
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct catalytic cross-coupling of alkenyllithium compounds†
Valentín
Hornillos
,
Massimo
Giannerini
,
Carlos
Vila
,
Martín
Fañanás-Mastral
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl; Fax: +31 50 363 4278; Tel: +31 50 3634296
First published on 28th November 2014
Abstract
A catalytic method for the direct cross-coupling of alkenyllithium reagents with aryl and alkenyl halides is described. The use of a catalyst comprising Pd2(dba)3/XPhos allows for the stereoselective preparation of a wide variety of substituted alkenes in high yields under mild conditions. In addition (1-ethoxyvinyl)lithium can be efficiently converted into substituted vinyl ethers which, after hydrolysis, give readily access to the corresponding methyl ketones in a one pot procedure.
Introduction
Palladium-catalysed cross-coupling of organometallic reagents with organic halides represents one of the most powerful methods for the construction of carbon–carbon bonds.1 Among these, reactions employing alkenylmetal reagents remain indispensable tools to access a broad range of highly substituted olefins and polyenes.2 With a variety of structurally diverse, functionalized organic halides available, the challenge of this transformation often remains the choice of the alkenyl nucleophile. Tin3 and boron4 reagents are frequently used for these coupling reactions. However, the high toxicity of organostannanes makes the Stille cross-coupling often less desirable. On the other hand, alkenyl boronic acids are prone to rapid protodeboronation or polymerization so requiring large excess of the coupling partner. Elegant solutions have been found via the conversion into more stable derivates such as boronic esters, MIDA boronates5 and trifluoroborates.6 Organosilicon compounds feature high stability and low toxicity although the use of fluoride anion for the activation is generally needed.7 Nonetheless, non-fluoride activators have recently been introduced to enhance the utility of these compounds.8,2d Alkenyl reagents for the Negishi cross-coupling (Zn, Al and Zr), have also been used in Pd-catalysed alkenylation reactions with a broad scope for the synthesis of dienes and polyenes.9 These alkenyl intermediates are routinely prepared by elementometalation9 from alkynes or transmetallation from the corresponding organolithium reagents, as is the case for boron, tin and silicon reagents. In contrast, the direct use of alkenyllithium compounds10 have, to the best of our knowledge, not been reported in Pd-catalysed cross-coupling reactions. Alkenyllithium reagents are easily accessible by lithium–halogen exchange or via direct metallation of the corresponding olefins without requiring purification prior to use.11 Their high reactivity has largely prohibited the use of these reagents in cross-coupling reactions due to the lack of selectivity. Pioneering studies by Murahashi and co-workers on the use of aryl and alkyllithium reagents in catalytic cross-coupling reactions revealed the limitations associated with their high reactivity.12,13 However, if controlled, this reactivity might be advantageous in facilitating the transmetallation with Pd allowing for milder reaction conditions which are beneficial to preserve the geometry of the alkene. In addition, their use would also drastically reduce the amount of byproducts generated with the light and non-toxic lithium halide being the only stoichiometric waste. Therefore, the development of a general method for the use of alkenyllithium reagents in catalytic cross-coupling reactions is highly desirable.Recently, our group reported the direct palladium catalysed cross-coupling of alkyl and (hetero)aryl lithium reagents with aryl and alkenyl (pseudo)halides, providing high yields and selectivity.14,15 The choice of the proper combination of catalyst and reaction conditions allows for efficient transmetallation, prevents the notorious lithium halogen exchange and homocoupling reactions and gives rise to the corresponding cross-coupled products in high yields. However, the use of alkenyllithium reagents in this transformation remains elusive.
Herein, we report a palladium-based method for the direct catalytic cross-coupling of alkenyllithium reagents and organic halides to afford substituted alkenes in high yield and isomeric purity under mild conditions in short reaction times (Scheme 1).
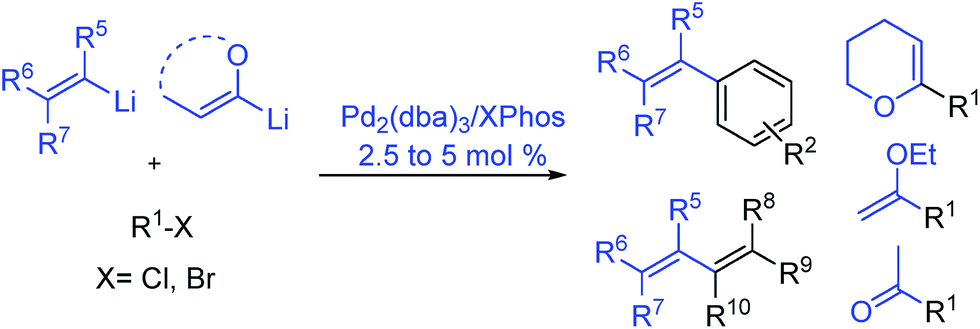 | ||
| Scheme 1 Palladium catalysed cross-coupling of alkenyllithium reagents. | ||
Results and discussion
Our investigation started with the reaction between (3-methylbut-2-en-2-yl)lithium, readily prepared from the corresponding bromide, and 4-methoxy-bromobenzene 1a, a reluctant arylbromide in many coupling reactions,16 in the presence of various Pd-based catalytic systems using toluene as solvent (Table 1).|
|
||||
|---|---|---|---|---|
| Entrya | [Pd] | Ligand | Conv. (%) |
2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4b :3:4b |
|
a Conditions: (3-methylbut-2-en-2-yl)lithium (0.40 mmol, 0.6 M in THF) was added to a solution of 1a (0.3 mmol) and catalyst in toluene (1 mL), 1 h addition time.
b
2a:3:4 ratios determined by GC analysis. dba = dibenzylideneacetone.
|
||||
| 1 | Pd[P(t-Bu)3]2 | Full | 44:56:0 |
|
| 2 | Pd2(dba)3 | L1, SPhos | 46 | 83:0:17 |
| 3 | Pd2(dba)3 | L2, CPhos | 38 | 87:0:13 |
| 4 | Pd2(dba)3 | L3, XPhos | Full | >99:0:0 |
| 5 | Pd-PEPPSI-IPent | Full | 96:0:4 |
|
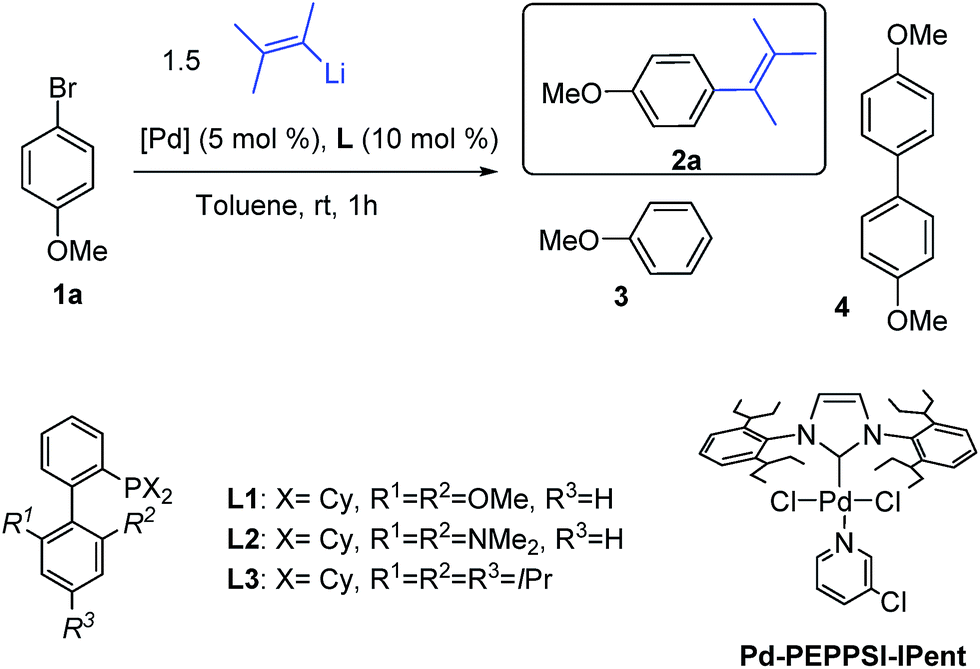
The use of Pd[P(t-Bu)3]217 (Table 1, entry 1), a catalyst we previously disclosed to be highly successful for the cross-coupling of alkyllithium reagents with aryl bromides14a,d led to the desired product 2a but in the presence of a large amount of dehalogenated side product 3. We then examined the use of Buchwald biaryl phosphine ligands18 in combination with Pd2(dba)3 (2.5 mol%). When ligands L1 and L2 were used, incomplete conversion was observed, along with homocoupling side product 4 (entries 2 and 3). To our delight, the use of XPhos L3 led to the cross-coupled product 2a, in 1 h at r.t., with excellent selectivity (>99%), avoiding dehalogenation and inhibiting the formation of the homocoupling product (entry 4). We also found that the air stable Pd-PEPPSI-IPent catalyst, introduced by Organ,19 displayed good selectivity although the presence of 4% of homocoupling was observed (entry 5).
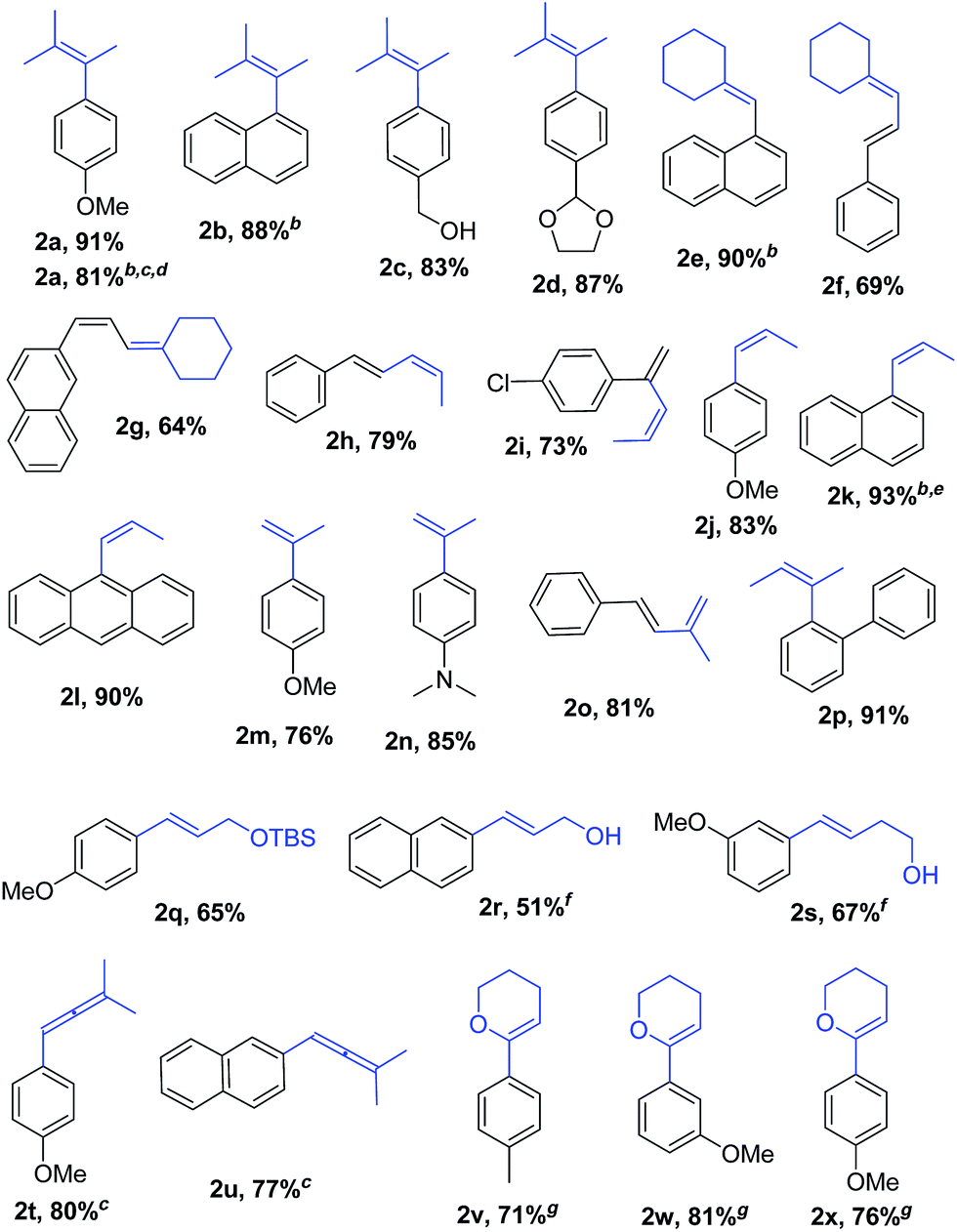
With Pd2(dba)3/Xphos as a highly efficient catalytic system, we set out to investigate the scope of this new reaction (Table 2)‡. A wide variety of alkenes were readily lithiated via direct metallation or by halogen–lithium exchange as shown in Scheme 2 (see ESI† for details).
|
|
|---|
|
a Conditions: RLi (1.3 equiv.) was added to a solution of organic bromide (0.3 mmol) and catalyst in toluene, 1 h addition time unless otherwise noted. Selectivity 2:3,4 > 90%. Isolated yields after column chromatography. For limitations of the method see Table S3, Schemes S2 and S3.
b X = Cl.
c The reaction was performed at 40 °C.
d 3.5 h addition time.
e The reaction was performed at 35 °C.
f After workup, product mixture was treated with TBAF to remove the silyl group prior to purification.
g 0.9 mmol scale. Reaction performed at 60 °C.
|
|

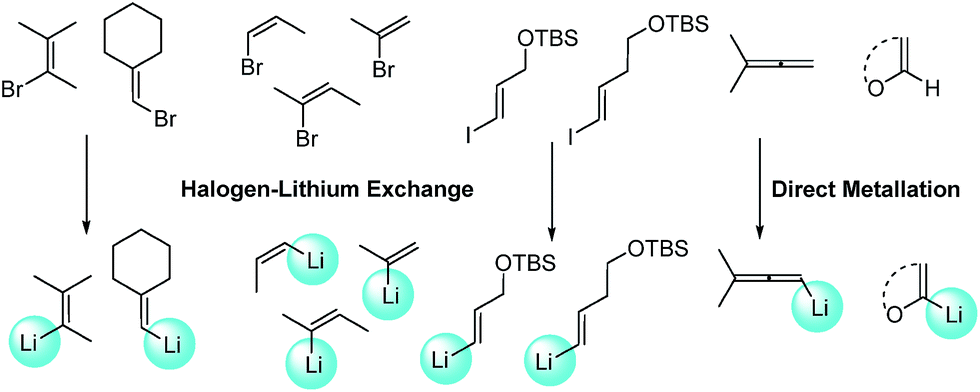 | ||
| Scheme 2 Synthesis of alkenyllithium reagents. | ||
The catalytic system proved to be also highly efficient in the reaction of (3-methylbut-2-en-2-yl)lithium with 1-chloronaphthalene 1b at r.t. (see also ESI, Table S1†). The corresponding tetrasubstituted alkene 2b was obtained in good yield with no trace of the 2-substituted regioisomer, indicating that benzyne intermediates via 1,2-elimination are not formed. To further explore the effectiveness of this catalytic systems we studied the more reluctant coupling partner 4-methoxy-chlorobenzene.14b Unfortunately, low conversion was found in the reaction at room temperature. However, the use of slightly higher temperatures (40 °C) and longer addition times (3.5 h) of the organolithium reagent allowed full conversion with high selectivity (see ESI, Table S2†). Substrate 1c, bearing an unprotected hydroxyl group, could also be coupled with this organolithium reagent (2.2 equiv.). The use of acetal-protected aldehyde 1d was also tolerated without cleavage of the protecting group. The cross-coupling of (cyclohexylidenemethyl)lithium and 1-chloronaphthalene proceeds efficiently giving alkene 2e in good yield. It is important to note that (E) and (Z) alkenyl bromides 1f and 1g reacted under the optimized conditions to give conjugated dienes 2f and 2g in good yield and with no presence of Fritsh–Butlenberg–Wiechell type rearrangement side products.20 In addition, both (E) and (Z)-olefins were coupled to form dienes 2f–2h with retention of stereochemistry. Illustrative is the use of (Z)-propenyllithium, obtained by treatment of (Z)-1-bromopropene with elemental lithium, which was smoothly coupled with retention of the (Z)-configuration, with a variety of alkenyl and aryl bromides providing the corresponding dienes 2h, 2i and alkenes 2j, 2k and 2l in good yields. Synthesis of diene 2h illustrates that both the alkenyl nucleophile and electrophile retain their geometrical configuration as confirmed by 1H NMR.21 In addition, vinyl bromide 1i reacted chemoselectively, leaving the chloride intact and available for further functionalization. It is interesting to note that (Z)-propenyllithium compared with the more hindered (3-methylbut-2-en-2-yl)lithium, required higher temperature in the coupling with 1-chloronaphthalene. This result could be due to a faster reductive elimination taking place from the more strained Pd(II) intermediates.22 This result is in accordance with similar observation using less hindered 2-chloronaphthalene. (see ESI, Scheme S1†).
2-Propenyllithium underwent cross-coupling reaction with 4-methoxy-bromobenzene and the more electron rich 4-bromo-N,N-dimethylaniline providing styrenes 2m and 2n in good yield without the need to increase temperature or reaction time. Retention of the olefin configuration was also observed in the reaction of this organolithium reagent with (E)-bromostyrene 2o. Moreover, a sterically hindered ortho-substituted arylbromide such as 2-bromo-1,1′-biphenyl 1p could be coupled in high yield and without loss of selectivity. Remarkably, alkenyllithium reagents bearing a protected alcohol functionality could also be used, providing the corresponding allylic TBS-protected alcohol 2q, allylic alcohol 2r and homoallylic alcohol 2s in fair to good yields with retention of the (E)-configuration. Interestingly, compound 2q is an intermediate in the synthesis of (−)-cytoxazone, a Novel Cytokine Modulator Isolated from Streptomyces sp.23
We envisioned extension of this coupling to allenyllithium24 compounds, providing a synthesis of substituted allenes.25 Under the optimized reaction conditions the coupling between (3-methylbuta-1,2-dienyl)lithium, readily available by direct metallation of the corresponding allene, and both 4-methoxy-bromobenzene and 2-chloronaphthalene proceeded in good yields and high regioselectivity and with less than 5% of regioisomeric alkyne arising from 1,3-lithium shift of the organolithium reagent.26
Next the use of functionalized (α-alkoxyvinyl)lithium reagents in this new cross-coupling transformation was investigated. An illustrative example is the reaction of (3,4-dihydro-2H-pyran-6-yl)lithium, obtained by direct metallation of commercially available dihydropyran with tBuLi.27a To our delight, reaction with p-methyl- and p- and m-methoxy-substituted bromoarenes provided compounds 2v, 2w and 2x in high yields and excellent selectivity (Table 2). Due to the reduced reactivity of this organometallic reagent, a slightly higher temperature was required (60 °C, 1 h). Nonetheless, established methods for the cross-coupling of glycal metal reagents require severe reaction conditions, long reaction times or they only were examined with the corresponding aryl iodides.27b,c
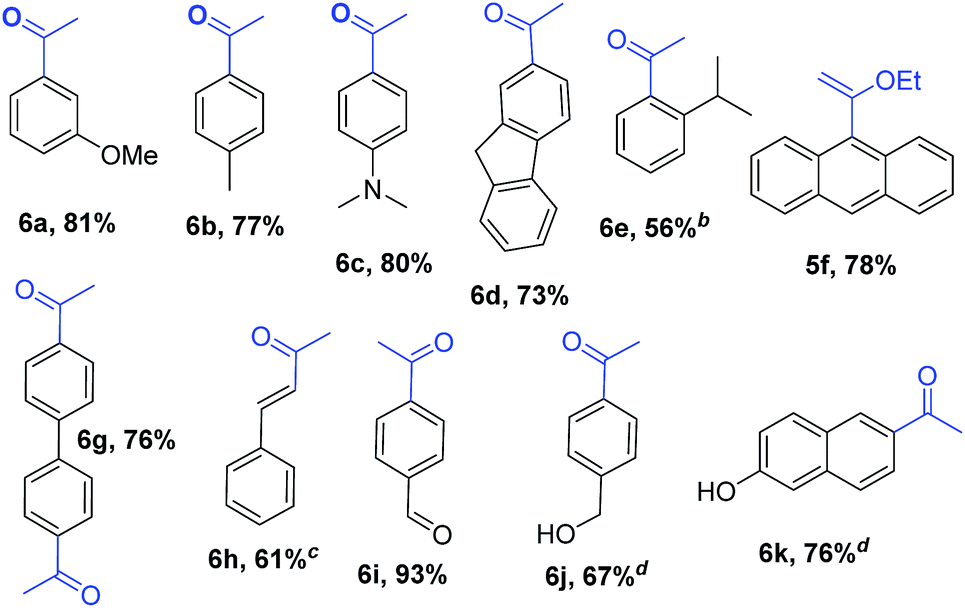
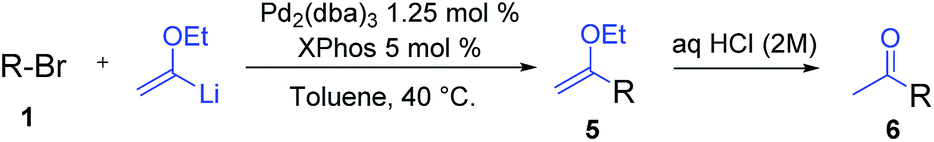
Encouraged by these results, we studied other α-alkoxyvinyl precursor such as the cheap and simple ethyl vinyl ether (Table 3). The use of the corresponding lithium reagent, readily obtained by direct lithiation, in the Pd-catalysed cross-coupling reaction could be used as a masked acetylating agent giving, after hydrolysis, the corresponding aryl methyl ketones.27b,28 This class of carbonyl derivates serves as versatile synthetic building blocks and are intermediates in the synthesis of drug candidates, fragrances and heterocycles.29 We performed this one pot transformation on larger scale (6 mmol), employing 2.5 mol% of catalyst to illustrate the synthetic utility of the method. As shown in Table 3, the method tolerates a variety of functional groups including acetals, ethers, amines alcohols and phenols. Electron rich substrates bearing amine, methyl and methoxy substituents underwent the expected coupling reaction at 40 °C in 2.5 h to give the corresponding ketones 6a–6c in good yields. Remarkably, bromofluorene was successfully employed, despite the acidity of the benzylic protons (pKa = 22). Although the formation of 6e demonstrates that hindered substrates are tolerated, we further validated this by coupling 9-bromoanthracene to give 5f in 78% yield. Facile multiple coupling is illustrated with the two fold acetylation of 4,4′-bis-bromobiphenyl, providing 6g in 76% yield. β-Bromo-styrene was coupled in moderate yield, with retention of the configuration. Acetal-protected p-bromobenzaldehyde was also tolerated in the reaction affording, after subsequent hydrolysis, 4-acetylbenzaldehyde 6i in excellent yield. Alcohols and phenols as Mg alkoxides are tolerated in this method to afford 6j and 6k. It should be noted that same of the compounds obtained such as 6a, 6d, 6e, 6i, or 6j would be challenging to synthesize through standard Friedel–Crafts acylation chemistry.30
|
|
|---|
| a Conditions: 3.0–6.0 mmol scale reaction. Aryl bromide (1 equiv.), (1-ethoxyvinyl)lithium (1.5 equiv., 0.60 M in THF). 2.5 h addition time. Toluene at 40 °C. Selectivity >90%. Yield values refer to isolated yields after purification. b 70% conversion. c 80% selectivity. d iPrMgCl (1.0 equiv., 2 M in Et2O) was added over 5 min prior to the organolithium. |
|
Conclusions
In summary, a fast and selective method has been developed for the Pd-catalysed alkenylation of aryl and alkenylhalides using organolithium reagents. This transformation leads to highly substituted alkenes under mild conditions and shows tolerance to various functional groups. Moreover, we describe the one pot synthesis of aryl methyl ketones involving coupling of lithiated alkenyl ethers.Acknowledgements
The Netherlands Organization for Scientific Research (NWO-CW), the National Research School Catalysis (NRSC-C), the European Research Council (ERC advanced grant 227897 to BLF), the Royal Netherland Academy of Arts and Sciences (KNAW) and the Ministry of Education Culture and Science (Gravitation program 024.601035) are acknowledged for financial support. C.V. was supported by Intra-European Marie Curie fellowship (FP7-PEOPLE-2011-IEF-300826).Notes and references
- (a) Metal-Catalysed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (b) Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (c) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E.-I. Negishi, Wiley-Interscience, New York, 2002 Search PubMed; (d) Topics in Organometallic Chemistry: Palladium in Organic Synthesis, ed. J. Tsuji, Springer, New York, 2005 Search PubMed; (e) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS PubMed; (f) X.-F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef CAS PubMed.
- (a) Science of synthesis, ed. G. Molander, J. P. Wolfe and M. Larhed, Thieme, 2013 Search PubMed; (b) S. E. Kelly,in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, pp. 729–817 Search PubMed; (c) Preparation of Alkenes, ed. J. M. J. Williams, Oxford University, Oxford, 1996 Search PubMed; (d) S. E. Denmark and C. R. Butler, Chem. Commun., 2009, 20 RSC.
- (a) A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6723 Search PubMed; (b) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed; (c) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651 CrossRef CAS PubMed; (d) Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, 2005 Search PubMed; (e) N. E. Leadbeater, Chem. Commun., 2005, 2881 RSC.
- (a) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704 CAS; (b) A. F. Littke, L. Schwartz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343 CrossRef CAS PubMed; (c) J. K. Stille, Angew. Chem., Int. Ed., 1986, 25, 508 CrossRef.
- (a) E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716 CrossRef CAS PubMed; (b) E. P. Gillis and M. D. Burke, Aldrichimica Acta, 2009, 42, 17 CAS; (c) E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484 CrossRef CAS PubMed.
- (a) G. A. Molander, T. Barcellos and K. M. Traister, Org. Lett., 2013, 15, 3342 CrossRef CAS PubMed; (b) E. Alacid and C. Nájera, J. Org. Chem., 2009, 74, 8191 CrossRef CAS PubMed; (c) G. A. Molander and L. A. Felix, J. Org. Chem., 2005, 70, 3950 CrossRef CAS PubMed; (d) G. A. Molander and C. R. Bernardi, J. Org. Chem., 2002, 67, 8424 CrossRef CAS PubMed; (e) G. A. Molander and M. R. Rivero, Org. Lett., 2002, 4, 107 CrossRef CAS PubMed.
- (a) T. Hiyama and Y. Nakao, Chem. Soc. Rev., 2011, 40, 4893 RSC; (b) S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114 RSC.
- (a) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486 CrossRef CAS PubMed; (b) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2012, 45, 864 CrossRef PubMed.
- (a) G. Wang, S. Mohan and E.-I. Negishi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11344 CrossRef CAS PubMed; (b) E.-I. Negishi, Q. Hu, Z. Huang, M. Qian and G. Wang, Aldrichimica Acta, 2005, 38, 71 CAS; (c) R. Álvarez, B. Vaz, H. Gronemeyer and A. R. de Lera, Chem. Rev., 2014, 114, 1 CrossRef PubMed.
- The use of organolithium and their subsequent transmetallation to softer organometallics to use in cross-coupling is well established: (a) E. J. Anctil and V. Snieckus, in Metal Catalysed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, vol. 1, pp. 761–813 Search PubMed; (b) J. Board, J. L. Cosman, T. Rantanen, S. P. Singh and V. Snieckus, Platinum Met. Rev., 2013, 57, 234 CrossRef.
- (a) Lithium Compounds in Organic Synthesis, ed. R. Luisi and V. Capriati, Wiley-VCH, Weinheim, 2014 Search PubMed; (b) The Chemistry of Organolithium Compounds, ed. Z. Rappoport and I. Marek, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS.
- (a) S. Murahashi, M. Yamamura, K. Yanagisawa, N. Mita and K. Kondo, J. Org. Chem., 1979, 44, 2408 CrossRef CAS; (b) S. Murahashi, J. Organomet. Chem., 2002, 653, 27 CrossRef CAS.
- For recent approaches to the indirect cross-coupling of organolithium reagents see: (a) A. B. Smith III, A. T. Hoye, D. Martinez-Solorio, W. Kim and R. Tong, J. Am. Chem. Soc., 2012, 51, 4533 CrossRef PubMed; (b) D. Martinez-Solorio, A. T. Hoye, M. H. Nguyen and A. B. Smith III, Org. Lett., 2013, 15, 2454 CrossRef CAS PubMed; (c) M. H. Nguyen and A. B. Smith III, Org. Lett., 2014, 16, 2070 CrossRef CAS PubMed , For an use of flow-chemistry technology for the Pd-catalysed cross-coupling with lithium reagents see: ; (d) A. Nagaki, A. Kenmoku, Y. Moriwaki, A. Hayashi and J. Yoshida, Angew. Chem., Int. Ed., 2010, 49, 7543 CrossRef CAS PubMed.
- (a) M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Nat. Chem., 2013, 5, 667 CrossRef CAS PubMed; (b) V. Hornillos, M. Giannerini, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Org. Lett., 2013, 15, 5114 CrossRef CAS PubMed; (c) M. Giannerini, V. Hornillos, C. Vila, M. Fañanás-Mastral and B. L. Feringa, Angew. Chem., Int. Ed., 2013, 52, 13329 CrossRef CAS PubMed; (d) C. Vila, M. Giannerini, V. Hornillos, M. Fañanás-Mastral and B. L. Feringa, Chem. Sci., 2014, 5, 1361 RSC; (e) C. Vila, V. Hornillos, M. Giannerini, M. Fañanás-Mastral and B. L. Feringa, Chem.–Eur. J., 2014, 20, 13078 CrossRef CAS PubMed.
- For highlights of the use of organolithium reagents in cross-coupling, see: (a) V. Pace and R. Luisi, ChemCatChem, 2014, 6, 1516 CrossRef CAS; (b) V. Capriati, F. M. Perna and A. Salomone, Dalton Trans., 2014, 43, 14204 RSC.
- S. E. Denmark, R. C. Smith, W. T Tau and J. M. Muhuhi, J. Am. Chem. Soc., 2009, 131, 3104 CrossRef CAS PubMed.
- G. C. Fu, Acc. Chem. Res., 2008, 41, 1555 CrossRef CAS PubMed.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed.
- C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef CAS PubMed.
- H. Rezaei, S. Yamanoi, F. Chemla and J. F. Normant, Org. Lett., 2010, 2, 419 CrossRef PubMed.
- The E–Z geometry of both the alkenyllithium reagents and alkenyl halides is fully preserved; the product geometry was unequivocally established by 1H NMR analysis and by comparison of the geometrical isomers with known compounds in the literature.
- J. F. Hartwig, Inorg. Chem., 2007, 46, 1936 CrossRef CAS PubMed.
- M. Seki and K. Mori, Eur. J. Org. Chem., 1999, 2965 CrossRef CAS.
- T. Jeffery-Luong and G. Linstrumelle, Synthesis, 1982, 738 CrossRef CAS.
- (a) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, vol. 1 and 2 Search PubMed; (b) For the use of allenyl metal reagents in cross-coupling reactions see: K. Radkowski, G. Seidel and A. Fürstner, Chem. Lett., 2011, 40, 950 CrossRef CAS and references cited therein.
- Unfortunately, mixtures of propargyl/1,2-allenyl species were observed when other allenyl lithium reagents or substrates were used (see ESI, Scheme S3†).
- (a) J. A. Soderquist and G. J.-H. Hsu, Organometallics, 1982, 1, 830 CrossRef CAS; (b) U. Lehmann, S. Awasthi and T. Minehan, Org. Lett., 2003, 5, 2405 CrossRef CAS PubMed; (c) S. E. Denmark and L. Neuville, Org. Lett., 2000, 2, 3221 CrossRef CAS PubMed.
- (a) M. Kosugi, T. Sumiya, Y. Obara, M. Suzuki, H. Sano and T. Migita, Bull. Chem. Soc. Jpn., 1987, 60, 767 CrossRef CAS; (b) H. B. Kwon, B. H. McKee and J. K. Stille, J. Org. Chem., 1990, 55, 3114 CrossRef CAS; (c) C. E. Russell and L. S. Hegedus, J. Am. Chem. Soc., 1983, 105, 6943 Search PubMed.
- H. Siegel and M. Eggersdorf, Ketones, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH, Germany, 2000, pp. 187–207 Search PubMed.
- S. D. Ramgren and N. K. Garg, Org. Lett., 2014, 16, 824 CrossRef CAS PubMed and references cited therein.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03117b |
| ‡ Representative procedure: In a dry Schlenk flask Pd2(dba)3 (2.5 mol%) and XPhos (10 mol%) were dissolved in toluene (2 mL/0.3 mmol of substrate) and the solution was stirred under nitrogen atmosphere at room temperature for 5 min. The substrate (1 equiv.) was added and the solution stirred at the indicated temperature. The corresponding lithium reagent solution (1.3 equiv., 0.6 or 0.68 M, see ESI† for details) was slowly added over 1 h by the use of a syringe pump. After the addition was completed a saturated solution of aqueous NH4Cl was added and the mixture was extracted with Et2O or AcOEt. The organic phases were combined and dried with anhydrous Na2SO4. Evaporation of the solvent under reduced pressure afforded the crude product that was then purified by column chromatography. |
| This journal is © The Royal Society of Chemistry 2015 |