
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A convergent rhodium-catalysed asymmetric synthesis of tetrahydroquinolines†
Ho Yin
Li
a,
Joachim
Horn
a,
Amanda
Campbell
b,
David
House
b,
Adam
Nelson
*ac and
Stephen P.
Marsden
*a
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: a.s.nelson@leeds.ac.uk; s.p.marsden@leeds.ac.uk
bGlaxoSmithKline Medicines Research Centre, Stevenage, SG1 2NY, UK
cAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
First published on 23rd July 2014
Abstract
Rh-catalysed conjugate additions of 2-aminophenyl boronic acid derivatives were exploited in diastereoselective and asymmetric syntheses of tetrahydroquinolines. In both cases, combinatorial variation of the substitution of the tetrahydroquinoline ring system was possible.
The tetrahydroquinoline ring system is an important synthetic target1 that is found in many bioactive compounds including natural products (e.g. dynemicin A2) and drugs (e.g. the thrombin inhibitor argatroban3a1 and the antiarrhythmic agent nicainoprol3b2). Established catalytic asymmetric synthetic approaches to tetrahydroquinolines include transition metal-catalysed hydrogenation and transfer-hydrogenation of quinolines,4 organocatalytic reduction of quinolines5 and dihydroquinolines,6 hetero-Diels–Alder reactions of aniline-derived imines with electron-rich dienophiles (Povarov reactions)7 and catalysed intramolecular hydride transfer/Mannich condensations.8
As part of a programme focused on the synthesis of diverse small molecule scaffolds,9 we have exploited Rh-catalysed conjugate additions10 in convergent heterocycle syntheses.9a,b For example, Rh-catalysed conjugate addition of 2-aminophenyl boronic acids 3 to enones 4 was followed by cyclisation‡ (→5) and oxidation to give quinolines 6 in good yield (Scheme 1).9a The reaction presumably proceeds by intramolecular condensation of the initial conjugate addition adduct to yield a 3,4-dihydroquinoline 5 and, hence, the corresponding quinoline 6.
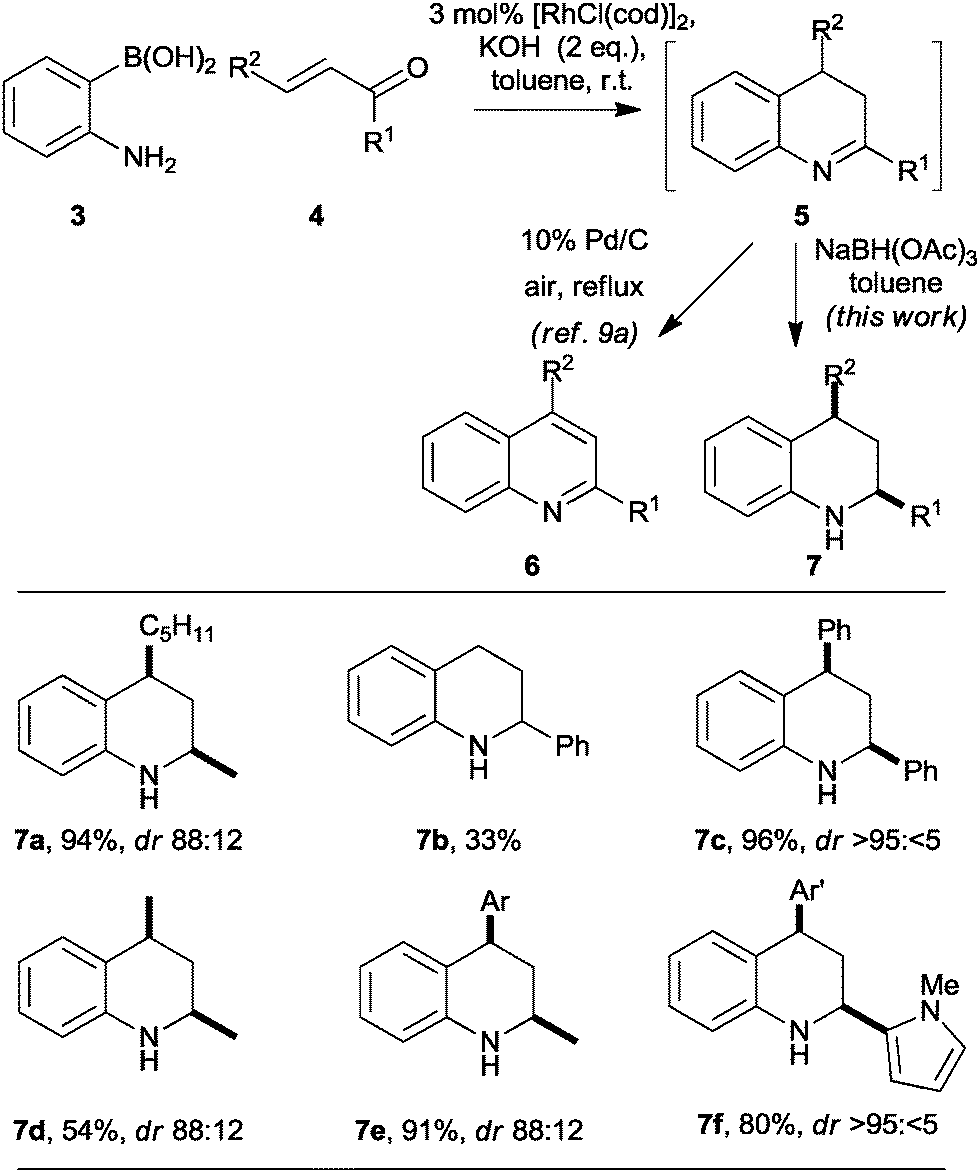 | ||
| Scheme 1 Stereoselective synthesis of tetrahydroquinolines. Diastereomeric ratios (dr) are reported for the purified products. Ar = p-methoxyphenyl; Ar′ = p-nitrophenyl. | ||
Although the general approach might, in principle, be exploited in asymmetric heterocycle synthesis, it had only been demonstrated in the synthesis of achiral9a,b or racemic9b heterocycles. We recognized that Rh-catalysed conjugate addition chemistry might enable a new convergent, and potentially asymmetric,11 synthesis of substituted tetrahydroquinolines 7 (Scheme 1). Initial studies focused on the convergent synthesis of the racemic tetrahydroquinoline 7a. Thus, after completion of the Rh-catalysed conjugate addition reaction, the reaction mixture was diluted with toluene and treated with an excess of sodium triacetoxyborohydride: the tetrahydroquinoline 7a was obtained in 94% yield with 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 diastereoselectivity.
:12 diastereoselectivity.
Our initial studies into the scope of the convergent synthesis of racemic tetrahydroquinolines 7 are summarised in Scheme 1. The synthesis of the 2-substituted tetrahydroquinoline 7b was lower yielding that that of the 2,4-disubstituted analogue 7a. However, with all of the α,β-disubstituted enones 4 studied, the reaction yielded the corresponding 2,4-disubstituted tetrahydroquinolines 7c–f in reasonable to excellent yield with both aliphatic and aromatic R1 and R2 substituents. In each case, the products were obtained with good to excellent diastereoselectivity in favour of the cis isomer.
To enable substitution of the benzenoid ring, we investigated the use of 2-aminophenylpinacolboronates 8, which may be prepared easily from the corresponding 2-bromoanilines (Scheme 2).12 The reaction between the parent pinacolboronate 8 (R3 = H) with chalcone was slower than that of the corresponding boronic acid 3. However, by increasing the catalyst loading (to 6 mol%), and the amount of base (to 2.5 eq.), the reaction was complete in a similar time, and a comparable yield of the tetrahydroquinoline 7c was obtained (compare Scheme 1 with Scheme 2). Remarkably, the synthesis of the 2-substituted tetrahydroquinoline 7b was much more effective with the pinacolboronate 8 (R3 = H) as the reactant, and a much improved 77% yield was observed (compare Scheme 1 with Scheme 2). In a similar vein, the 2,4-disubstituted tetrahydroquinolines 7g–i were obtained in good yield and with high diastereoselectivity. The accessibility of substituted 2-aminophenyl pinacolboronates enabled the synthesis of tetrahydroquinolines 7j–m in which the benzenoid ring had been substituted.
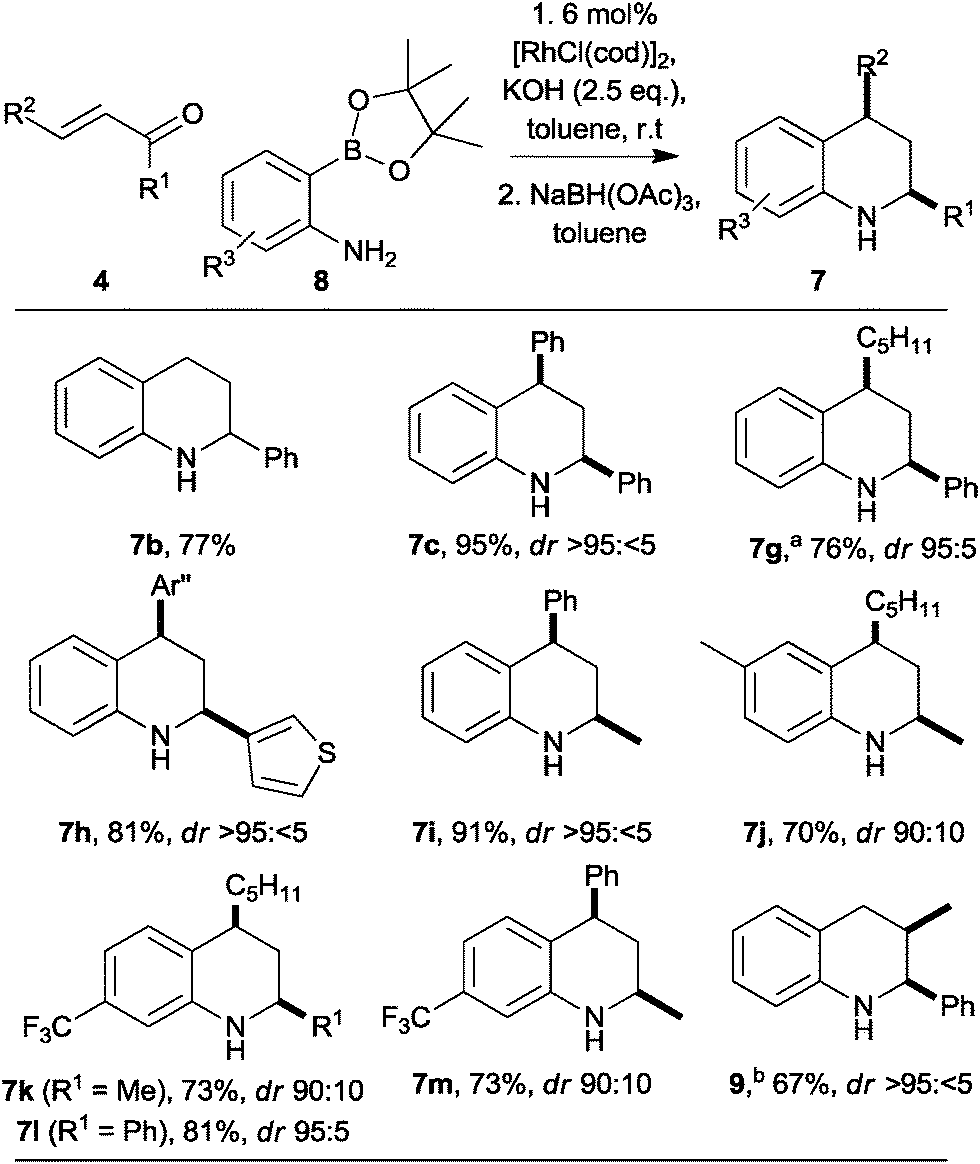 | ||
| Scheme 2 Stereoselective synthesis of tetrahydroquinolines. Diastereomeric ratios (dr) for purified products. a Performed at 50 °C. b Performed at 40 °C with 2-methyl-1-phenylprop-2-en-one; reduction conditions: LiAlH4, toluene, r.t. Ar′′ = p-chlorophenyl. | ||
The syntheses of the 2,4-disubstituted tetrahydroquinolines 7 were all highly diastereoselective in favour of the cis diastereoisomer. The 1,3-diaxial orientations of H-2 and H-4 were determined by careful analysis of vicinal coupling constants13 and, for 3h and 3l, observation of strong mutual nOe interactions; in addition, the cis diastereoisomer of 3d is a known compound.14 The stereoselectivity may be explained in terms of axial attack15 of the reducing agent on the 3,4-dihydroquinoline intermediate.
Extension to the synthesis of a 2,3-disubstituted tetrahydroquinoline was also possible (Scheme 2). Thus, with 2-methyl-1-phenyl prop-2-en-one, the known16 tetrahydroquinoline 9 (3JH2,H3 = 3.5 Hz) was obtained in 67% yield as a >95:<5 mixture of diastereoisomers. As previously observed with the β-unsubstituted enone (→7b; compare Scheme 1 with Scheme 2), the yield was higher with the pinacolboronate 8 (R3 = H) as the reactant (67%) than with the corresponding boronic acid 3 (58%).
We next focused on the development of an asymmetric tetrahydroquinoline synthesis. In studies directed towards an asymmetric synthesis of tetrahydroquinolones, we had found that addition of the pinacolboronate 8 (R3 = H) to methyl cinnamate gave racemic products with a wide range of chiral ligands; however, the corresponding Boc-protected substrate 10 (R3 = H) gave, with (R,R,S,S)-Duanphos as ligand,17,18 a low yield of the corresponding tetrahydroquinolone in >98% ee (ESI†). These initial results prompted us to investigate the addition of the Boc-protected pinacolboronates 10 to unsaturated ketones. In each case, the intermediate conjugate addition products were treated with triethylsilane in TFA to effect deprotection, cyclisation and reduction, and the enantiomeric excess of the corresponding tetrahydroquinolines 8 was determined by chiral HPLC (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Product | Yield/% | dr | eea/% |
| a Determined by chiral analytical HPLC. b The ee of the corresponding 3,5-dinitrobenzamide derivative was determined. c 3 mol% [Rh(nbd)Cl]2 and 6 mol% (S,S,R,R)-Duanphos were used. d 3 mol% [Rh(nbd)Cl]2 and 6 mol% (R,R,S,S)-Duanphos were used. | ||||
| 1 | 7a | 76 | 94:6 |
>98b |
| 2 | 7c | 60 | >95:5 |
>98 |
| 3 | 7h | 45 | >95:5 |
>98b |
| 4 | 7i | 65 | >95:5 |
>98 |
| 5 | 7k | 72 | >95:5 |
>98 |
| 6 | 7l | 65 | 92:8 |
92 |
| 7 | 7m | 72 | >95:5 |
>98 |
| 8c | ent-7c | 78 | >95:5 |
98 |
| 9d | 7g | 62 | >95:5 |
87 |
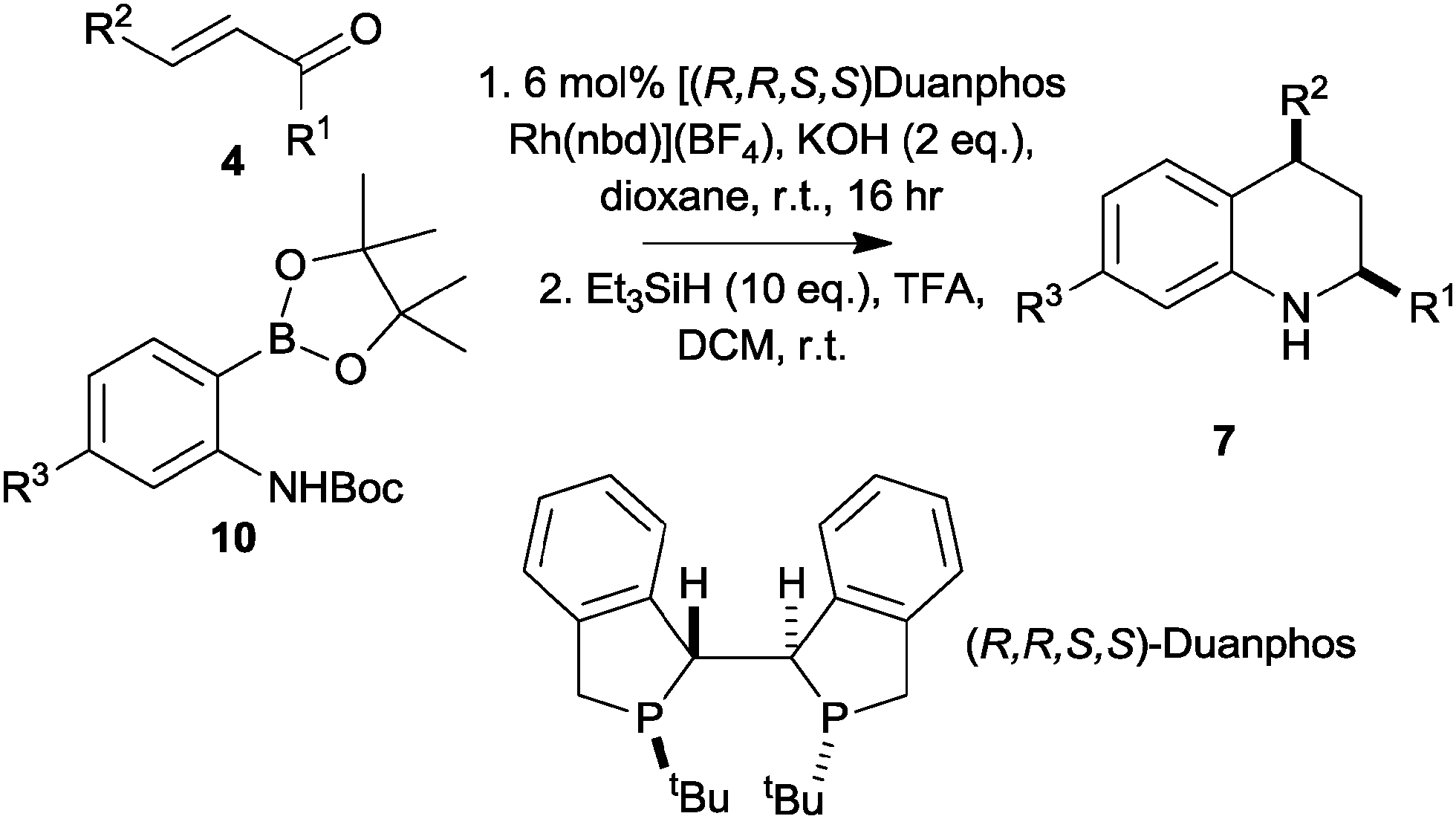
The reactions of the Boc-protected pinacolboronate 10 (R3 = H) with a range of α,β-unsaturated ketones, catalysed by 6 mol% (R,R,S,S)-Duanphos[Rh(nbd)][BF4], were successful with both aromatic and aliphatic R1 and R2 groups (entries 1–4, Table 1). The use of the Boc-protected pinacolboronate 10 (R3 = CF3) was also successful, and allowed variation of the substitution of the benzenoid ring (entries 5–7). The stereoselectivity of the reactions was remarkable: the products 7 were obtained with very high cis diastereoselectivity and with good to excellent enantiomeric excess. The absolute configuration of the tetrahydroquinolines 7c and 7l was determined by comparison of their experimental and simulated vibrational circular dichroism spectra.19 This outcome is consistent with the observed sense of induction in reported asymmetric conjugate addition reactions using this catalyst system.18
We also investigated the effect of generating the chiral catalyst in situ. Thus, the enantiomerically-enriched tetrahydroquinolines ent-7c and 7g could be prepared using the combination of 3 mol% [Rh(nbd)Cl]2 and 6 mol% of either (S,S,R,R) or (R,R,S,S)-Duanphos (entries 8 and 9, Table 1). The yield of the tetrahydroquinoline 7c was higher under these conditions than with 6 mol% (R,R,S,S)-Duanphos[Rh(nbd)][BF4] (compare entries 2 and 8).
A novel convergent and stereoselective synthesis of tetrahydroquinolines exploited the Rh-catalysed addition of 2-aminophenyl boronate derivatives to α,β-unsaturated ketones as the key step. Remarkably, it was possible to develop a highly enantioselective variant of the reaction that exploited the specific combination of Duanphos as the chiral ligand and Boc-protected pinacolboronates as the reactants. The synthetic approach was modular, and will likely be adapted to synthesis of a range of other benzo-fused heterocyclic ring systems. We thank EPSRC and GlaxoSmithKline for funding, Douglas Minick for conducting VCD experiments, and Amgen and ChiralQuest for generous gifts of metal complexes.
Notes and references
- A. R. Katritzky, S. Rachwal and B. Rachwal, Tetrahedron, 1996, 52, 15031 CrossRef CAS.
- M. Konishi, H. Ohkuma, K. Matsumoto, T. Tsuno, H. Kamei, T. Miyaki, T. Oki, H. Kawaguchi, G. D. VanDuyne and J. Clardy, J. Antibiot., 1989, 42, 1449 CrossRef CAS.
- (a) L. R. Bush, Cardiovasc. Drug Rev., 1991, 9, 247 CrossRef CAS PubMed; (b) S. Imanishi, T. Kimura and M. Arita, Cardiovasc. Drug Rev., 1991, 9, 223 CrossRef CAS PubMed.
- For examples, see: (a) W.-B. Wang, S.-M. Lu, P.-Y. Yang, X.-W. Han and Y.-G. Zhou, J. Am. Chem. Soc., 2003, 125, 10536 CrossRef CAS PubMed; (b) M. T. Reetz and X. Li, Chem. Commun., 2006, 2159 RSC; (c) W.-J. Tang, S.-F. Zhu, L.-J. Xu, Q.-L. Zhou, Q.-H. Fan, H.-F. Zhou, K. Lam and A. S. C. Chan, Chem. Commun., 2007, 613 RSC; (d) C. Wang, C. Li, X. Wu, A. Pettman and J. Xiao, Angew. Chem., Int. Ed., 2009, 48, 6524 CrossRef CAS PubMed; (e) T. Wang, L.-G. Zhuo, Z. Li, F. Chen, Z. Ding, Y. He, Q.-H. Fan, J. Xiang, Z.-X. Yu and A. S. C. Chan, J. Am. Chem. Soc., 2011, 133, 9878 CrossRef CAS PubMed; (f) H. Zhou, Z. Li, Z. Wang, T. Wang, L. Xu, Y. He, Q.-H. Fan, J. Pan, L. Gu and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 8464 CrossRef CAS PubMed.
- (a) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef CAS PubMed; (b) Q.-S. Guo, D.-M. Du and J. Xu, Angew. Chem., Int. Ed., 2008, 47, 759 CrossRef CAS PubMed; (c) M. Rueping, T. Theissman, S. Raja and J. W. Bats, Adv. Synth. Catal., 2008, 350, 1001 CrossRef CAS; (d) M. Rueping and T. Theissman, Chem. Sci., 2010, 1, 473 RSC; (e) M. Rueping, T. Theissman, M. Stoeckel and A. P. Antonchick, Org. Biomol. Chem., 2011, 9, 6844 RSC; (f) Q. A. Chen, K. Gao, Y. Duan, Z. S. Ye, L. Shi, Y. Yang and Y.-G. Zhou, J. Am. Chem. Soc., 2012, 134, 2442 CrossRef CAS PubMed; (g) X.-F. Tu and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 11346 CrossRef CAS PubMed.
- Z.-Y. Han, H. Xiao, X.-H. Chen and L.-Z. Gong, J. Am. Chem. Soc., 2009, 131, 9182 CrossRef CAS PubMed.
- (a) H. Ishitani and S. Kobayashi, Tetrahedron Lett., 1996, 37, 1973 CrossRef . For select recent examples, see: ; (b) T. Akiyama, H. Morita and K. Fuchibe, J. Am. Chem. Soc., 2006, 128, 13070 CrossRef CAS PubMed; (c) H. Liu, G. Dagousset, G. Masson, P. Retailleau and J. Zhu, J. Am. Chem. Soc., 2009, 131, 4598 CrossRef CAS PubMed; (d) G. Bergonzini, L. Gramigna, A. Mazzanti, M. Fochi, L. Bernardi and A. Ricci, Chem. Commun., 2010, 46, 327 RSC; (e) M.-S. Xie, X.-H. Chen, Y. Zhu, B. Gao, L.-L. Lin, X.-H. Liu and X.-M. Feng, Angew. Chem., Int. Ed., 2010, 49, 3799 CrossRef CAS PubMed; (f) H. Xu, S. J. Zuend, M. G. Woll, Y. Tao and E. N. Jacobsen, Science, 2010, 327, 986 CrossRef CAS PubMed; (g) G. Dagousset, J. P. Zhu and G. Masson, J. Am. Chem. Soc., 2011, 133, 14804 CrossRef CAS PubMed; (h) C. Min, N. Mittal, D. X. Sun and D. Seidel, Angew. Chem., Int. Ed., 2013, 52, 14084 CrossRef CAS PubMed; (i) C. S. Luo and Y. Huang, J. Am. Chem. Soc., 2013, 135, 8193 CrossRef CAS PubMed; (j) J. Calleja, A. B. Gonzalez-Perez, A. R. de Lera, R. Alvarez, F. J. Fananas and F. Rodriguez, Chem. Sci., 2014, 5, 996 RSC.
- (a) S. Murarka, I. Deb, C. Zhang and D. Seidel, J. Am. Chem. Soc., 2009, 131, 13226 CrossRef CAS PubMed; (b) K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166 CrossRef CAS PubMed; (c) Y.-K. Kang, S. M. Kim and D. Y. Kim, J. Am. Chem. Soc., 2010, 132, 11847 CrossRef CAS PubMed.
- (a) J. Horn, S. P. Marsden, A. Nelson, D. House and G. G. Weingarten, Org. Lett., 2008, 10, 4117 CrossRef CAS PubMed; (b) J. Horn, H. Y. Li, S. P. Marsden, A. Nelson, R. J. Shearer, A. J. Campbell, D. House and G. G. Weingarten, Tetrahedron, 2009, 65, 9002 CrossRef CAS PubMed; (c) P. Tosatti, J. Horn, A. J. Campbell, D. House, A. Nelson and S. P. Marsden, Adv. Synth. Catal., 2010, 352, 3153 CrossRef CAS; (d) P. Tosatti, A. J. Campbell, D. House, A. Nelson and S. P. Marsden, J. Org. Chem., 2011, 76, 5495 CrossRef CAS PubMed.
- (a) M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229 CrossRef CAS . For reviews, see: ; (b) T. Hayashi, Synlett, 2001, 879 CrossRef CAS; (c) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS PubMed; (d) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2093 RSC.
- (a) Y. Takaya, M. Ogasawara, T. Hayashi, M. Sakai and N. Miyaura, J. Am. Chem. Soc., 1998, 120, 5579 CrossRef CAS . For reviews, see ref. 10c,d and: ; (b) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed; (c) D. Mueller and A. Alexakis, Chem. Commun., 2012, 48, 12037 RSC.
- O. Baudoin, D. Guénard and F. Guéritte, J. Org. Chem., 2000, 65, 9268 CrossRef CAS.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870 CrossRef CAS.
- M. Ueda, S. Kawai, M. Hayashi, T. Naito and O. Miyata, J. Org. Chem., 2010, 75, 914 CrossRef CAS PubMed.
- R. V. Stevens, in Strategies and Tactics in Organic Synthesis, ed. T. Lindberg, Academic Press, 1984, vol. 1 Search PubMed.
- S.-I. Murahashi, Y. Imada and Y. Hirai, Bull. Chem. Soc. Jpn., 1989, 62, 2968 CrossRef CAS.
- (a) D. Liu and X. Zhang, Eur. J. Org. Chem., 2005, 646 CrossRef CAS; (b) I. C. Lennon, Chim. Oggi, 2010, 28, 46 CAS.
- For an example of a Rh-catalysed conjugate addition with Duanphos as ligand, see: J. L. Zigterman, J. C. S. Woo, S. D. Walker, J. S. Tedrow, C. J. Borths, E. E. Bunel and M. M. Faul, J. Org. Chem., 2007, 72, 8870 CrossRef CAS PubMed.
- (a) P. J. Stephens, in Computational Medicinal Chemistry for Drug Discovery, ed. P. Bultinick, H. de Winter, W. Langenaeker and J. P. Tollenare, Marcel Dekker, New York, 2004, pp. 699–725 Search PubMed; (b) T. Kuppens, P. Bultinick and W. Langenaeker, Drug Discovery Today: Technol., 2004, 1, 269 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc04940c |
| ‡ See ref. 9a for evidence for the formation of 5 (R1 = Me; R2 = C5H11). |
| This journal is © The Royal Society of Chemistry 2014 |