
Gold(I)-catalyzed asymmetric [3 + 2]-cycloadditions of γ-1-ethoxyethoxy-propiolates and aldehydes†
Feng
Liu
a,
Yidong
Wang
a,
Weiming
Ye
a and
Junliang
Zhang
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Process, Department of Chemistry, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, China. E-mail: jlzhang@chem.ecnu.edu.cn; Web: http://faculty.ecnu.edu.cn/s/1811/main.jspy
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P.R. China
First published on 14th January 2015
Abstract
A gold-catalyzed asymmetric intermolecular [3 + 2]-cycloaddition of γ-1-ethoxyethoxy-propiolate and aldehydes by the use of the chiral phosphoramidite (S,R,R)-5a is developed, which provides facile access to highly substituted 2,5-dihydrofurans in up to 84% yield with up to 97% ee. Control experiments support the result that this transformation indeed proceeds via an all-carbon gold 1,3-dipole with an open carbocation rather than a SN2 type reaction.
The development of homogeneous gold catalyzed cycloadditions has been remarkably rapid in the past few years,1 due to their good tolerance to air and moisture and high capability to activate C–C π-bonds. The inherent linear binding mode keeps the chiral ligand of the gold complex away from the generated stereogenic center, resulting in the development of enantioselective gold catalysis,2 which still poses considerable difficulty, especially for the intermolecular cases,3 although some strategies have been developed to address this challenging issue.
Substituted 2,5-dihydrofurans represent a class of important structural scaffolds, which are not only frequently found in natural products and biologically active molecules but also found to be useful building blocks.4 In 2008, The group of L. Zhang developed an elegant method for the synthesis of substituted 2,5-dihydrofurans from readily available γ-1-ethoxyethoxy-propiolates and aldehydes or ketones via a gold-catalyzed intermolecular [3 + 2]-cycloaddition.5 However, the enantioselective variant of this intermolecular cycloaddition and the mechanism (1,3-dipolar cycloaddition vs. tandem SN2/cyclization) of this reaction have not been well addressed so far. Due to the continual interest in developing asymmetric gold-catalysis,6 we have developed herein an enantioselective gold(I)-catalyzed intermolecular [3 + 2]-cycloaddition by the application of chiral Binol-derived phosphoramidite as the ligand. The control experiments support the result that the reaction pathway of this reaction is through a 1,3-dipole rather than tandem SN2/cyclization.7
γ-1-Ethoxyethoxy-propiolate 1a and 4-methoxybenzaldehyde 2a were chosen as model substrates to test a series of chiral ligands. Given the success of chiral bisphosphines in gold-catalyzed enantioselective reactions, our initial efforts focused on the use of chiral bisphosphines such as chiral Bipheps as ligands (see ESI†), only the privileged ligand (R)-MeO-dtbm-biphep 6 was found to be the most efficient in enantioinduction to give 65% ee. We next turned to examine chiral phosphoramidites as ligands (Fig. 1), which can easily undergo substitution at many positions.8 We were pleased to find that a moderate degree of enantioinduction (68% ee) could be obtained with the Binol-derived phosphoramidite (R,R,R)-4a as the ligand (Table 1, entry 1). With this encouraging result, other modified chiral phosphoramidites by introduction of various substituents at 3 and 3′-positions of the BINOL moieties were then prepared and investigated (Table 1, entries 3–6). 85% ee was observed by the use of (R,R,R)-4c with a 3,3′-phenyl group as the ligand albeit with only 33% yield (Table 1, entry 4), while the corresponding (S)-BINOL derived (S,R,R)-4c induced a slightly lower enantioselectivity (77% ee, Table 1, entry 7). Attempts to improve the enantioselectivity failed by introducing other substituents such as methyl (4b), bulky Ph3Si (4d) and 3,5-bis-(trifluoro-methyl)phenyl (4e) at 3 and 3′-positions of the chiral ligand. The spirobiindane derived phosphoramidite (S)-SIPHOS-PE could not give high ee either (73% ee, Table 1, entry 8) and even its diastereomer (R)-SIPHOS-PE gives lower ee (43% ee, Table 1, entry 9), which indicates that not only the chirality of the axial backbone but also the stereogenic center affects the enantioselectivity. The knowledge gained from our previous work6a that the bite angle also plays a crucial role in gold-catalyzed asymmetrical reaction encouraged us to test the octahydrobinol-derived phosphoramidites 5a and 5b with different bite angles (Table 1, entries 10–12). After some attempts, the methyl substituted (R,R,R)-5b resulted in a substantially higher enantioselectivity than the corresponding 4b (82% ee vs. 59% ee, Table 1, entries 11 vs. 3). To our delight, (S,R,R)-5a delivered excellent enantioselectivity (90% ee, Table 1, entry 12). Notably, (S,R,R)-5a and (R,R,R)-5a derived gold complexes result in different enantiomers of 2,5-dihydrofurans, indicating that the axial chirality dictates the absolute stereochemistry of the [3 + 2] cycloadduct. With the use of (S,R,R)-5a as a chiral ligand, a series of silver salts such as AgOTf, AgSbF6, AgBF4, AgO2C4F7 and NaBAr4 (Ar = 3,5-CF3C6H3) were then examined, among which AgNTf2 afforded the best result. There is a slight improvement in the enantioselectivity when the reaction is run at 0 °C. The addition of 3 Å MS led to a remarkable improvement in the yield. When 4 equiv. of anisaldehyde 2a were used, the desired product 3aa was isolated in 78% yield with 92% ee (entry 18). These results clearly validated the strategy and suggested that further improvement might be feasible not only by introducing substituents at 3 and 3′-positions but also through the modification of the bite angle. Other γ-1-ethoxyethoxy-propiolate esters such as methyl (1b), ethyl (1c) and tert-butyl (1d) are applicable to the reaction conditions except for the benzyl ester (1e), leading to a similar level of enantioselectivity (Table 1, entries 19–22).
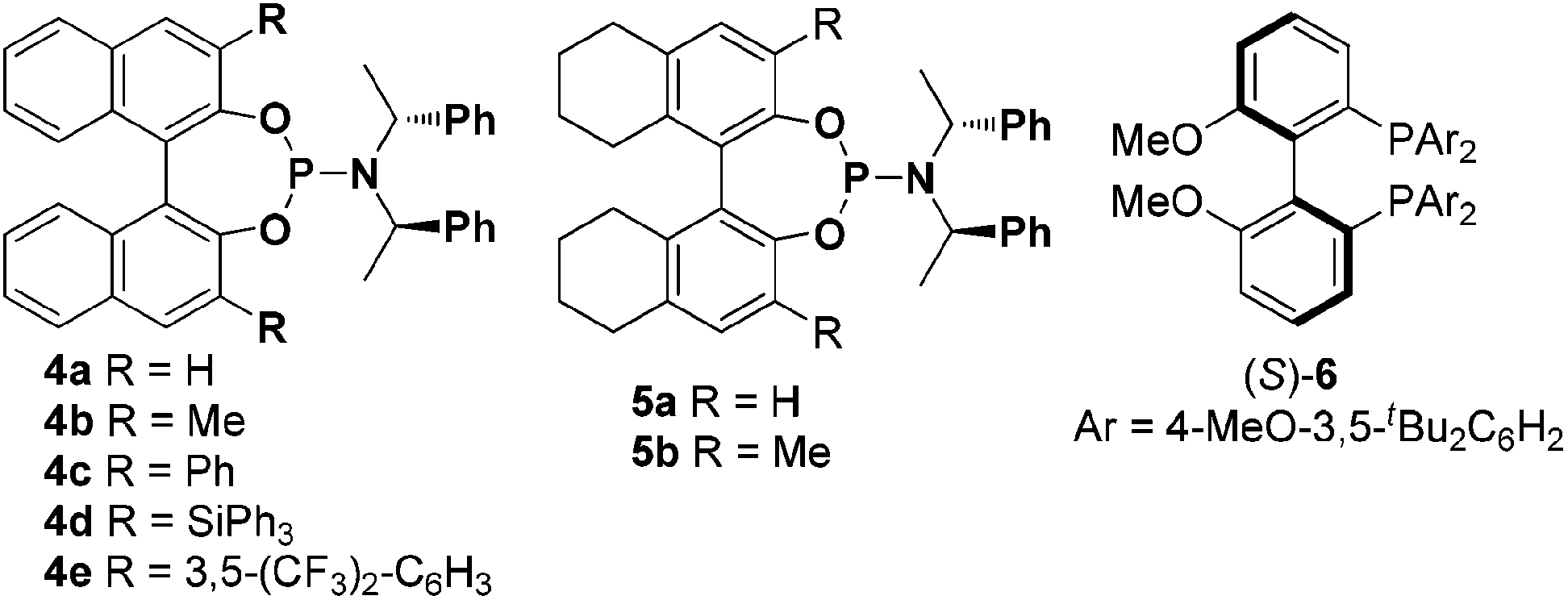 | ||
| Fig. 1 Screened chiral ligands. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 R | Ligand | Solvent | Product | |
| 3 (yield %) | ee% | ||||
a Unless otherwise noted, substrate 1 (0.4 mmol), 4-methoxybenzaldehyde 2a (2 equiv.) and LAuNTf2 (5 mol%, LAuCl/AgX = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were used in 8 mL of solvent at 25 °C for 1 h. Isolated yields are shown and the enantiomeric excess is determined by chiral HPLC.
b The reaction is performed at 0 °C.
c 3 Å MS (150 mg) was added.
d 4 equiv. of 2a were used. :1) were used in 8 mL of solvent at 25 °C for 1 h. Isolated yields are shown and the enantiomeric excess is determined by chiral HPLC.
b The reaction is performed at 0 °C.
c 3 Å MS (150 mg) was added.
d 4 equiv. of 2a were used.
|
|||||
| 1 | i Pr(1a) | (R,R,R)-4a | CH2Cl2 | 3aa (46) | −66 |
| 2 | 1a | (S,R,R)-4a | CH2Cl2 | 3aa (48) | 68 |
| 3 | 1a | (S,R,R)-4b | CH2Cl2 | 3aa (30) | 59 |
| 4 | 1a | (S,R,R)-4c | CH2Cl2 | 3aa (33) | 85 |
| 5 | 1a | (R,R,R)-4d | CH2Cl2 | 3aa (35) | −66 |
| 6 | 1a | (R,R,R)-4e | CH2Cl2 | 3aa (23) | −77 |
| 7 | 1a | (R,R,R)-4c | CH2Cl2 | 3aa (56) | −77 |
| 8 | 1a | (S)-SIPHOS-PE | CH2Cl2 | 3aa (54) | 73 |
| 9 | 1a | (R)-SIPHOS-PE | CH2Cl2 | 3aa (55) | −43 |
| 10 | 1a | (R,R,R)-5a | CH2Cl2 | 3aa (46) | −69 |
| 11 | 1a | (R,R,R)-5b | CH2Cl2 | 3aa (60) | −82 |
| 12 | 1a | (S,R,R)-5a | CH2Cl2 | 3aa (51) | 90 |
| 13 | 1a | (S,R,R)-5a | DCE | 3aa (48) | 87 |
| 14 | 1a | (S,R,R)-5a | Toluene | 3aa (24) | 84 |
| 15 | 1a | (S,R,R)-5a | CHCl3 | 3aa (45) | 90 |
| 16b | 1a | (S,R,R)-5a | CH2Cl2 | 3aa (50) | 92 |
| 17b,c | 1a | (S,R,R)-5a | CH2Cl2 | 3aa (73) | 93 |
| 18b,c,d | 1a | (S,R,R)-5a | CH2Cl2 | 3aa (78) | 92 |
| 19 | Me (1b) | (S,R,R)-5a | CH2Cl2 | 3ba (53) | 87 |
| 20 | Et (1c) | (S,R,R)-5a | CH2Cl2 | 3ca (61) | 89 |
| 21 | t Bu (1d) | (S,R,R)-5a | CH2Cl2 | 3da (64) | 83 |
| 22 | Bn (1e) | (S,R,R)-5a | CH2Cl2 | 3ea (47) | 77 |
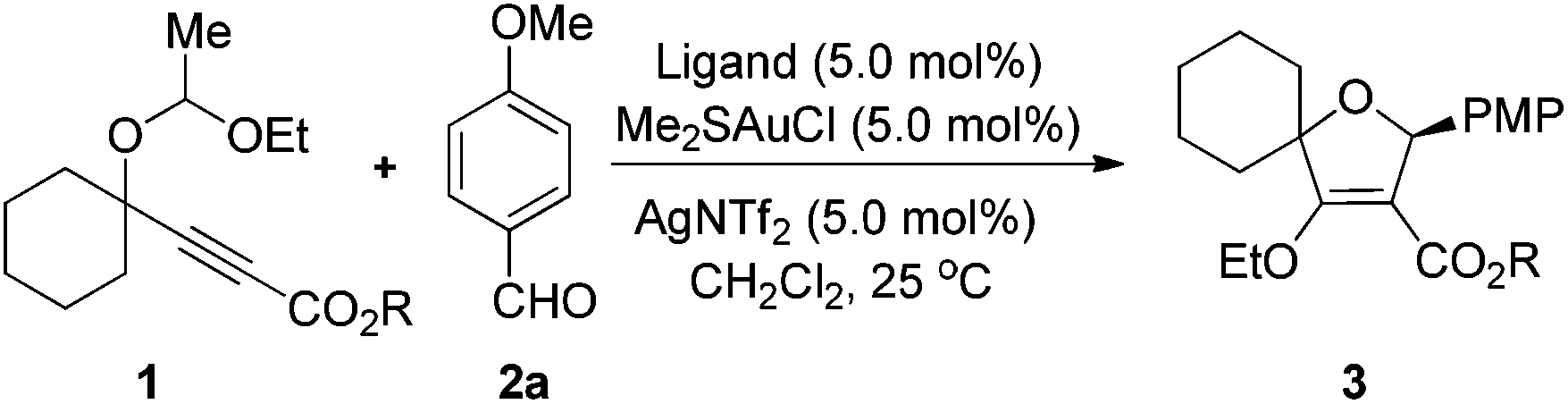
With the optimized conditions in hand, we explored the substrate scope of the reaction by variation of two reaction components (Table 2). In general, para-substituted aryl aldehydes gave better ees than ortho-substituted ones. For example, the reaction of para-methoxybenzaldehyde 2a with 1a gave the product 3aa with 93% ee, while only 77% ee of 3ad was obtained from the corresponding ortho-methoxy benzaldehyde 2d. 3ae with 84% ee could be obtained in 78% yield from the corresponding 2,4-bismethoxybenzaldehydes 2e and 1a. The highest selectivity was observed in the case of p-Me2N–C6H4CHO as the substrate, where 3ab and 3fb were isolated in 97% ee. Gratifyingly, cinnamaldehyde is also compatible with this transformation to produce the corresponding products 3ac and 3fc in good yields with reasonable enantioselectivities. Heterocyclic 1-H-pyrrole-2-carbaldehyde also enables the highly enantioselective synthesis of pyrrol-2-yl-substituted dihydrofurans 3af and 3ff. Acyclic 1f and cyclic 1h with a five-membered cyclopentane ring are applicable to this transformation very well. However, substrate 1g with a seven-membered cycle only affords the product 3ga in moderate yield and enantioselectivity, indicating that the ring size also affects the enantioselectivity.
 | (1) |
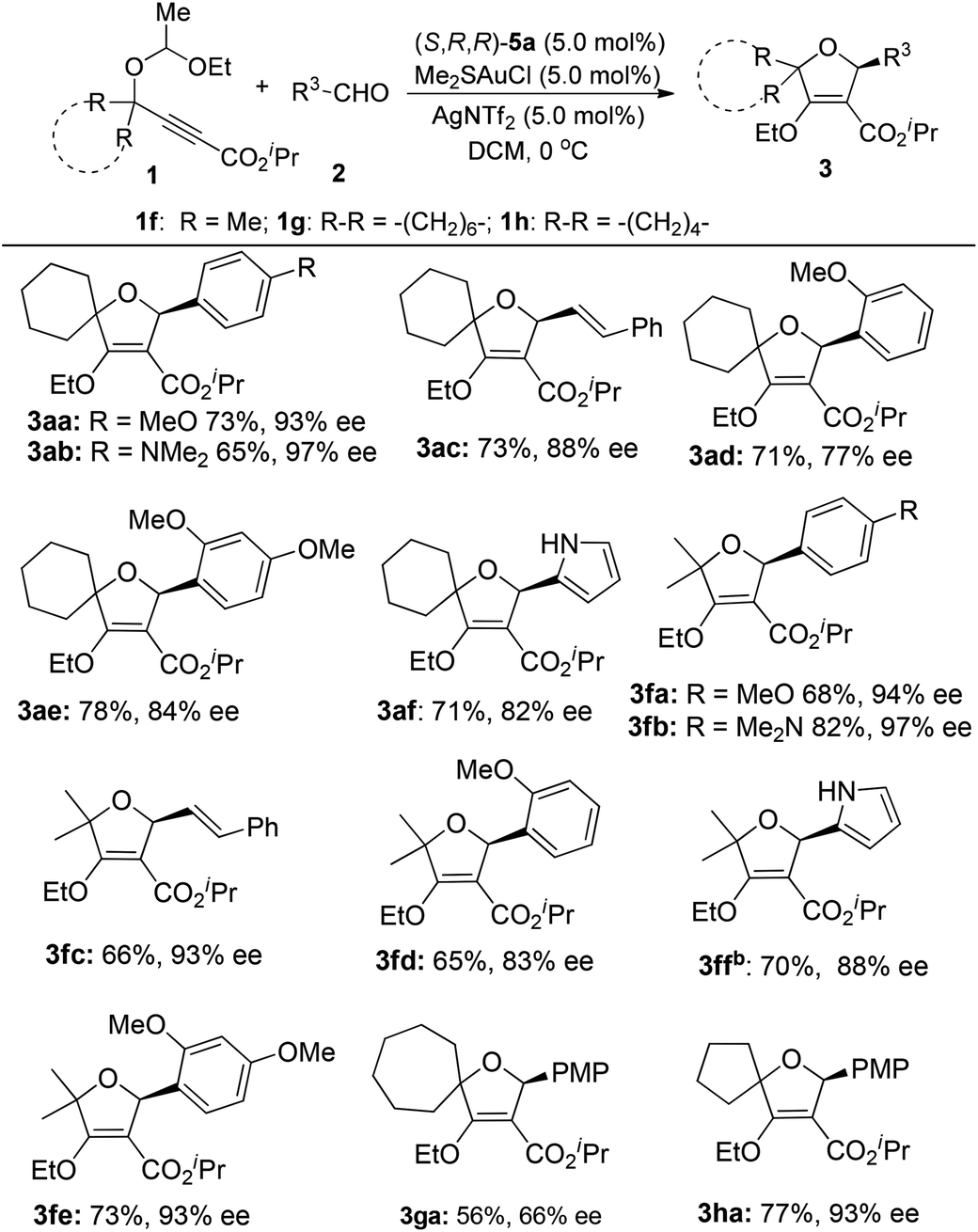
When we tried to extend the scope of aldehydes to indole-3-carbaldehydes, it was found that the present reaction conditions using (S,R,R)-5a as the chiral ligand were not applicable any more (eqn (1)). A significant erosion in the enantiomeric excess was observed (70% ee). This result indicated that the enantioselectivity of this reaction is kind of substrate dependent. After further investigation, we were pleased to find that this issue can be well addressed by the use of (R,R,R)-5b as the chiral ligand, resulting in the product 3ag as the opposite enantioisomer in 62% yield with 92% ee. Inspired by this result, the reactions of various indole-3-carbaldehydes with 1a and 1f were then examined under the catalysis of the (R,R,R)-5b/gold complex (Table 3). The reaction of 1a with N-allyl indole-3-carbaldehyde 2h afforded the cycloadduct 3ah in 70% yield with 87% ee. Similarly, the reaction of 1a with 5-methoxy indole-3-carbaldehydes 2i and 2j also worked well to provide products 3ai and 3aj in reasonable yields with 90% and 92% ee, respectively.
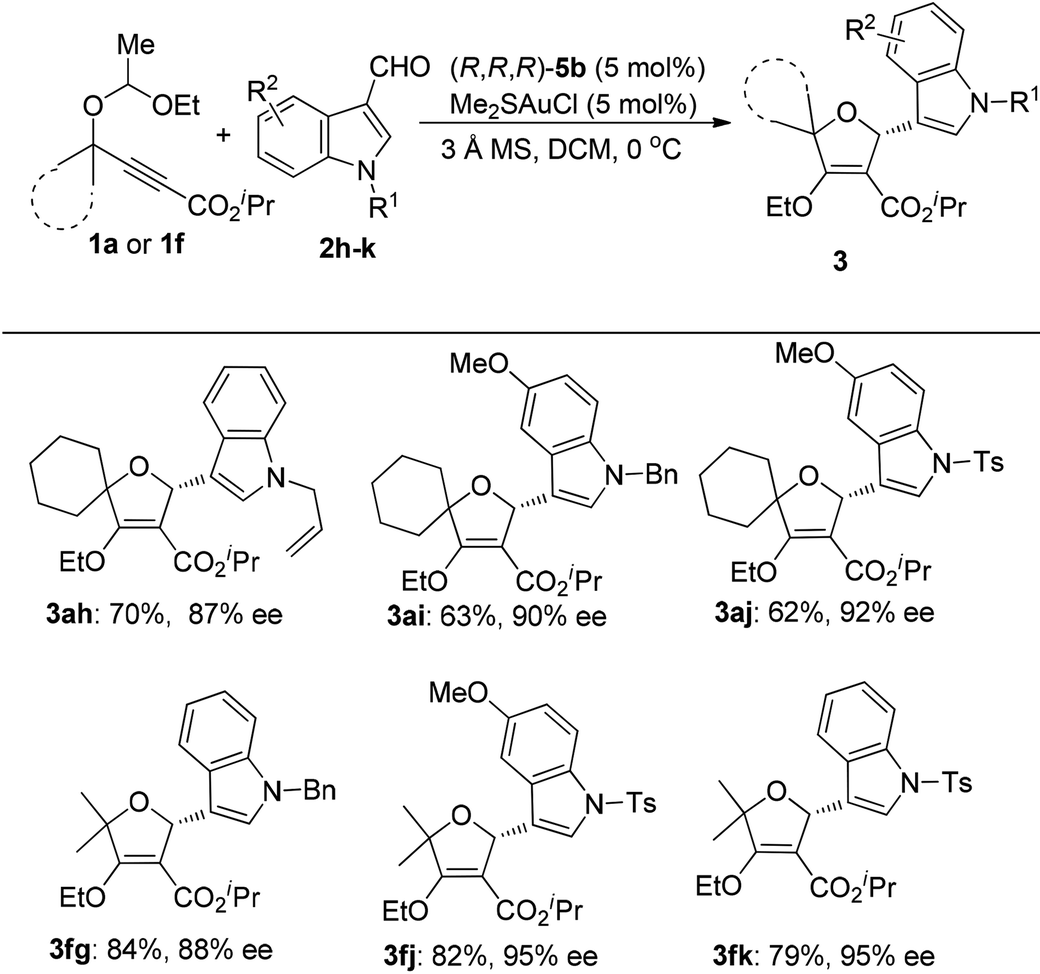
|
This catalyst system could also be well applied to the reactions of acyclic 1f and indole-3-carbaldehydes, delivering the desired products 3fg, 3fj and 3fk in high yields with 88–95% ees. The absolute configuration of the products was confirmed by single-crystal X-ray diffraction analysis of compound 3fk.9 The absolute chemistry of other compounds was rationalized from compound 3fk. The X-ray crystal structure of (R,R,R)-5bAuCl was also obtained and is shown in ESI.†
In order to gain insight into the reaction mechanism, racemic and enantioenriched secondary alcohol derived substrates were subjected to the reaction conditions. Notably, the reaction of racemic 1i with 2a gave the desired -2,5-dihydrofuran 3ia with two stereocenters in 44% yield with only 16% ee and 6.0:1 dr under the catalysis of the (S,R,R)-5a derived gold(I) complex, however, the diastereo- and enantioselectivity could be dramatically improved when (S)-MeO-dtbm-biphep ((S)-6) was employed as the chiral ligand. The yield would be improved to 64% with 5 equivalents of an aldehyde (Scheme 1).
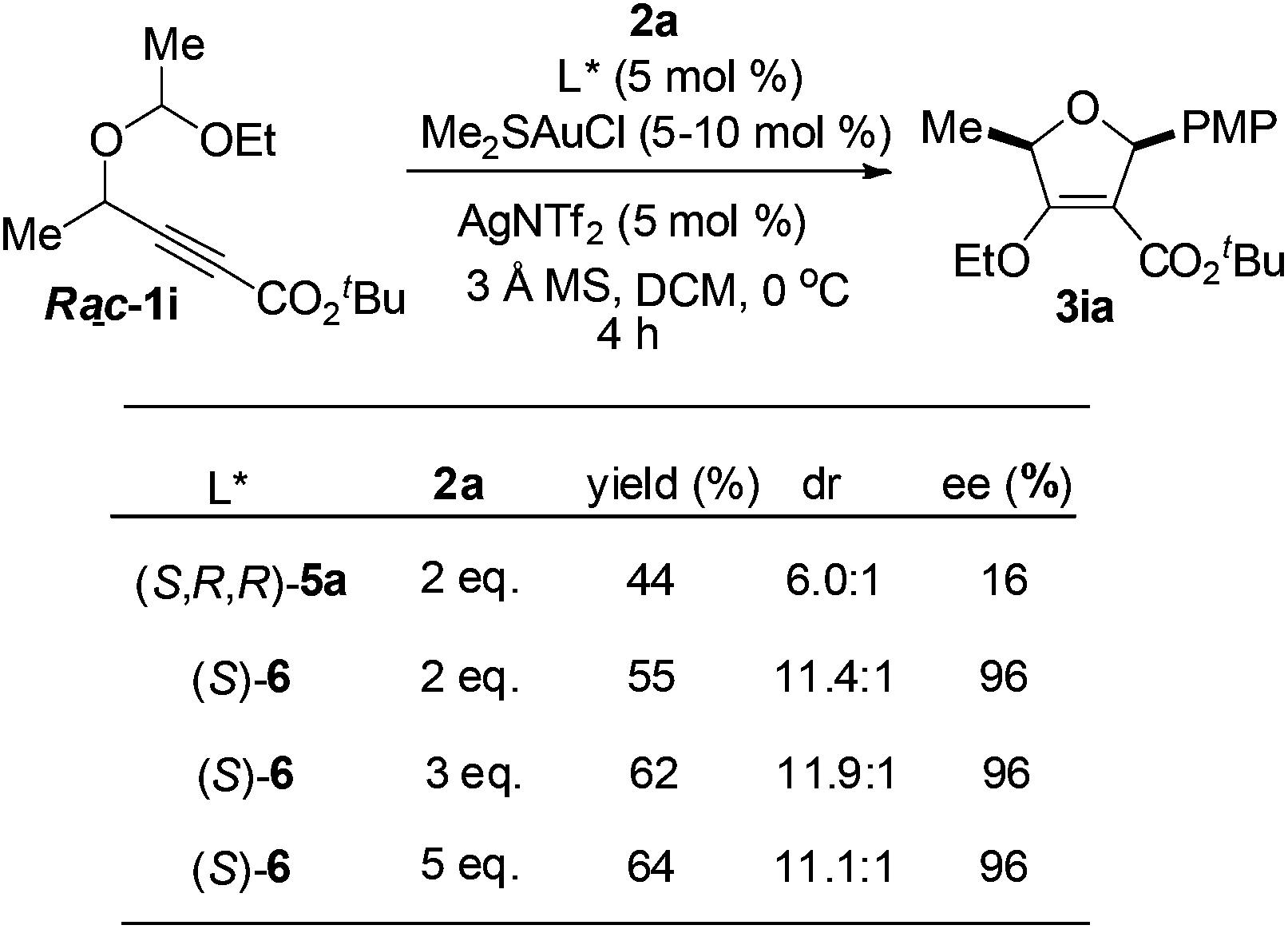 | ||
| Scheme 1 Asymmetric cycloaddition of (±)-1i with 2a. | ||
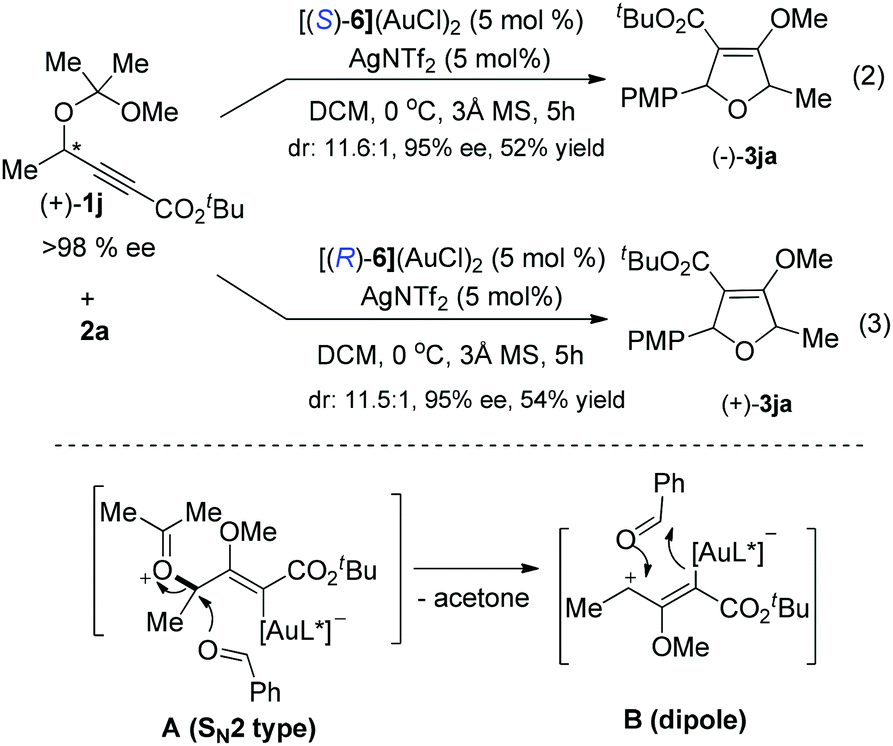
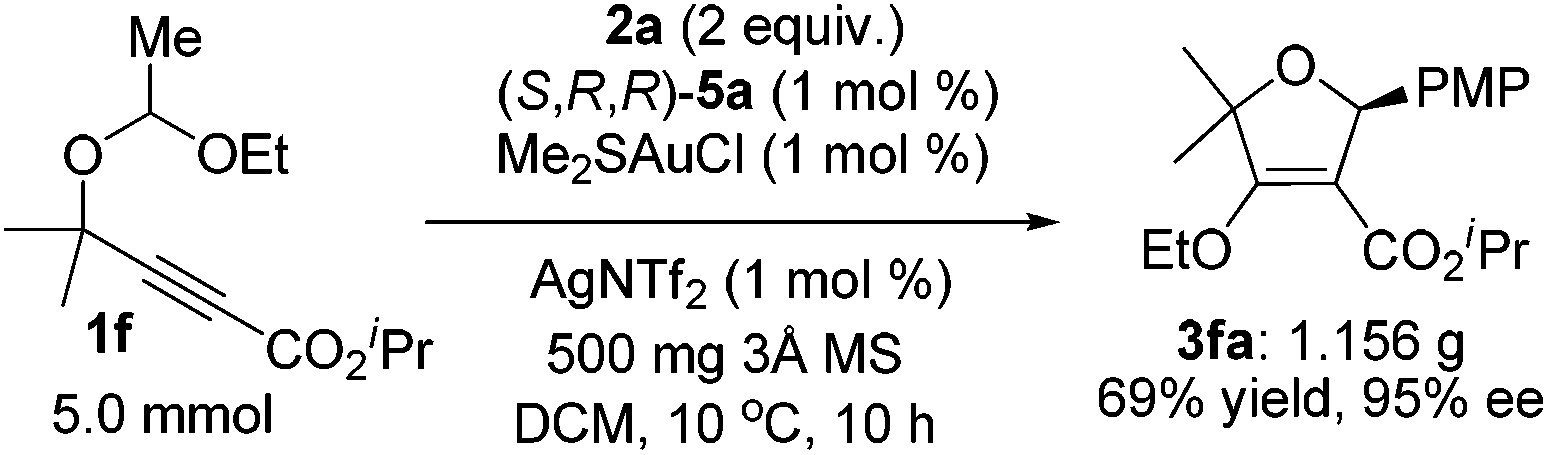 | (4) |
Furthermore, this asymmetric gold(I)-catalyzed reaction is scalable up to the gram-scale without any loss of selectivity and efficiency. The catalyst loading could be reduced to only 1 mol% on a 5 mmol scale to furnish 1.156 g of 3fa in 69% yield with 95% ee at 10 °C for 10 h (eqn (4)), indicating that this transformation has a potential practical use in organic synthesis.
A catalytic cycle is proposed as depicted in Scheme 2. The reaction is believed to be initiated by the anti-attack on a gold-activated C–C multiple bond (a π-complex) by the intramolecular oxygen atom of the acetal moiety, furnishing intermediate Int-A. Then a 1,3-dipole intermediate Int-B would be produced through the elimination of one molecule of acetaldehyde. Next, the aryl aldehyde attacks the open carbocation to form the intermediate Int-C. Finally, the product is delivered through cyclization and elimination of the catalytic gold species. The observed absolute stereochemistry can be explained by the proposed induction models shown. The transition state Ts-A is more favorably formed than Ts-B to preferentially give the desired product with observed stereochemistry, because of the steric repulsion between the Ar group and the methyl group of the chiral catalyst.
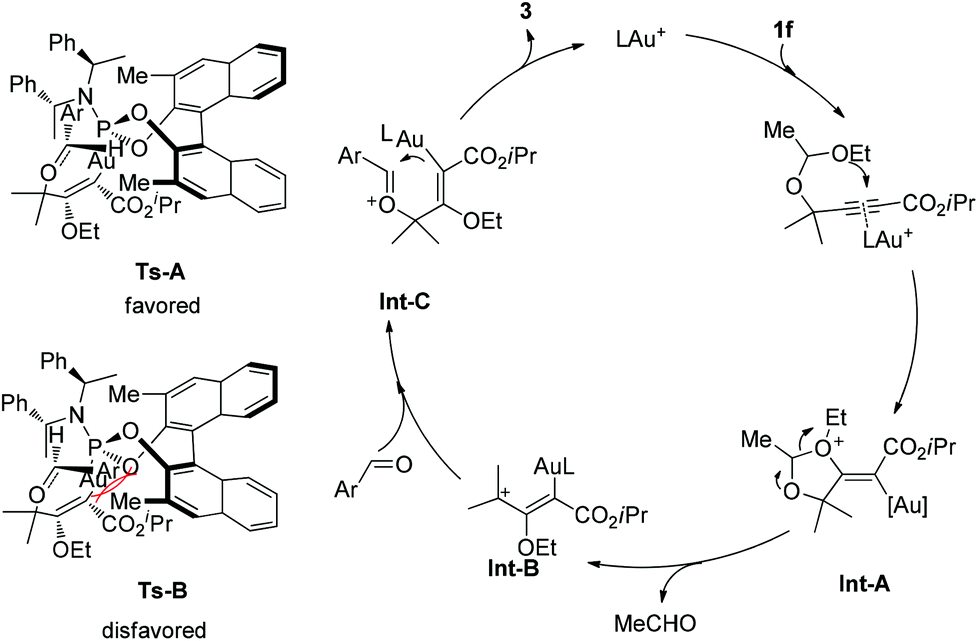 | ||
| Scheme 2 Possible mechanistic pathway and an induction model. | ||
Conclusions
In summary, we have described a highly enantioselective gold-catalyzed [3 + 2] cycloaddition of in situ generated all-carbon gold-1,3-dipoles with aldehydes under mild conditions. The method has enabled the enantioselective synthesis of a wide variety of functionalized tri- and tetra-substituted 2,5-dihydrofurans in moderate to high yields with good diastereoselectivities and enantioselectivities. A broad array of substrates is applied to this transformation by careful choice of octahydrobinol-derived phosphoramidite or MeO-dtbm-biphep derived gold(I) complexes as catalysts. The control experiments support that this transformation indeed proceeds via an all-carbon gold 1,3-dipole with an open carbocation rather than a SN2 type reaction. The easy scale-up character also adds value to this transformation to make it a potential practically useful method. Further studies including synthetic applications and extension of the scope of dipolarophiles are currently underway and will be reported in due course.Notes and references
- For reviews on gold-catalyzed cycloadditions, see: (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (b) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (c) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed; (d) S. M. Abu Sohel and R.-S. Liu, Chem. Soc. Rev., 2009, 38, 2269 RSC; (e) A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; (f) A. Corma, A. Leyva-Perez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (g) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef CAS PubMed; (h) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (i) M. Bandini, Chem. Soc. Rev., 2011, 40, 1358 RSC; (j) B.-L. Lu, L. Dai and M. Shi, Chem. Soc. Rev., 2012, 41, 3318 RSC; (k) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS; (l) Y.-W. Sun, Q. Xu and M. Shi, Beilstein J. Org. Chem., 2013, 9, 2224 CrossRef PubMed; (m) F. López and J. L. Mascareñas, Beilstein J. Org. Chem., 2013, 9, 2250 CrossRef PubMed; (n) G. Cera and M. Bandini, Isr. J. Chem., 2013, 53, 848 CrossRef CAS; (o) D.-F. Chen, Z.-Y. Han, X.-L. Zhou and L.-Z. Gong, Acc. Chem. Res., 2014, 47, 2365 CrossRef CAS PubMed; (p) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS PubMed; (q) C. Obradors and A. M. Echavarren, Acc. Chem. Res., 2014, 47, 902 CrossRef CAS PubMed; (r) L. Fensterbank and M. Malacria, Acc. Chem. Res., 2014, 47, 953 CrossRef CAS PubMed.
- For recent reviews or accounts on enantioselective gold-catalysis, see: (a) Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889 CrossRef CAS PubMed; (b) M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2011, 47, 6536 RSC; (c) A.-L. Lee, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2011, 107, 369 RSC; (d) A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657 CrossRef CAS PubMed; (e) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994 CrossRef CAS PubMed; (f) M. Rudolph and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC; (g) A. Marinetti, H. Jullien and A. Voituriez, Chem. Soc. Rev., 2012, 41, 4884 RSC; (h) S. Sengupta and X. Shi, ChemCatChem, 2010, 2, 609 CrossRef CAS; (i) C. C. J. Loh and D. Enders, Chem. – Eur. J., 2012, 18, 10212 CrossRef CAS PubMed; (j) D. Garayalde and C. Nevado, ACS Catal., 2012, 2, 1462 CrossRef CAS.
- For recent examples of gold-catalyzed asymmetric intermolecular cycloadditions, please see: (a) Z.-M. Zhang, P. Chen, W. Li, Y.-F. Niu, X.-L. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 4350 CrossRef CAS PubMed; (b) J. F. Briones and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 13314 CrossRef CAS PubMed; (c) B. Liu, K.-N. Li, S.-W. Luo, J.-Z. Pang and L.-Z. Gong, J. Am. Chem. Soc., 2013, 135, 3323 CrossRef CAS PubMed; (d) G.-H. Li, W. Zhou, X.-X. Li, Q.-W. Bi, Z. Wang, Z.-G. Zhao, W.-X. Hu and Z. Chen, Chem. Commun., 2013, 49, 4770 RSC; (e) S. Suárez-Pantiga, C. Hernández-Díaz, E. Rubio and J. M. González, Angew. Chem., Int. Ed., 2012, 51, 11552 CrossRef PubMed; (f) S. A. Gawade, B. S. Hunia and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 7835 CrossRef CAS PubMed; (g) K. L. Butler, M. Tragni and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2012, 51, 5175 CrossRef CAS PubMed; (h) S. Padilla, J. Adrio and J. C. Carretero, J. Org. Chem., 2012, 77, 4161 CrossRef CAS PubMed; (i) J. F. Briones and H. W. L. Davies, J. Am. Chem. Soc., 2012, 134, 11916 CrossRef CAS PubMed; (j) S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912 CrossRef CAS PubMed.
- (a) M. Brichacek, L. A. Batory, N. A. McGrath and J. T. Njardarson, Tetrahedron, 2010, 66, 4832 CrossRef CAS; (b) A. Karadeolian and M. A. Kerr, Angew. Chem., Int. Ed., 2010, 49, 1133 CrossRef CAS PubMed; (c) J. Lee and J. S. Panek, Org. Lett., 2011, 13, 502 CrossRef CAS PubMed.
- L. Zhang, J. Am. Chem. Soc., 2008, 130, 12598 CrossRef PubMed.
- (a) F. Liu, D. Qian, L. Li, X. Zhao and J. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6669 CrossRef CAS PubMed; (b) G. Zhou, F. Liu and J. Zhang, Chem. – Eur. J., 2011, 17, 3101 CrossRef CAS PubMed.
- For an example of tandem SN2 and cyclization, see: Y. Zhang and J. Zhang, Chem. Commun., 2012, 48, 4710 RSC.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486 CrossRef CAS PubMed.
- CCDC 926425 (3fk) and 926426 ((R,R,R)-5bAuCl).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 926425 and 926426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00261j |
| This journal is © the Partner Organisations 2015 |