Design of a new series of chiral phosphite–olefin ligands and their application in asymmetric catalysis†
Yue-Na
Yu
and
Ming-Hua
Xu
*
Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, 50807388, Shanghai 201203, China. E-mail: xumh@simm.ac.cn; Fax: +86 21-50807388
First published on 5th June 2014
Abstract
A novel type of open-chain chiral chelating olefin bearing a simple monophosphite unit with a binaphthyl backbone was designed and synthesized, which could serve as an elegant chiral bidentate ligand in the rhodium-catalyzed asymmetric 1,2-additions of organoboron reagents to α-diketones and N-sulfonyl aldimines.
Over the past few decades, transition-metal-catalyzed asymmetric transformations have proved to be very powerful strategies to access various enantioenriched compounds from achiral substrates.1 Consequently, development of effective chiral ligands for asymmetric catalysis became one of the most important and challenging goals of research in organic synthesis.2 In the past ten years, the design and use of chiral olefins as steering ligands has emerged as a fascinating new field.3 A diverse range of chelating olefins including hybrid ones have been introduced for enantioselective asymmetric reactions.3,4–7 Among these, phosphorus-based olefins represent a particularly interesting class of ligands since the first demonstration by Grützmacher,4 as they combine the strongly coordinating phosphorus atom and the weakly coordinating olefin into one ligand molecule. They have been utilized in transition-metal-catalyzed asymmetric hydrogenations, conjugate additions, allylic alkylations, aminations, and intramolecular hydroacylations.4–7 Notably, Hayashi's chiral phosphine–olefin (2)5 and Carreira's phosphoramidite–olefin (4)6 showed the great potential of this ligand class in asymmetric catalysis. However, in view of the currently reported phosphorus–olefins in the literature,4–7 they generally possess a complicated scaffold with backbone chirality (Fig. 1), which in many cases is not synthetically easily accessible. Moreover, most ligands suffered from unsatisfactory catalytic performances.1,7b,d–i Thus, the design and development of simple and efficient chiral phosphorus-based olefins for enantioselective catalysis remain highly desirable. Herein, we describe our discovery of a new series of chiral phosphite–olefin ligands that can effectively promote asymmetric 1,2-addition under rhodium catalysis.
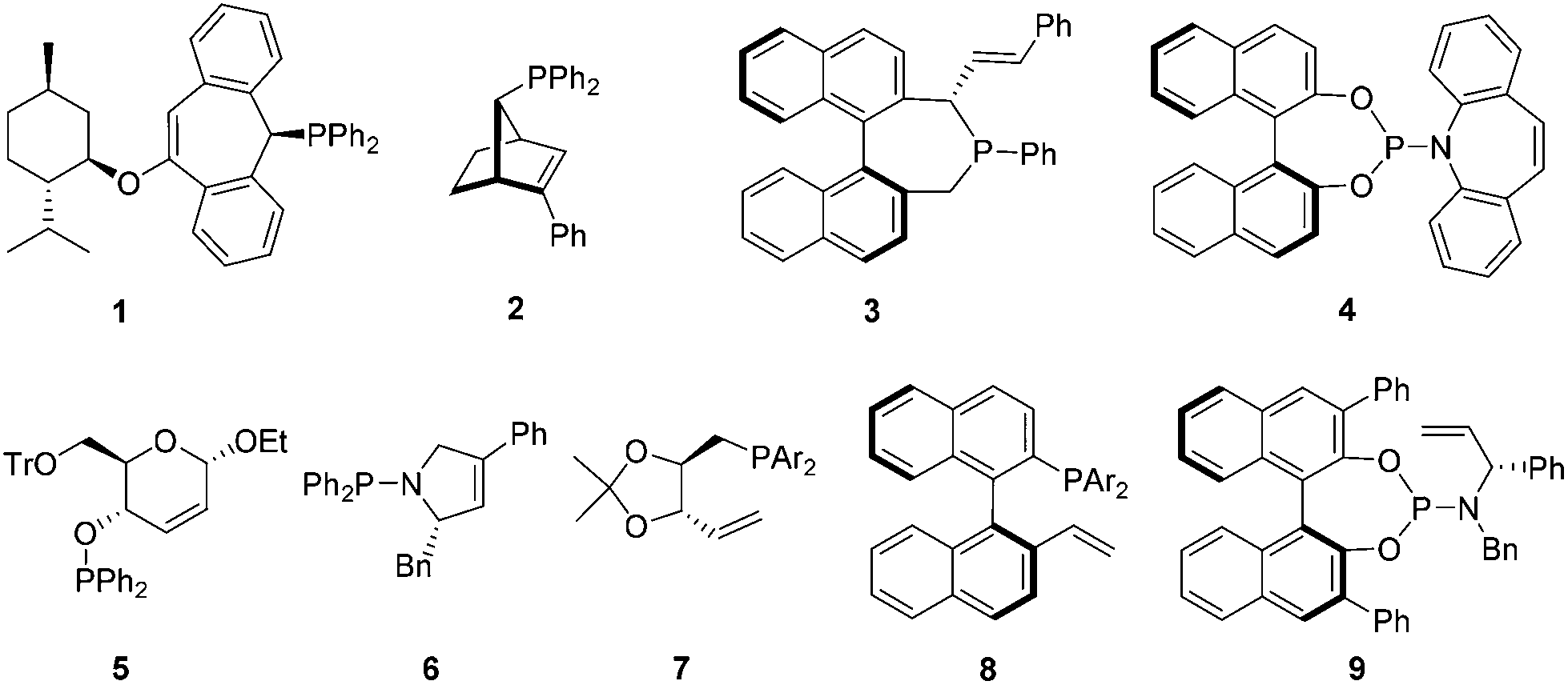 | ||
| Fig. 1 Representative chiral phosphorus–olefin hybrid ligands. | ||
Recently we disclosed our successful design of a new exciting class of chiral sulfur-based olefin ligands and their application in a series of rhodium-catalyzed asymmetric transformations.8 Unlike most related olefin ligands possessing sophisticated chiral cyclic scaffolds, these sulfur–olefins are easily prepared by taking advantage of stereogenic sulfonamide/sulfoxide as a single chiral directing group. With this innovative design principle in mind, we further conceived the idea of designing a new series of chiral olefin ligands consisting of a non-cyclic alkene framework bearing an appropriate chiral phosphorus-containing unit (Fig. 2). Chiral monophosphites which can be synthesized with relative ease have been known to be one class of “privileged ligands” in asymmetric catalysis.9 However, the use of a monophosphite in the chiral olefin hybrid ligand remains unprecedented. We therefore envisioned the possibility of incorporating an axially chiral monophosphite unit, which should provide a chiral environment suitable for chiral induction.
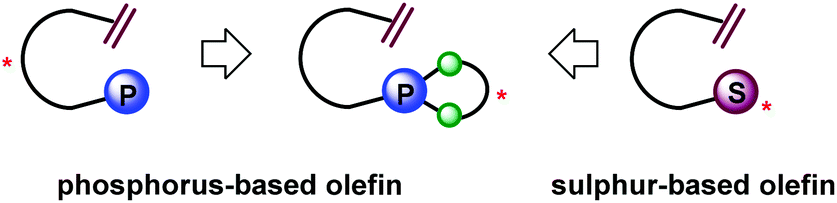 | ||
| Fig. 2 Open-chain P–olefin design without main backbone chirality. | ||
In order to realize this idea of open-chain chiral phosphite–olefin, we began our investigation by constructing the ligand molecule by attaching both a phosphite unit and olefin groups into a benzene ring in a 1,2-fashion. The axially chiral unit is a configurationly stable binaphthyl derivative. Initially, compound L1 was prepared using cheap salicylaldehyde as the starting material, which underwent a Horner–Wadsworth–Emmons reaction with readily available benzyl phosphonate, followed by a straightforward two-step assembly of chiral monophosphite (Scheme 1). Crystallographic analysis of L1 unequivocally confirmed the compound structure.10
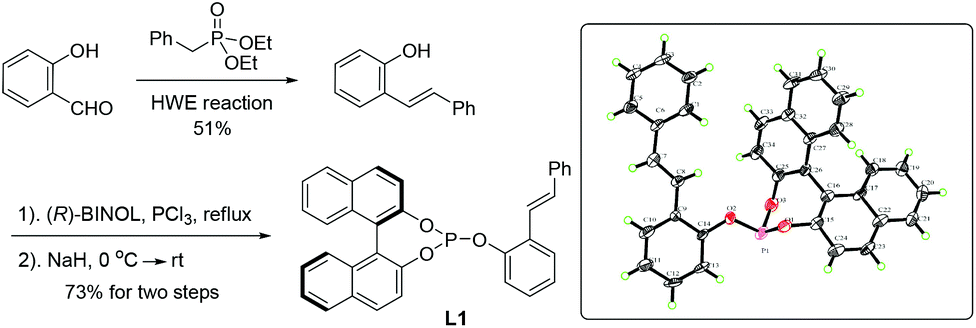 | ||
| Scheme 1 Synthesis of the phosphite–olefin ligand. | ||
To examine the catalytic potential of the designed ligand, we chose to explore Rh-catalyzed 1,2-addition of aryl boronic acids to α-diketone substrates in an attempt to generate optically active tertiary α-hydroxyketone derivatives.11 As shown in Table 1, the reaction is carried out in the presence of 1.5 mol% of [Rh(coe)2Cl]2 and 3 mol% of L1 under aqueous K3PO4–THF at room temperature for 12 h. To our delight, the transformation proceeded smoothly and gave the corresponding product in moderate yield (70%) with an excellent enantioselectivity (93% ee). To ensure that the double bond is involved closely in coordination with rhodium in the catalytic process, we designed compounds L2 and L3 which lack the olefin donor for control experiments. Indeed, complexes of L2 and L3 with [Rh(coe)2Cl]2 exhibited completely different catalytic properties as compared to L1 and led to the product with a contrary absolute configuration in low yield and ee. These results clearly indicated that it is necessary for the olefin donor to be present on the ligand. Encouraged by the promising result, a series of structurally diverse phosphite–olefin ligands (L4–L9) was accordingly prepared and evaluated in the same rhodium-catalyzed 1,2-addition (Table 1). Interestingly, the use of ligands L1, L4, and L5, which contain a different aromatic moiety attached to the double bond, gave the same level of enantioselectivity (91–93% ee); ligand L6 bearing a meta-methyl to the olefin group showed slightly decreased catalytic reactivity and selectivity. Further ligand architecture modification was carried out by introducing two phenyl substituents onto the 3- and 3′-positions of the binaphthyl unit. Unexpectedly, sterically more hindered ligand L7 performed equally to L1. Besides, ligands L8–9 bearing a terminal olefin led to the product 11a with a significant loss in both yield and enantioselectivity. The performances of L8 and L9 clearly indicate that substituents on the olefin are essential for obtaining high catalytic activity and stereoselectivity. Taking into account the catalytic performance and ease of synthesis, the simplest phosphite–olefin L1 was chosen as the ideal ligand for further intensive study.
|
|
|---|

|
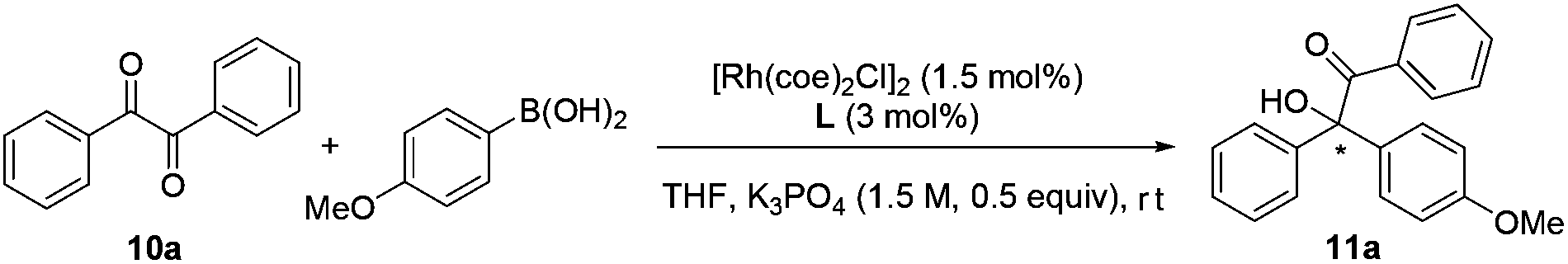
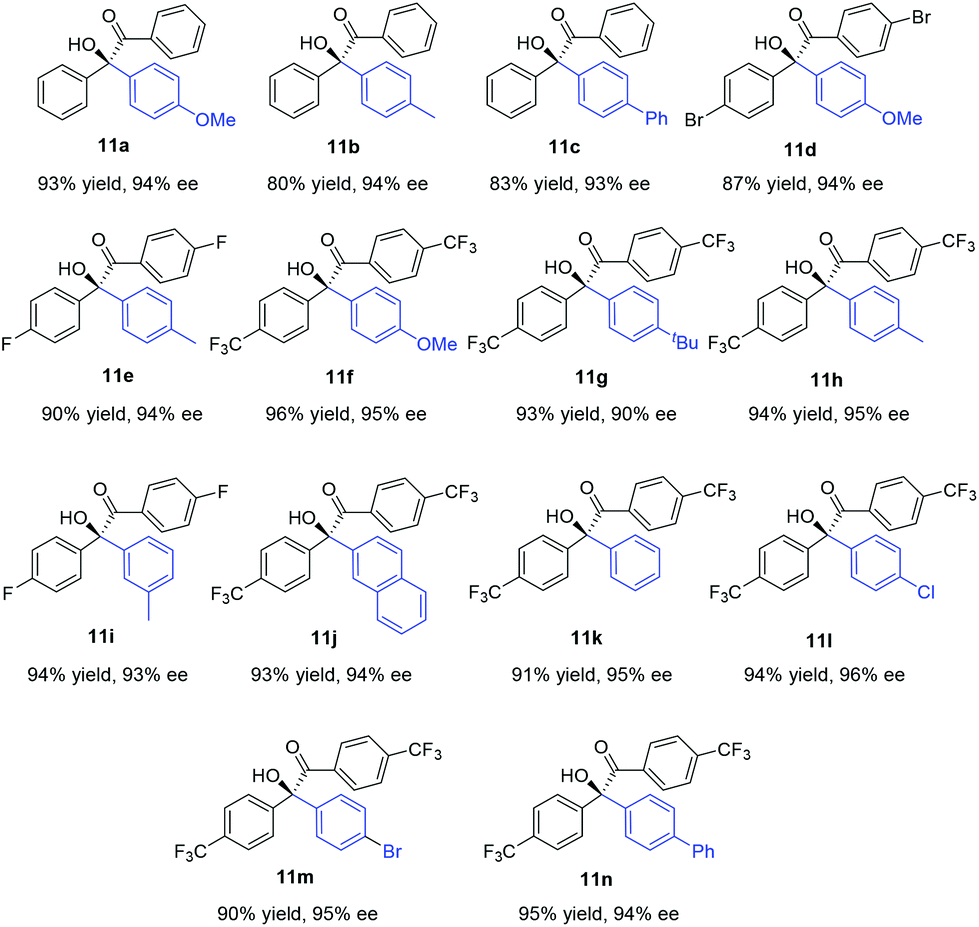
Under these optimal conditions, we turned our attention to investigate the reaction substrate scope using L1 as a chiral ligand. As summarized in Table 2, a wide range of arylboronic acids with varying electronic and steric demands were successfully examined with α-diketones (10). Gratifyingly, all transformations proceeded efficiently to give the desired products in good yields and excellent selectivities (90–96% ee). The electronic properties of the arylboronic acids did not appear to affect the reactivity and enantioselectivity of the reaction (11f–n). Sterically hindered arylboronic acids could also be successfully employed (11i and 11j). Since aromatic compounds bearing trifluoromethyl play a more and more important role in pharmaceutical science,12 investigation of CF3-substituted benzil substrate was conducted. Notably, a variety of enantiomerically enriched trifluoromethyl-containing α-hydroxyketones (11f–n) were readily obtained, which may provide new opportunities for drug discovery and development.
|
|
|---|
| a The reaction was carried out with α-diketone (0.25 mmol) and 2 equiv. of arylboronic acid, in the presence of 1.5 mol% of [Rh(coe)2Cl]2, 3 mol% of L1, and 2.5 M aq. K3PO4 (0.83 equiv.) in 1.0 mL of toluene for 12 h. b Yield of the isolated product. c Determined by chiral HPLC analysis. d The absolute configuration of 11a was determined by comparing the HPLC chromatogram with known data.8d |
|
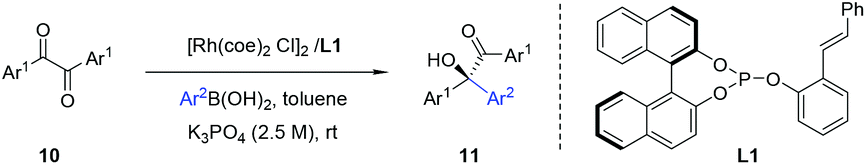
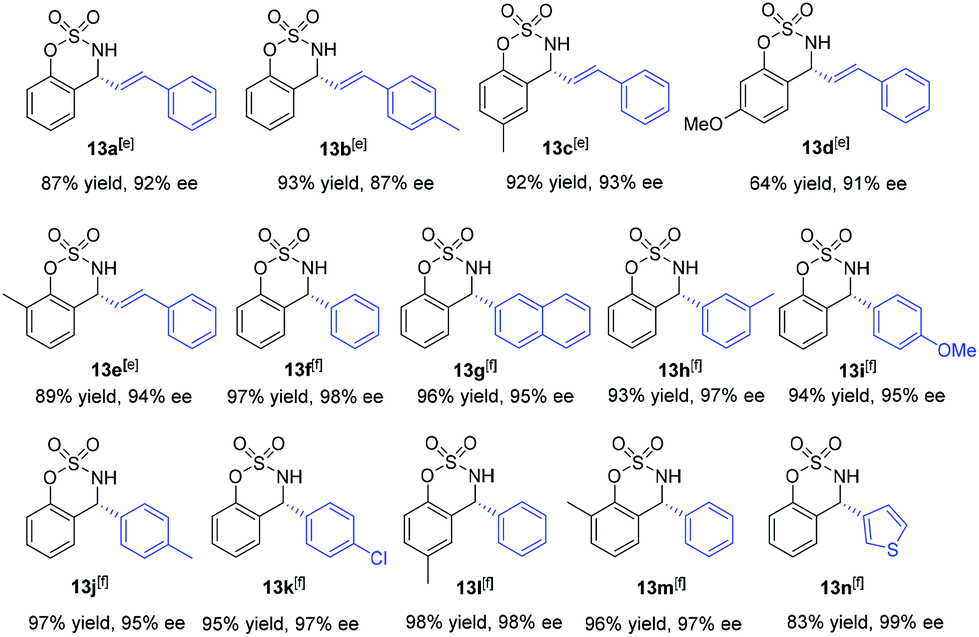
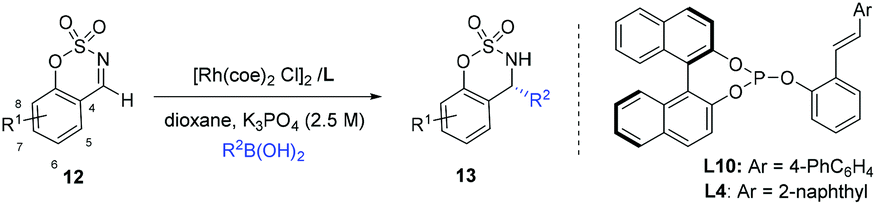
In our earlier work, we have successfully applied S/olefin hybrid ligands in rhodium-catalyzed asymmetric arylation of cyclic N-sulfonyl aldimines.8h However, the use of the sulfur–olefin ligand for asymmetric styrylation of cyclic aldimines could only provide less satisfactory results with moderate enantiocontrol (<60% yield, ∼80% ee). Recently, Lam and coworkers reported an efficient styrylation of cyclic N-sulfonyl aldimines through rhodium–diene-catalyzed asymmetric addition of potassium alkenyltrifluoroborates.13 Although certain successful results have been attained in this field, enantioselective addition of readily accessible styrylboronic acid to this kind of imines remains undeveloped. Encouraged by the results on α-diketone addition, we decided to expand the investigation of our new catalytic system to the asymmetric 1,2-addition of imines. The reaction of N-sulfonyl aldimine 12 (R1 = H) with commercially available trans-β-styreneboronic acid catalyzed by Rh/L1 was initially examined. Gratifyingly, under identical conditions, the transformation proceeded smoothly and gave a promising enantioselectivity (75% ee) as well as high yield (87%). To achieve better results, an extensive screening of ligands and reaction conditions was carried out again. Fortunately, the reaction could lead to an improved ee (92%) of the desired sulfamidate product 13a when L10 was employed as the ligand in K3PO4–dioxane at room temperature. Subsequently, diverse cyclic aldimines and styreneboronic acids were evaluated to define the reaction scope. Substrates with 7-substituted arene provided lower reactivity and enantioselectivity compared to those bearing 6- and 8-substituted arenes (13c, 13d, 13e). The para-Me substituent on the phenyl ring of the styrylboronic acid led to a decrease of the enantioselectivity (13b). In addition, we were pleased to find that these substrates underwent efficient arylation with arylboron reagents and afforded the corresponding products in high yields with excellent enantioselectivities (13f–n, 95–99% ee) under similar reaction conditions. Notably, heteroaryl 3-thiophenylboronic acid could also be successfully employed (13n) (Table 3).
|
|
|---|
| a The reaction was carried out with aldimines (0.25 mmol) and 2 equiv. of boronic acid, in the presence of 2.5 mol% of [Rh(coe)2Cl]2, 5 mol% of ligand, and 2.5 M aq. K3PO4 (0.83 equiv.) in 1.0 mL of dioxane for 3–6 h, unless otherwise noted. b Yield of the isolated product. c Determined by chiral HPLC analysis. d The absolute configuration at the newly created stereocenter was determined to be R by comparing with reported HPLC chromatogram data. e Using L10 as a ligand at room temperature for 12 h. f Using L4 as a ligand at 50 °C. |
|
In summary, we have developed a novel class of chiral phosphorus-based olefin ligands which combines the features of easy accessibility and fascinating ligand efficiency. Key to the success of this design was the use of simple monophosphites with a chiral binaphthyl structure as both coordinating and directing groups, avoiding the construction of complicated scaffolds with main backbone chirality between phosphorus and olefin binding sites. It represents the first example of using chiral phosphite–olefin ligands for asymmetric catalysis. Their catalytic performance has been demonstrated in Rh-catalyzed asymmetric 1,2-addition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X (X: O, N) double bond with the efficient synthesis of various enantioenriched α-hydroxyketones (up to 96% ee) and cyclic sulfamidates (up to 99% ee) in high yields under mild conditions. Further investigations for more effective chiral phosphite–olefin ligands as well as expansion of their other applications in asymmetric catalysis are currently underway.
X (X: O, N) double bond with the efficient synthesis of various enantioenriched α-hydroxyketones (up to 96% ee) and cyclic sulfamidates (up to 99% ee) in high yields under mild conditions. Further investigations for more effective chiral phosphite–olefin ligands as well as expansion of their other applications in asymmetric catalysis are currently underway.
We are grateful for financial support from the National Natural Science Foundation of China (21325209).
Notes and references
- (a) Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, ed. H.-U. Blaser and E. Schmidt, Wiley-VCH, New York, 2004 Search PubMed; (b) New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley, New Jersey, 2007 Search PubMed; (c) Catalysis in Asymmetric Synthesis, ed. V. Caprio and J. M. J. Williams, Wiley, 2nd edn, 2009 Search PubMed; (d) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New Jersey, 3rd edn, 2010 Search PubMed; (e) Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, 2010 Search PubMed.
- (a) Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) Chiral Auxiliaries and Ligands in Asymmetric Synthesis, ed. J. Seyden-Penne, Wiley-Interscience, 1995 Search PubMed.
- For reviews, see: (a) F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 3364 CrossRef CAS PubMed; (b) J. B. Johnson and T. Rovis, Angew. Chem., Int. Ed., 2008, 47, 840 CrossRef CAS PubMed; (c) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482 CrossRef CAS PubMed; (d) R. Shintani and T. Hayashi, Aldrichimica Acta, 2009, 42, 31 CAS; (e) C.-G. Feng, M.-H. Xu and G.-Q. Lin, Synlett, 2011, 1345 CAS; (f) X. Feng and H. Du, Asian J. Org. Chem., 2012, 1, 204 CrossRef CAS; (g) Y. Li and M.-H. Xu, Chem. Commun., 2014, 50, 3771 RSC.
- (a) P. Maire, S. Deblon, F. Breher, J. Geier, C. Böhler, H. Rügger, H. Schönberg and H. Grützmacher, Chem. – Eur. J., 2004, 10, 4198 CrossRef CAS PubMed; (b) C. Thoumazet, L. Ricard, H. Grützmacher and F. P. Floch, Chem. Commun., 2005, 1592 RSC; (c) E. Piras, F. Läng, H. Rüegger, D. Stein, M. Wörle and H. Grützmacher, Chem. – Eur. J., 2006, 12, 5849 CrossRef CAS PubMed.
- (a) R. Shintani, W.-L. Duan, T. Nagano, A. Okada and T. Hayashi, Angew. Chem., Int. Ed., 2005, 44, 4611 CrossRef CAS PubMed; (b) W. Duan, H. Iwamura, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 2130 CrossRef CAS PubMed.
- For selected examples, see: (a) C. Defieber, M. A. Ariger, P. Moriel and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 3139 CrossRef CAS PubMed; (b) R. Mariz, A. Briceño, R. Dorta and R. Dorta, Organometallics, 2008, 27, 6605 CrossRef CAS; (c) M. Roggen and E. M. Carreira, J. Am. Chem. Soc., 2010, 132, 11917 CrossRef CAS PubMed; (d) E. Drinkel, A. Briceňo, R. Dorta and R. Dorta, Organometallics, 2010, 29, 2503 CrossRef CAS; (e) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 5568 CrossRef CAS PubMed; (f) T. J. Hoffman and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 10670 CrossRef CAS PubMed; (g) M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 8652 CrossRef PubMed; (h) M. A. Schafroth, D. Sarlah, S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2012, 134, 20276 CrossRef CAS PubMed; (i) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2013, 135, 994 CrossRef CAS PubMed; (j) J. Y. Hamilton, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3006 CrossRef CAS PubMed; (k) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020 CrossRef CAS PubMed.
- For ligand 3, see: (a) P. Kasák, V. B. Arion and M. Widhalm, Tetrahedron: Asymmetry, 2006, 17, 3084 CrossRef PubMed; For ligand 5, see: (b) T. Minuth and M. M. K. Boysen, Org. Lett., 2009, 11, 4212 CrossRef CAS PubMed; For ligand 6, see: (c) R. Shintani, Y. Narui, S. Hayashi and T. Hayashi, Chem. Commun., 2011, 47, 6123 RSC; For ligand 7, see: (d) Z. Liu and H. Du, Org. Lett., 2010, 12, 3054 CrossRef CAS PubMed; For ligand 8, see: (e) Z. Cao, Y. Liu, Z. Liu, X. Feng, M. Zhuang and H. Du, Org. Lett., 2011, 13, 2164 CrossRef CAS PubMed; For ligand 9, see: (f) Z. Liu, Z. Cao and H. Du, Org. Biomol. Chem., 2011, 9, 5369 RSC; (g) Y. Liu and H. Du, Org. Lett., 2013, 4, 740 CrossRef PubMed; For other examples, see: ligands: (h) P. Štěpnička and I. Císařová, Inorg. Chem., 2006, 45, 8785 CrossRef PubMed; (i) R. T. Stemmler and C. Bolm, Synlett, 2007, 1365 CAS.
- (a) S.-S. Jin, H. Wang and M.-H. Xu, Chem. Commun., 2011, 47, 7230 RSC; (b) W.-Y. Qi, T.-S. Zhu and M.-H. Xu, Org. Lett., 2011, 13, 3410 CrossRef CAS PubMed; (c) S.-S. Jin, H. Wang, T.-S. Zhu and M.-H. Xu, Org. Biomol. Chem., 2012, 10, 1764 RSC; (d) T.-S. Zhu, S.-S. Jin and M.-H. Xu, Angew. Chem., Int. Ed., 2012, 51, 780 CrossRef CAS PubMed; (e) H. Wang, T.-S. Zhu and M.-H. Xu, Org. Biomol. Chem., 2012, 10, 9158 RSC; (f) T.-S. Zhu, J.-P. Chen and M.-H. Xu, Chem. – Eur. J., 2013, 19, 865 CrossRef CAS PubMed; (g) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (h) H. Wang and M.-H. Xu, Synthesis, 2013, 2125 CAS; (i) Y. Li, D.-X. Zhu and M.-H. Xu, Chem. Commun., 2013, 49, 11659 RSC; For a focus review on sulfur–olefins, see: ref. 3g.
- For reviews on monophosphite ligands, see: (a) M. Diéguez, O. Pàmies and C. Claver, Tetrahedron: Asymmetry, 2004, 15, 2113 CrossRef PubMed; (b) A. Gual, C. Godard, S. Castillón and C. Claver, Tetrahedron: Asymmetry, 2010, 21, 1135 CrossRef CAS PubMed; (c) P. W. N. M. Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077 CrossRef PubMed; (d) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef PubMed.
- CCDC 995509 (L1) contains the supplementary crystallographic data.
- For an early example of the non-asymmetric arylation of α-diketones using arylstannanes, see: S. Oi, M. Moro, H. Fukuhara, T. Kawanishi and Y. Inoue, Tetrahedron, 2003, 59, 4351 CrossRef CAS.
- (a) P. Kirsch and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4216 CrossRef CAS; (b) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS.
- Y. Luo, A. J. Carnell and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 6762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 995509. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00135d |
| This journal is © the Partner Organisations 2014 |