, have been synthesized and characterized. We present the first mechanochemical cocrystallization of N-bromosuccinimide.
:1 and 2:1, have been synthesized and characterized. We present the first mechanochemical cocrystallization of N-bromosuccinimide.
Halogen bonds are non-covalent interactions that form between polarisable and electron-deficient halogen atoms (Br, I) and electron-rich Lewis bases (nucleophilic atoms such as O, N, S, etc.).1 They have been utilised as alternatives to hydrogen bonds in crystal engineering of organic and metal–organic materials.2 An overview of the currently available literature reveals that studies of halogen bonding have largely focused on cocrystals of perfluorohalocarbons as classic halogen bond donors.3 Numerous reports have been published over the past decade dealing with crystal engineering and synthesis of multicomponent materials containing iodo-substituted perfluorinated donors.4 Recently, Rissanen and Raatikainen have introduced a new family of halogen bond donors.5 They have demonstrated how N-halosuccinimides can be used for constructing halogen bonded molecular crystals. Also, in a recent study, Fourmigué and Espinosa described N-iodoimide derivatives as strong halogen bond donors.6N-halosuccinimides are commercially available inexpensive compounds, commonly used as halogenation agents in organic synthesis.7 They possess an extremely polarised halogen atom but have not yet been fully recognized and studied as potential halogen bond donors.8 A cursory search of the Cambridge Structural Database8b (CSD) based on the ability of N-halosuccinimides to act as a halogen bond donor has resulted in 13 hits; 10 of those have been derived from N-iodosuccinimide (nis) and 3 from N-bromosuccinimide (nbs).
Herein we report a mechanochemical approach9 to the construction of halogen-bonded cocrystals of nbs. We have selected 4,4′-bipyridine (bpy) as a good counterpart to nbs, a molecule that is a commonly used ditopic hydrogen or halogen bond acceptor (Fig. 1). Compared to the traditional cocrystallization method, synthesis from a solution, mechanochemical synthesis has been proven superior and is preferable because it sidesteps finding an appropriate solvent or solvent mixture.10,11 In general it is much faster, therefore, unexpected products resulting from unwanted reactions such as oxidation, halogenation or solvolysis are avoided. To the best of our knowledge, this is the first mechanosynthesis of multicomponent halogen-bonded solids using nbsvia neat grinding (NG) and liquid-assisted grinding (LAG).12 However, numerous reports have been published dealing with mechanochemical approaches to halogenation using nbs and nis compounds.13
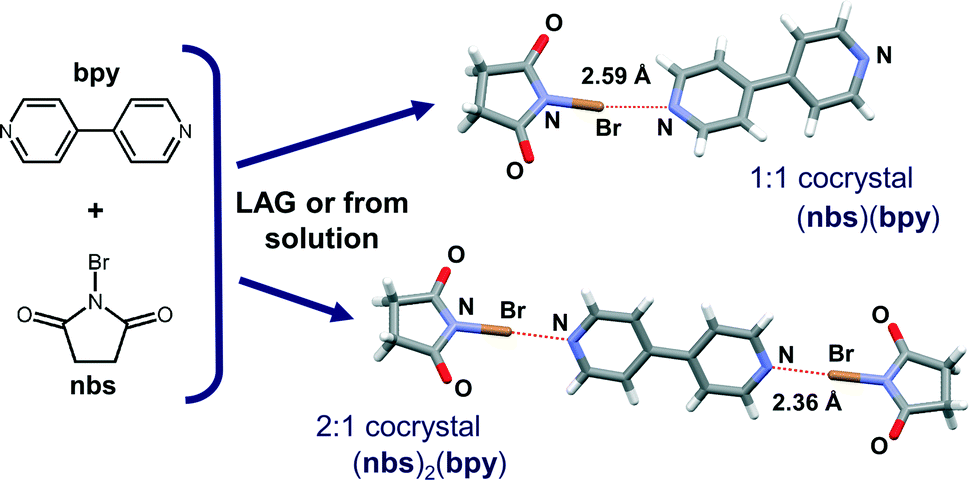 | ||
| Fig. 1 Structures of reactants, N-bromosuccinimide (nbs) and 4,4-bipyridine (bpy), and the cocrystal structures obtained as a result of the single crystal X-ray diffraction experiment. | ||
In order to explore the stoichiometric ratio of nbs and bpy as well as the reactivity in the solid state, we first attempted the mechanochemical synthesis of the cocrystals by NG and LAG of nbs with bpy in the stoichiometric ratios 1:1 and 2:1, respectively. Milling was conducted in a Retsch MM200 mill using a stainless steel milling assembly (see the ESI†) under normal laboratory conditions (temperature ca. 25 °C, 40–50% RH). To observe the grinding experiments, as well as to facilitate the characterisation of the new cocrystals by single-crystal X-ray diffraction, mechanochemical experiments were accompanied by crystallization from the solution. All reactants and products have been characterised by means of powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC). NG of nbs with bpy in a stoichiometric ratio of 1:1 for 15 min afforded a (nbs)(bpy) cocrystal, as proven by PXRD, but the NG experiment in a stoichiometric ratio of 2:1 for 15 min afforded a mixture of products, where the (nbs)(bpy) cocrystal was present as the major product and the (nbs)2(bpy) cocrystal only in traces (see the ESI†). When the same NG experiment was carried out for 80 min, the reaction was still not quantitatively complete – this time the experiment resulted in a mixture with the (nbs)2(bpy) cocrystal as the major product (see the ESI†). Considering that a liquid phase can significantly enhance the scope and rate of the mechanochemical experiment12,14 we turned to LAG. An addition of a small quantity of acetonitrile to the reaction mixtures,15 followed by grinding for 15 min, quantitatively provided both 1:1 and 2:1 cocrystals with PXRD patterns identical to those of the cocrystals prepared by the solution method (Fig. 2). In the solution experiments, nbs and bpy have been dissolved in hot acetonitrile. The solutions were left at room temperature, and the products, the 1:1 and 2:1 cocrystals, crystallized upon cooling. Single crystals of both cocrystals suitable for X-ray diffraction were obtained by concomitant crystallization from acetone. The measured PXRD patterns of (nbs)(bpy) and (nbs)2(bpy) obtained by both methods, grinding and from solution, are in good agreement with those calculated from the single crystal data (Fig. 2, see the ESI†). The (bpy)(nbs) and (nbs)2(bpy) cocrystals crystallize in the monoclinic system with four formula units per unit cell. The molecular structures of the cocrystals with the atom numbering scheme are given in Fig. S1 (see the ESI†). In the crystal structure of (bpy)(nbs), each nbs molecule is associated with a bpy molecule via an N⋯Br halogen bond. The Br⋯N distance of 2.586(2) Å is 23.9% less than the sum of the van der Waals (VDW) radii of nitrogen and bromine (3.4 Å) and almost linear, with an N–Br⋯N angle of 171.20(6)°. Only one of the two nitrogen atoms in each bpy molecule is involved in bonding with an nbs molecule (Fig. 3). Furthermore, crystal structure determination of (nbs)2(bpy) revealed two pairs of symmetry equivalent (CO)2N–Br⋯N halogen bonds with contact distances of 2.358(3) and 2.374(2) Å, which are 30.6 and 30.2% less than the sum of the VDW radii, respectively. Both contacts are highly linear, with an N–Br⋯N angle of 176.5(1)° and 179.0(1)°. For both cocrystals, the N–Br bond in the nbs molecules is extended compared to that observed for the pure nbs crystal16 (0.03 Å for the 1:1 cocrystal; 0.07 Å and 0.08 Å for the 2:1 cocrystal). All these observations indicate the presence of a strong halogen bond with a partially covalent character and with charge transfer from the N–Br bond to the Br⋯N contact.17 Considering that an nbs molecule possesses carbonyl groups and is also a strong hydrogen bond acceptor, components in both cocrystals create a specific halogen and hydrogen bonding pattern. The crystal structure of (nbs)(bpy) can be described by the association of 1:1 halogen bonded complexes into a hydrogen bonded 3D network via the C–H⋯O hydrogen bonds between nbs and bpy (C9⋯O1 contact of 3.542(3) Å, C14⋯O2 contact of 3.342(3) Å and C11⋯O2 contact of 3.356(3) Å) and via the C–H⋯O hydrogen bonds between nbs molecules (C3⋯O1 contact of 3.412(3) Å and C2⋯O1 contact of 3.384(3) Å). A notable detail of the cocrystal structure is the fact that each nbs molecule is connected to seven neighbouring molecules (three nbs and four bpy) as a halogen bond donor, ditopic hydrogen bond donor and ditopic hydrogen bond acceptor (Fig. 3). As in the 1:1 cocrystal, the crystal structure of (nbs)2(bpy) can be described by the association of 2:1 halogen bonded complexes into a hydrogen bonded 3D network via the C–H⋯O hydrogen bonds. The crystal structure contains two symmetrically nonequivalent nbs and bpy molecules (Fig. 4). One bpy molecule is connected with four neighbouring nbs molecules via the described halogen bond and bifurcated C–H⋯O hydrogen bonds (C15⋯O3 contact of 3.477(4) Å and C17⋯O3 contact of 3.341(5) Å). The second bpy molecule is connected with six neighbouring nbs molecules via the described halogen bond, C–H⋯O hydrogen bonds (C9⋯O4 contact of 3.515(4) Å), and via the bifurcated C–H⋯O hydrogen bonds (C10⋯O2 contact of 3.403(4) Å and C12⋯O2 contact of 3.434(4) Å). The overall structure results from the combination of such interactions and it is additionally stabilized by the C–H⋯O hydrogen bonds between nbs molecules (C3⋯O4 contact of 3.406(5) Å). Furthermore, the described interactions in both cocrystals can be observed by close analysis of the two-dimensional fingerprint plot derived from the Hirshfeld surface18 of the cocrystal components (see the ESI†). Thermal analysis of the prepared cocrystals by DSC reveals that both compounds are of similar thermal stability. The DSC curves of both cocrystals have the same profile and show one weak endothermic change peak and one strong exothermic change peak. They occur for (nbs)(bpy) at 116 °C (3.5 kJ mol−1) and 131 °C (58.4 kJ mol−1), and for (nbs)2(bpy) at 103 °C (3.7 kJ mol−1) and 136 °C (114.6 kJ mol−1) (see the ESI†). For both cocrystals, the exothermic change peak corresponds to the cocrystal decomposition and is at a lower temperature than that of the pure nbs reactant (208 °C, 42.9 kJ mol−1).
 | ||
| Fig. 2 Comparison of reactants, mechanochemical products, crystallization products and calculated powder patterns. | ||
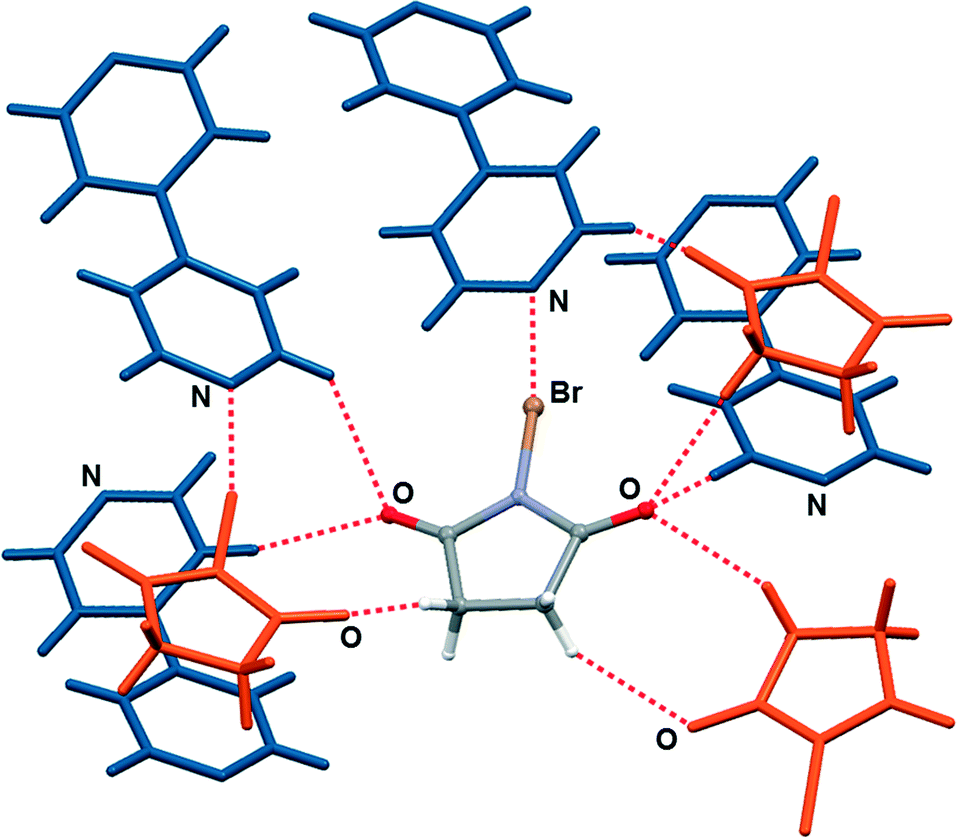 | ||
| Fig. 3 Halogen and hydrogen bonding of nbs molecule with neighbouring molecules in the crystal structure of (nbs)(bpy). | ||
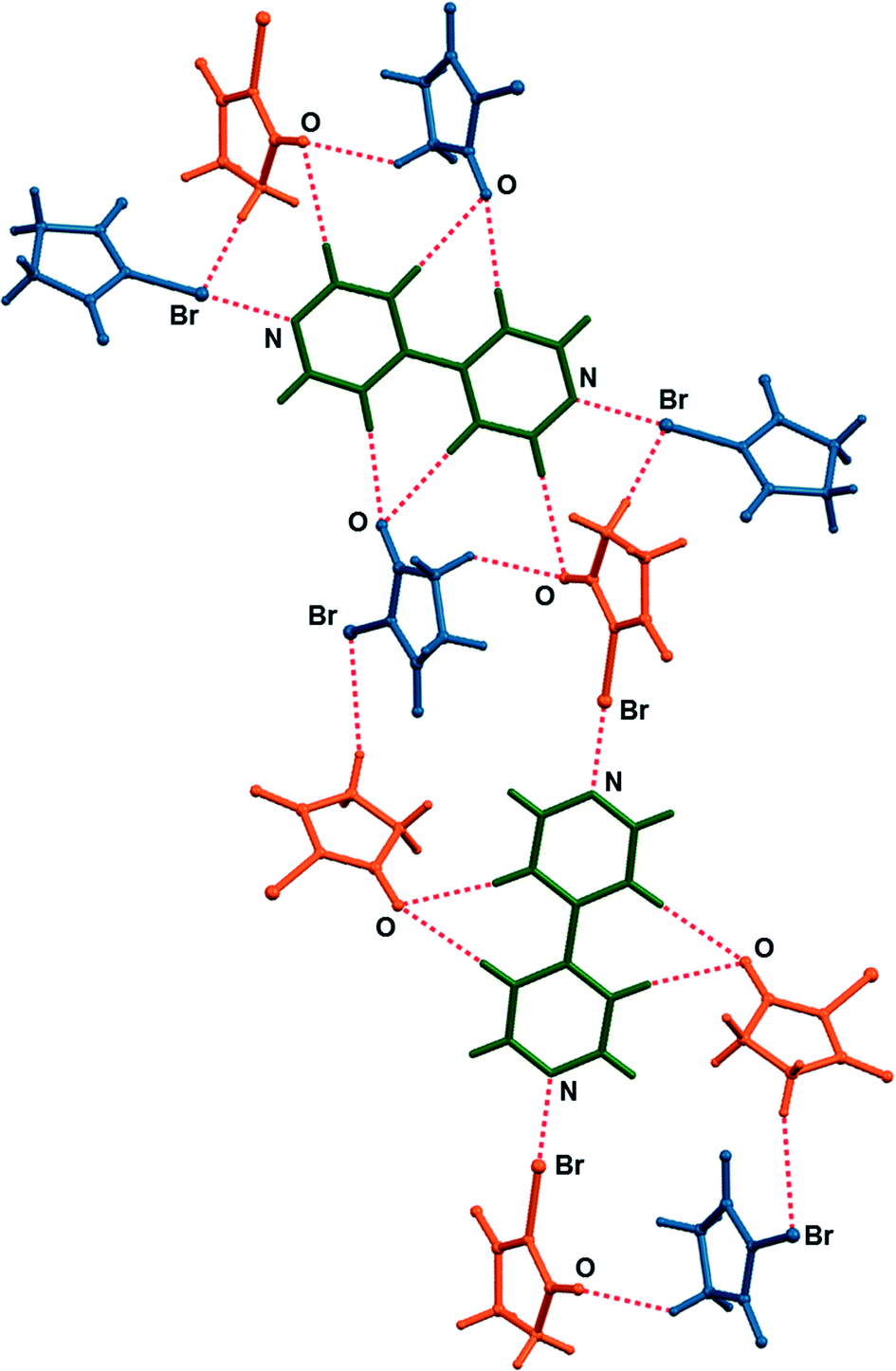 | ||
| Fig. 4 Network of halogen and hydrogen bonded molecules in the crystal structure of (nbs)2(bpy). | ||
Conclusions
In summary, we have demonstrated how nbs, a commercially available compound commonly used as a brominating agent in organic synthesis, can be used in cocrystal synthesis. To the best of our knowledge, this is the first demonstration of mechanosynthesis of multicomponent halogen-bonded solids using nbs as a halogen bond donor. The mechanochemical approach exhibits notable advantages over conventional solution-based synthesis of such compounds, not only through reducing the time of preparation and isolation, but also by evading the formation of potentially unwanted halogenation products. Crystal structure analysis of (nbs)(bpy) and (nbs)2(bpy) demonstrated that the presence of a polarised halogen atom and two carbonyl groups on the nbs molecule leads to a specific halogen and hydrogen bonding pattern in their crystal structure. The high stability of both materials, as a result of halogen and hydrogen bonding patterns, was proven by a relatively high decomposition temperature. Also, crystal structure analysis revealed indications of a very strong halogen bond with a partially covalent character and with charge transfer from the N–Br bond to the Br⋯N contact. We believe that the described results will have significant impact on the design and synthesis of future halogen-bonded materials containing N-haloimides as well as on halogen bond-driven crystal engineering in general.Acknowledgements
This research was supported by the Ministry of Science and Technology of the Republic of Croatia under the project 119-1193079-3069 and by the Croatian Science Foundation under the project IP-2014-09-7367. We are grateful to Prof. Vladimir Stilinović and Vinko Nemec for productive discussions and helpful suggestions.Notes and references
- (a) A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686 CrossRef CAS PubMed; (b) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed.
- (a) P. Sgarbossa, R. Bertani, V. Di Noto, M. Piga, G. A. Giffin, G. Terraneo, T. Pilati, P. Metrangolo and G. Resnati, Cryst. Growth Des., 2012, 12, 297 CrossRef CAS; (b) M. T. Johnson, Z. Džolić, M. Cetina, O. F. Wendt, L. Öhrström and K. Rissanen, Cryst. Growth Des., 2012, 12, 362 CrossRef CAS PubMed; (c) C. Merkens, F. Pan and U. Englert, CrystEngComm, 2013, 15, 8153 RSC; (d) D. Cinčić, T. Friščić and W. Jones, Chem. Mater., 2008, 20, 6623 CrossRef; (e) D. Cinčić, T. Friščić and W. Jones, New J. Chem., 2008, 32, 1776 RSC; (f) O. S. Bushuyev, D. Tan, C. J. Barrett and T. Friščić, CrystEngComm, 2015, 17, 73 RSC; (g) D. Cinčić and T. Friščić, CrystEngComm, 2015, 17, 7870 RSC.
- (a) R. W. Troff, T. Mäkelä, F. Topić, A. Valkonen, K. Raatikainen and K. Rissanen, Eur. J. Org. Chem., 2013, 1617 CrossRef CAS; (b) M. Fourmigué, Curr. Opin. Solid State Mater. Sci., 2009, 13, 36 CrossRef; (c) C. B. Aakeröy, T. K. Wijethunga, M. A. Haj, J. Desper and C. Moore, CrystEngComm, 2014, 16, 7218 RSC; (d) D. Cinčić, T. Friščić and W. Jones, CrystEngComm, 2011, 13, 3224 RSC.
- (a) T. Shirman, D. Freeman, Y. D. Posner, I. Feldman, A. Facchetti and M. E. van der Boom, J. Am. Chem. Soc., 2008, 130, 8162 CrossRef CAS PubMed; (b) D. W. Bruce, P. Metrangolo, F. Meyer, T. Pilati, C. Präsang, G. Resnati, G. Terraneo, S. G. Wainwright and A. C. Whitwood, Chem. – Eur. J., 2010, 16, 9511 CrossRef CAS PubMed; (c) D. Cinčić, T. Friščić and W. Jones, Chem. – Eur. J., 2008, 14, 747 CrossRef PubMed; (d) D. Cinčić, T. Friščić and W. Jones, J. Am. Chem. Soc., 2008, 130, 7542 CrossRef PubMed.
- (a) K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972 RSC; (b) K. Raatikainen and K. Rissanen, Chem. Sci., 2012, 3, 1235 RSC.
- O. Makhotkina, J. Lieffrig, O. Jeannin, M. Fourmigué, E. Aubert and E. Espinosa, Cryst. Growth Des., 2015, 15, 3464 CAS.
- (a) H. J. Dauben Jr. and L. L. McCoy, J. Am. Chem. Soc., 1959, 81, 4863 CrossRef; (b) C. Djerassi, Chem. Rev., 1948, 43, 271 CrossRef CAS PubMed.
- (a) E. H. Crowston, A. M. Lobo, S. Prabhakar, H. S. Rzepa and D. J. Williams, J. Chem. Soc., Chem. Commun., 1984, 276 RSC; (b) F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- (a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (b) T. Friščić, Chem. Soc. Rev., 2012, 41, 3493 RSC.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621 Search PubMed.
- (a) D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638 RSC; (b) D.-K. Bučar, S. Filip, M. Arhangelskis, G. O. Lloyd and W. Jones, CrystEngComm, 2013, 15, 6289 RSC; (c) V. Nemec, N. Škvorc and D. Cinčić, CrystEngComm, 2015, 17, 6274 RSC.
- (a) T. Friščić, A. V. Trask, W. Jones and W. D. S. Motherwell, Angew. Chem., Int. Ed., 2006, 45, 7546 CrossRef PubMed; (b) D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002 CrossRef CAS PubMed; (c) V. Štrukil, L. Fábián, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksić and T. Friščić, Chem. Commun., 2010, 46, 9191 RSC.
- (a) J. A. R. P. Sarma and A. Nagaraju, Perkin 1 (2000), 2000, 2, 1113 Search PubMed; (b) X.-L. Wu, J.-J. Xia and G.-W. Wang, Org. Biomol. Chem., 2008, 6, 548 RSC; (c) G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668 RSC; (d) I. Pravst, M. Zupan and S. Stavber, Tetrahedron, 2008, 64, 5191 CrossRef CAS.
- (a) D. Cinčić, I. Brekalo and B. Kaitner, Chem. Commun., 2012, 48, 11683 RSC; (b) D. Cinčić, M. Juribašić, D. Babić, K. Molčanov, P. Šket, J. Plavec and M. Ćurić, Chem. Commun., 2011, 47, 11543 RSC.
- The amount of liquid was 40 μL per 200 mg of reactants, corresponding to a liquid-to-solid ratio of η = 0.2 μL mg−1, see: T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418 RSC.
- O. Jabay, H. Pritzkow and J. Jander, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1977, 32, 1416 Search PubMed.
- C. Wang, D. Danovich, Y. Mo and S. Shaik, J. Chem. Theory Comput., 2014, 10, 3726 CrossRef CAS PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for solution-based synthesis, instrumental experimental details and PXRD, and DSC data. CCDC 1465378 and 1465379 contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00638h |
| This journal is © The Royal Society of Chemistry 2016 |