
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and in silico studies of new quinazolinone derivatives as antitumor PARP-1 inhibitors†
Sayed K. Ramadan a,
Eman Z. Elrazazb,
Khaled A. M. Abouzid*bc and
Abeer M. El-Naggar*a
a,
Eman Z. Elrazazb,
Khaled A. M. Abouzid*bc and
Abeer M. El-Naggar*a
aDepartment of Chemistry, Faculty of Science, Ain Shams University, Abbassia, 11566 Cairo, Egypt. E-mail: elsayedam@sci.asu.edu.eg
bDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Ain Shams University, Abbassia, 11566 Cairo, Egypt. E-mail: khaled.abouzid@pharma.asu.edu.eg
cDepartment of Organic and Medicinal Chemistry, Faculty of Pharmacy, University of Sadat City, Sadat City, Egypt
First published on 10th August 2020
Abstract
Herein, we report an eco-friendly synthesis of a new series of quinazolinone-based derivatives as potential PARP-1 inhibitors. The 4-quinazolinone scaffold was utilized as a bioisostere to the phthalazinone core of the reference compound Olaparib. Most of the synthesized compounds displayed appreciable inhibitory activity against PARP-1. Compound 12c showed inhibitory activity at IC50 = 30.38 nM comparable to Olaparib, which has IC50 = 27.89 nM. Cell cycle analysis was performed for compounds 12a and 12c, and both exhibited cell growth arrest at G2/M phase in the MCF-7 cell line. In addition, both compounds increased the programmed apoptosis compared to the control. Furthermore, molecular docking of the final compounds into the PARP-1 active site was executed to explore their probable binding modes. Also, a computational QSAR and in silico ADMET study was performed. The results of this study revealed that some of the newly synthesized compounds could serve as a new framework to discover new PARP-1 inhibitors with anti-cancer activity.
1. Introduction
Recently, great attention has been given to the PARP-1 enzyme as a promising anti-cancer therapeutic target1 as it is a member of the PARP family of proteins, which play a crucial role in the repair of single-stranded DNA breaks via the base excision repair pathway.2 In addition, PARP-1 has a major role in a variety of cellular processes, like cellular differentiation, gene transcription, inflammation, mitosis, and cell death, which contribute to the antitumor activity of PARP-1 inhibitors.3 PARP-1 inhibition has a synthetic lethality in the presence of mutations of BRCA 1/2, which are essential proteins in homologous recombination (HR) of DNA double strand breaks, due to the dependence of the mutant cancer cells on PARP-1 for DNA repair and cell survival.4,5 Thus, compounds acting as PARP-1 inhibitors may lead to selective cell death, particularly in ovarian and breast cancers.6 Several PARP-1 inhibitors have already been FDA approved, including AG014699 (Rucaparib), AZD2281 (Olaparib), MK4827 (Niraparib), and BMN673 (Talazoparib) (Fig. 1).7–11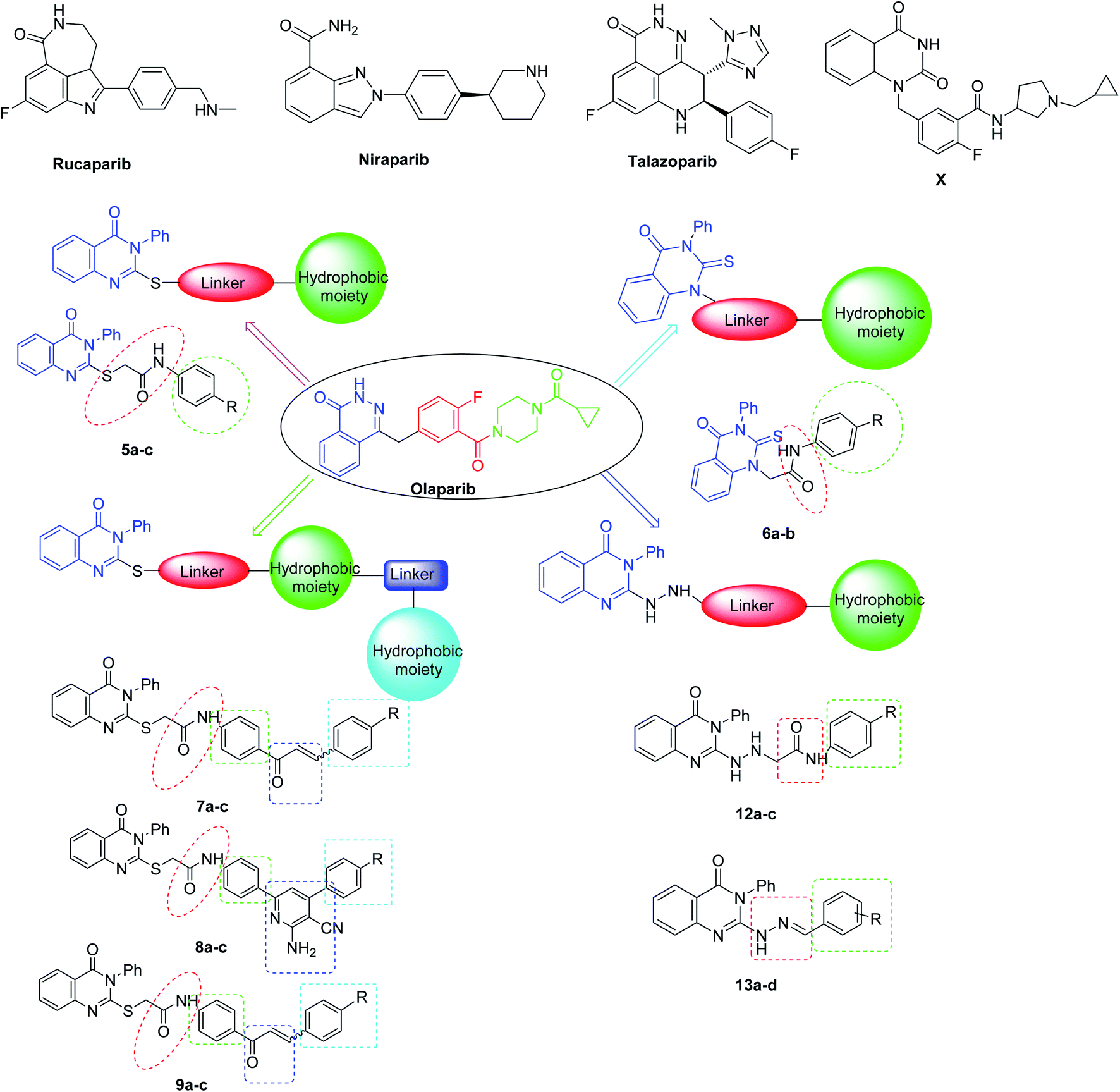 | ||
| Fig. 1 Chemical structures of the approved PARP-1 inhibitors (Rucaparib, Niraparib, and Talazoparib and Olaparib) and the target derivatives [(5a–c), (6a–c), (7a,b), (8a–c), (9a–c), (12a–c), and (13a–c)]. | ||
As a response to DNA damage, PARP-1 catalyzes the cleavage of nicotinamide adenine dinucleotide NAD+ into nicotinamide and ADP ribose units, which are transferred to different acceptor proteins participating in DNA damage repair processes, including itself, histones and formed ADP-ribose polymers (PAR). So, PARP-1 has two binding sites; one is occupied by the nicotinamide-ribose (NI site) and the other is the adenine-ribose binding site (AD site).12
2. Rationale and design
The majority of the reported PARP-1 inhibitors occupy the NI site through hydrogen bonding and pi–pi stacking interactions, and some of them exhibit additional interactions in the AD site, which is large enough to accommodate diverse structures and thus increases their potency, as for Olaparib.13,14 Since the quinazoline nucleus represents a privileged scaffold in many anticancer drugs,15 in this study, we employed the quinazolinone scaffold as a bioisostere of the phthalazinone core of Olaparib to occupy the NI site of PARP-1, since many quinazoline-based compounds are known as anticancer agents.16 Also, a series of quinazolinone derivatives were synthesized as PARP-1 inhibitors. Among this series, compound X displayed the highest inhibitory activity17,18 (Fig. 1). Therefore, we decided to introduce various substitutions at position-2 of the quinazolinone ring to explore additional interactions with the AD site; the linker aims to mimic the carbonyl group of Olaparib and engage with the enzyme through hydrogen bonding and a hydrophobic moiety was added to mimic the cyclopropyl group of Olaparib. The newly synthesized compounds were tested for their inhibitory activities against the PARP-1 enzyme (Fig. 1).3. Results and discussion
3.1. Chemistry
Our continuing efforts were directed toward the design and construction of new heterocyclic compounds with anticipated pharmacological activities19,20 using green chemistry tools.21,22 Therefore, in this study, we report a highly efficient method for the synthesis of target potentially bioactive quinazoline derivatives (3–14) using both conventional and microwave irradiation methods, as outlined in Schemes 1–3. It was observed that the microwave approach proved to be extremely fast, providing good to excellent yields (80–93%) as compared with the conventional method (54–68%). Here, the most noticeable advancement was the speed at which the reaction proceeded as the reaction was completed within 4–6 min, being 40–42 times faster than the conventional method. These results are summarized in Table 1.| Comp. no. | M. W | Conv. | Comp. no. | M. W | Conv. | ||||
|---|---|---|---|---|---|---|---|---|---|
| TM (min) | YM (%) | Tc (min) | Yc (%) | TM (min) | YM (%) | Tc (min) | Yc (%) | ||
| 5a | 3 | 85.57 | 360 | 67.44 | 8c | 6 | 88.45 | 540 | 69.57 |
| 5b | 4 | 93.53 | 480 | 59.71 | 9a | 4 | 91.43 | 480 | 54.44 |
| 5c | 3 | 86.51 | 420 | 69.55 | 9b | 4 | 92.34 | 420 | 65.59 |
| 6a | 3 | 85.47 | 360 | 57.73 | 9c | 4 | 91.39 | 480 | 68.49 |
| 6b | 6 | 93.50 | 480 | 59.81 | 12a | 4 | 89.60 | 360 | 63.47 |
| 6c | 3 | 86.58 | 420 | 69.66 | 12b | 6 | 88.47 | 540 | 54.55 |
| 7a | 3 | 85.51 | 360 | 57.65 | 12c | 4 | 91.44 | 480 | 65.59 |
| 7b | 4 | 93.61 | 480 | 59.60 | 13a | 4 | 92.54 | 420 | 67.66 |
| 7c | 3 | 86.59 | 420 | 63.63 | 13b | 5 | 88.50 | 460 | 66.58 |
| 8a | 4 | 91.48 | 480 | 57.65 | 13c | 4 | 89.49 | 360 | 67.45 |
| 8b | 4 | 89.55 | 360 | 68.54 | 13d | 6 | 88.48 | 340 | 68.46 |
Herein, the key starting material, 2-mercapto-3-phenylquinazolinone 3, was prepared via treating anthranilic acid with phenyl isothiocyanate in boiling ethanol containing a few drops of triethylamine.23 2-Chloro-N-(substituted)acetamide derivatives (4a–c) were prepared by treatment of methyl 4-aminobenzoate, 4-aminoacetophenone and sulfanilamide with chloroacetyl chloride under basic conditions at room temperature.
Alkylation of quinazoline 3 with 2-chloro-N-(substituted)acetamide derivatives 4a–c was carried out afterwards to achieve both S-alkylated and N-alkylated products 5a–c and 6a–c, respectively (Scheme 1). The structures of compounds 5a–c, and 6a–c were inferred from their elemental analyses, IR, NMR and MS spectra and were compatible with the proposed structures. Thus, the IR spectra exhibited two absorption bands at around ν = 3414 and 3228 cm−1 due to NH groups and two strong carbonyl absorption bands ranging from 1725–1685 cm−1. Their 1H NMR spectra provided exchangeable singlets of NH groups around the region δ = 13.00–10.30 ppm.
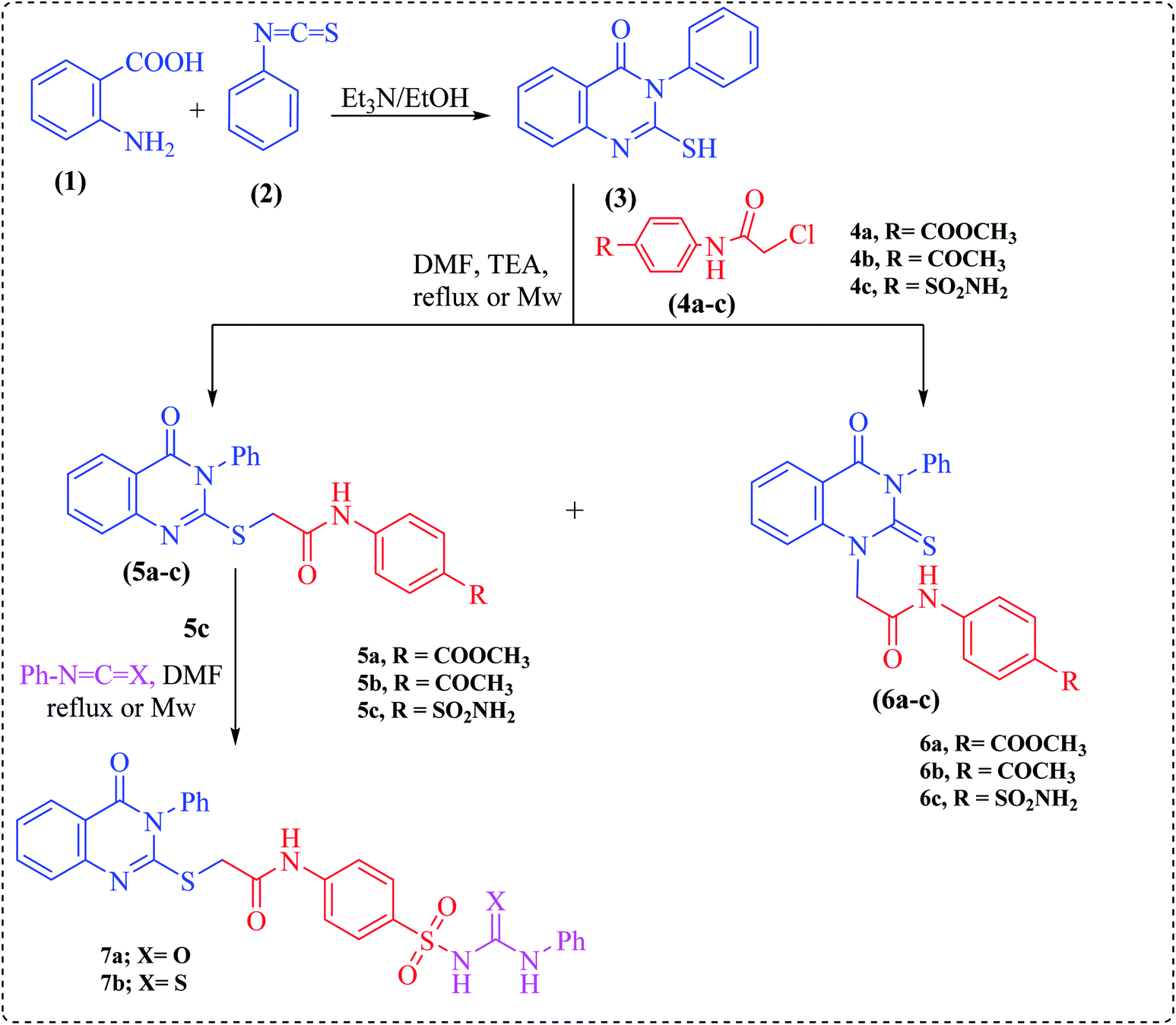 | ||
| Scheme 1 Synthesis of compounds 3–7. | ||
In turn, the sulfonamide derivative 5c was further reacted with phenyl isocyanate or phenyl isothiocyanate in DMF under microwave irradiation at 200 W power for 2 min to afford urea or thiourea derivatives 7a and b, respectively (Scheme 1). The structures of 7a and b were deduced by the absence of the NH2 absorption band characteristic for compound 5c in the IR and 1H NMR spectra.
One-pot multicomponent reaction of the acetyl derivative 5b with malononitrile and the appropriate aromatic aldehydes, namely 4-methoxy-, 4-chloro- or 4-nitrobenzaldehyde, in dioxane containing ammonium acetate under microwave irradiation at 200 W power for 2–3 min furnished the corresponding enaminonitrile derivatives, namely N-(4-(6-amino-4-(4-substituted phenyl)-5-cyanopyridin-2-yl)phenyl)-2-(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-ylthio)-acetamide (8a–c), respectively. The structures of these compounds were established based on their analytical and spectral data. Thus, the IR spectra showed the existence of cyano group absorption bands at around ν = 2226–2216 cm−1. Their 1H NMR spectra exhibited singlets in the region of δ = 13.00–10.30 ppm, assigned to two exchangeable protons, which are attributed to the NH2 protons of the pyridine moiety.
On the other hand, chalcones and their derivatives are known to be important intermediates in organic synthesis because they serve as building block synthons for a wide variety of heterocyclic compounds which are of physiological importance.24–26 The presence of an enone functionality in the chalcone moiety confers biological activity, like anti-inflammatory,27 antifungal,28 antioxidant,29 antimalarial,30 antituberculosis,31 anti-HIV32 and antitumor33 activities. Indeed, Claisen–Schmidt condensation of ketone 5b with the same aromatic aldehydes as in Scheme 2, in the presence of NaOH, under microwave irradiation, afforded the corresponding α,β-unsaturated ketones (chalcones), namely N-(4-(3-(4-substituted phenyl)acryloyl)phenyl)-2-(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-ylthio)acetamide (9a–c), respectively. The structures of these chalcones were substantiated by their analytical and spectral data. The 1H NMR spectra displayed the absence of the singlet of methyl protons in compound 5b (cf. Experimental).
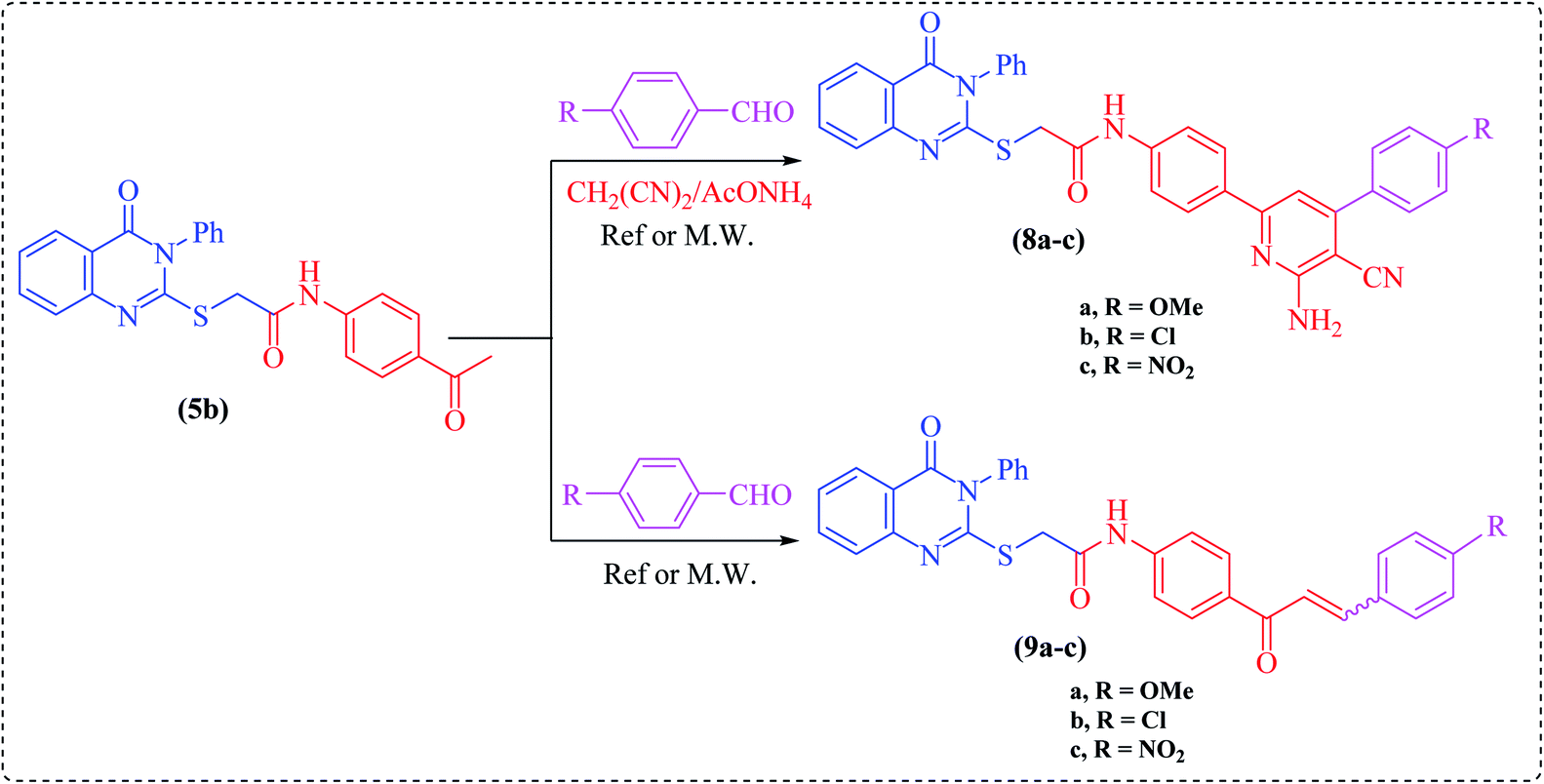 | ||
| Scheme 2 Synthesis of compounds 8–9. | ||
Noteworthily, hydrazinolysis of compound 5a using hydrazine hydrate in n-butanol under reflux conditions failed to produce the expected hydrazide 10, but afforded 2-hydrazinyl-3-phenylquinazoline derivative 11 (Scheme 3). The structure of 11 was confirmed by spectral data and by direct comparison with an authentic sample prepared by treating the starting quinazoline 3 with hydrazine hydrate in refluxing dioxane.23 Subsequently, the reaction of hydrazinoquinazoline 11 with 2-chloro-N-acetamide derivatives 4a–c in DMF containing two drops of triethylamine under microwave irradiation furnished the hydrazinylacetamide derivatives, namely N-(4-substituted phenyl)-2-(2-(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)hydrazinyl)-acetamide (12a–c). The structures of all newly synthesized compounds were confirmed on the basis of spectral and elemental analyses, which were in full agreement with the proposed structures.
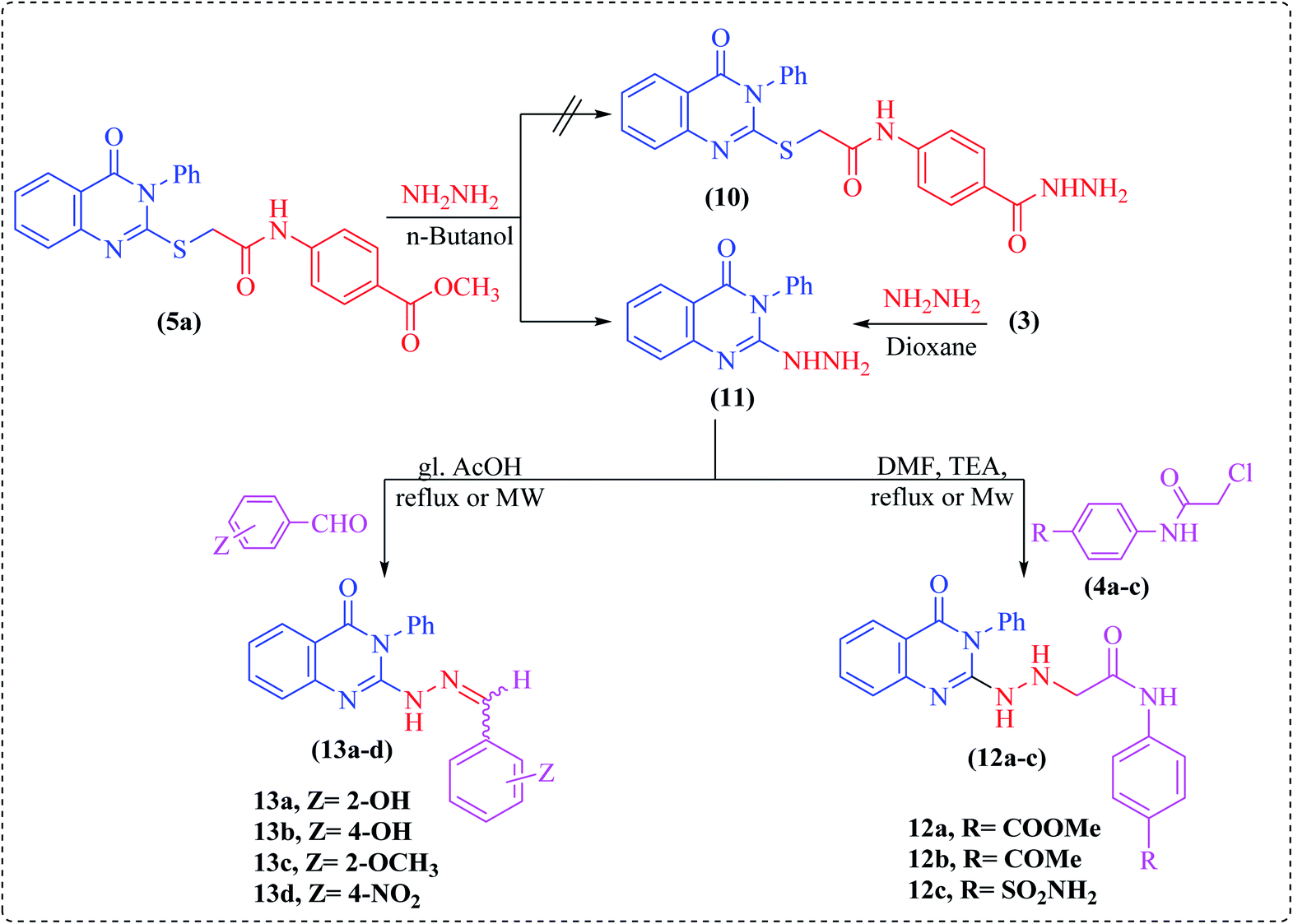 | ||
| Scheme 3 Synthesis of compounds 11–13. | ||
Finally, condensation of compound 11 with different aromatic aldehydes, namely 2-hydroxy-, 4-hydroxy-, 2-methoxy-, or 4-nitrobenzaldehyde under microwave irradiation or using the conventional method in glacial acetic acid produced the corresponding hydrazone derivatives 13a–d (Scheme 3). In the 1H NMR spectra of all compounds, a singlet peak corresponding to the imine proton (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) was observed as a sharp singlet downfield of the aryl proton peaks, as well as a broad singlet for the NH group, while the singlet of NH2 protons was absent (cf. Experimental).
CH) was observed as a sharp singlet downfield of the aryl proton peaks, as well as a broad singlet for the NH group, while the singlet of NH2 protons was absent (cf. Experimental).
3.2. Biological evaluation
| Compd | IC50 (nM) | Compd | IC50 (nM) |
|---|---|---|---|
| a Data are displayed as mean ± S. E. M. n = 3 (three independent repeats). Statistical analysis was carried out using one-way ANOVA followed by Dunnett post hoc test.b Statistically significant from Olaparib at p < 0.05. | |||
| Olaparib | 30.38 ± 1.61 | 9a | 101.6b ± 5.88 |
| 5a | 336.0b ± 13.08 | 9b | 174.3b ± 8.72 |
| 5b | 218.3b ± 10.34 | 9c | 313.7b ± 15.35 |
| 5c | 354.1b ± 18.28 | 12a | 39.07 ± 3.89 |
| 6a | 55.96 ± 2.95 | 12b | 89.69b ± 5.67 |
| 6b | 64.67 ± 3.38 | 12c | 27.89 ± 3.45 |
| 7a | 128.4b ± 8.57 | 13a | 102.3b ± 6.84 |
| 7b | 79.06b ± 4.31 | 13b | 44.16 ± 3.12 |
| 8a | 32.49 ± 2.64 | 13c | 45.64 ± 2.86 |
| 8b | 154.66b ± 8.76 | 13d | 485.3b ± 26.0 |
| 8c | 403.2b ± 21.27 | ||
The higher potency of compounds 6a–b when compared to compounds 5a–b revealed that substitution at position-1 of the quinazoline was better than at position-2. Also, the extension of compound 5c to give compounds 7a and 7b significantly increased the potency, suggesting extra hydrophobic binding in the AD site of PARP-1. In compounds 8a–c, the methoxy derivative displayed higher activity than the chloro and nitro derivatives, suggesting that the presence of an electron donating group is more preferred than an electron withdrawing group. This was also observed in compounds 9a–c. Interestingly, series 12a–c showed very good potency in comparison to Olaparib with the sulfonamide derivative 12c showing the highest activity among all of the synthesized compounds. Of the hydrazone derivatives 13a–d, compound 13b showed higher activity then 13a, suggesting that substitution in position-4 of the phenyl ring is better than at position-2. Also, the methoxy derivative 13c showed better potency than the hydroxyl derivative 13a and the nitro substitution in 13d greatly decreased the potency, suggesting that electron donating groups were preferred over electron withdrawing groups.
| Compound | DNA content% | |||
|---|---|---|---|---|
| % G0–G1 | % S | % G2–M | % Pre G1 | |
| 12a/MCF7 | 38.51 | 23.43 | 38.06 | 26.55 |
| 12c/MCF7 | 43.28 | 33.23 | 23.49 | 19.31 |
| Stauroporine/MCF7 | 37.19 | 26.22 | 36.59 | 31.74 |
| cont. MCF7 | 59.26 | 34.17 | 6.57 | 1.57 |
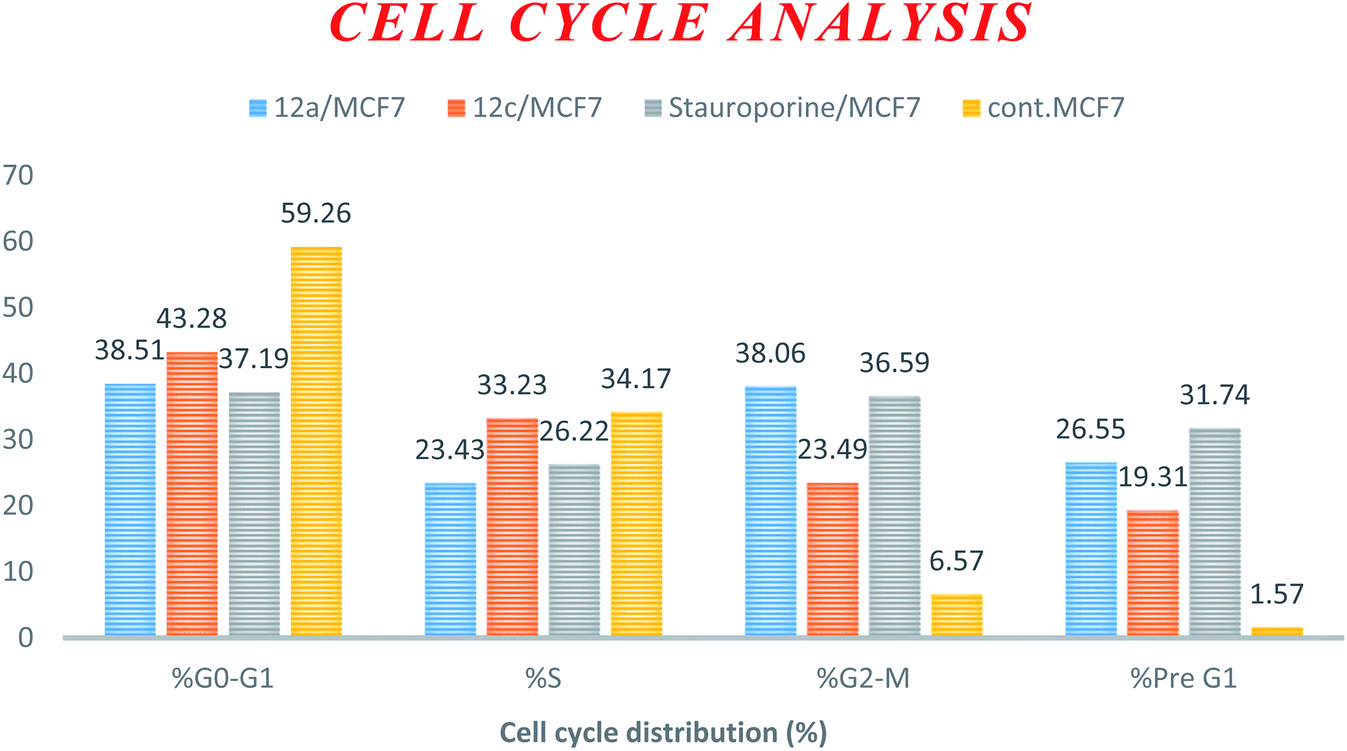 | ||
| Fig. 2 Cell cycle analysis and apoptosis effect in the MCF-7 cell line when treated with compounds 12a and 12c. | ||
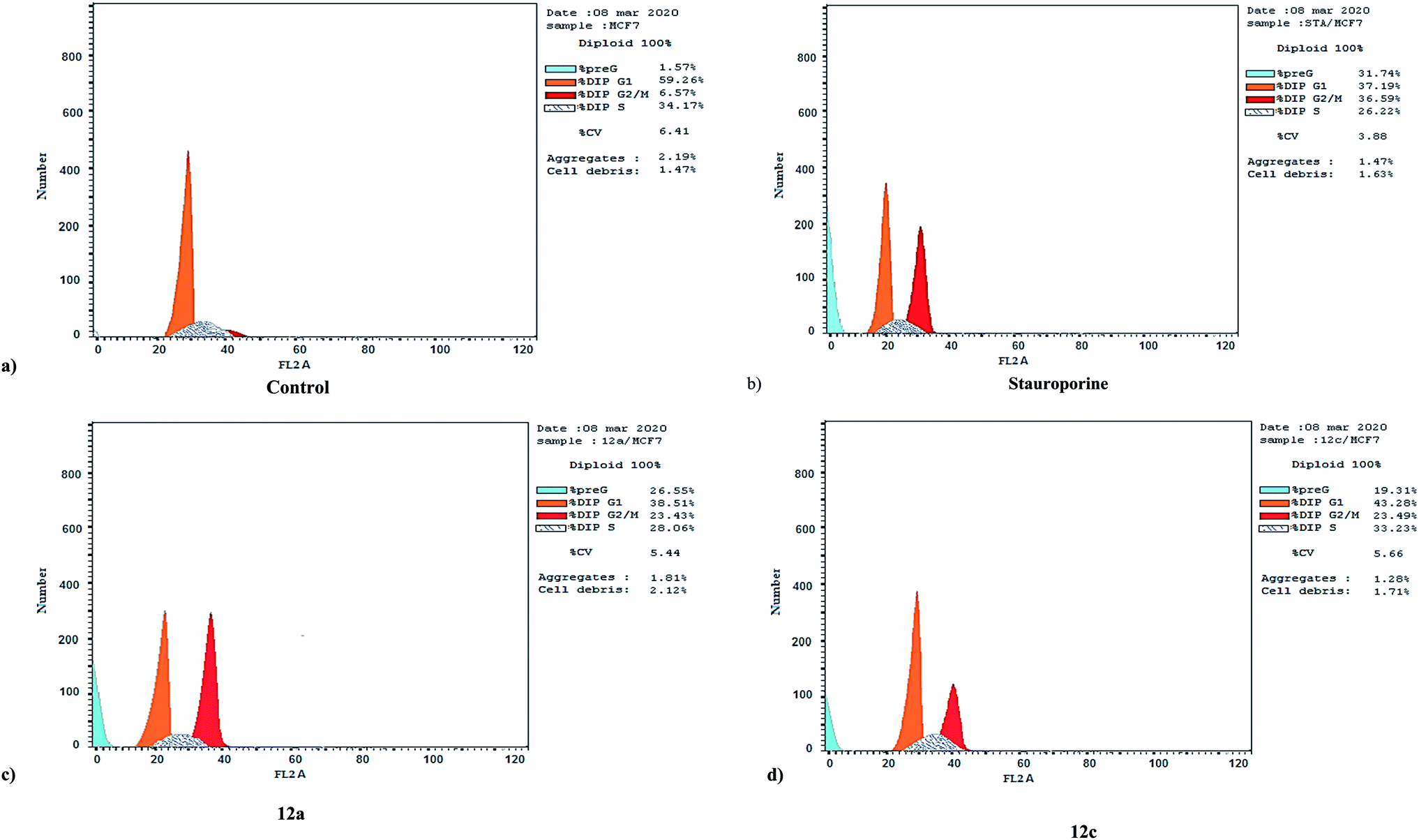 | ||
| Fig. 3 MCF-7 cell distribution upon treatment with compounds 12a and 12c. | ||
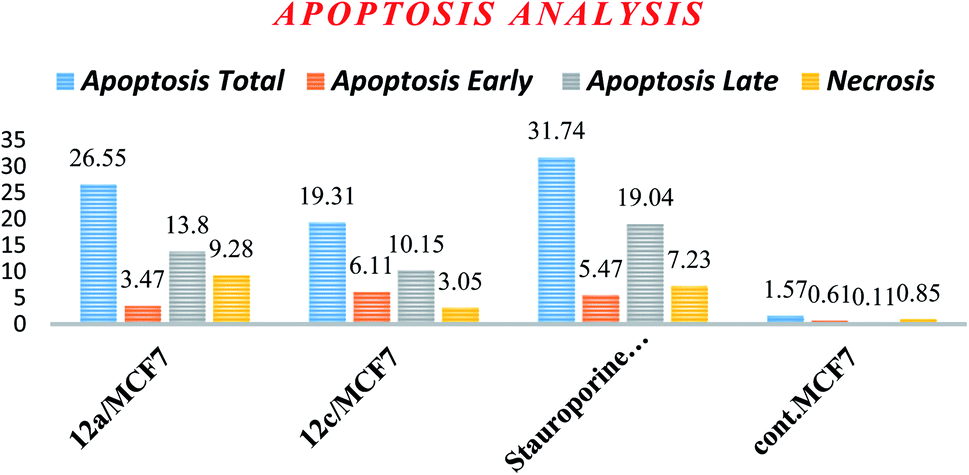 | ||
| Fig. 4 Percentage of cell death induced by compounds 12a and 12c in MCF-7 cells. | ||
| Apoptosis | Necrosis | |||
|---|---|---|---|---|
| Total | Early | Late | ||
| 12a/MCF7 | 26.55 | 3.47 | 13.8 | 9.28 |
| 12c/MCF7 | 19.31 | 6.11 | 10.15 | 3.05 |
| Stauroporine/MCF7 | 31.74 | 5.47 | 19.04 | 7.23 |
| cont. MCF7 | 1.57 | 0.61 | 0.11 | 0.85 |
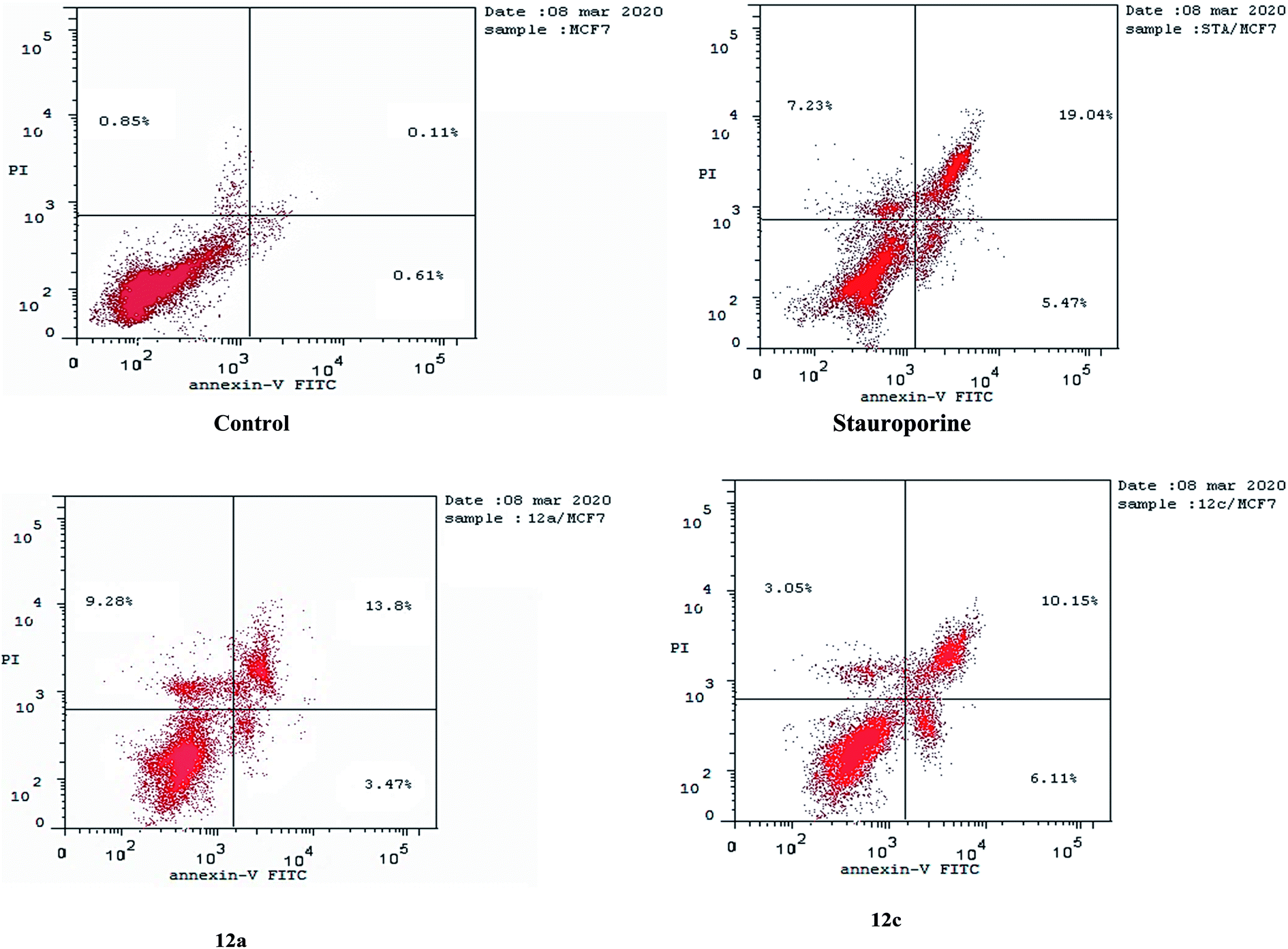 | ||
| Fig. 5 Apoptosis induced in MCF-7 cells by compound 12a and 12c. | ||
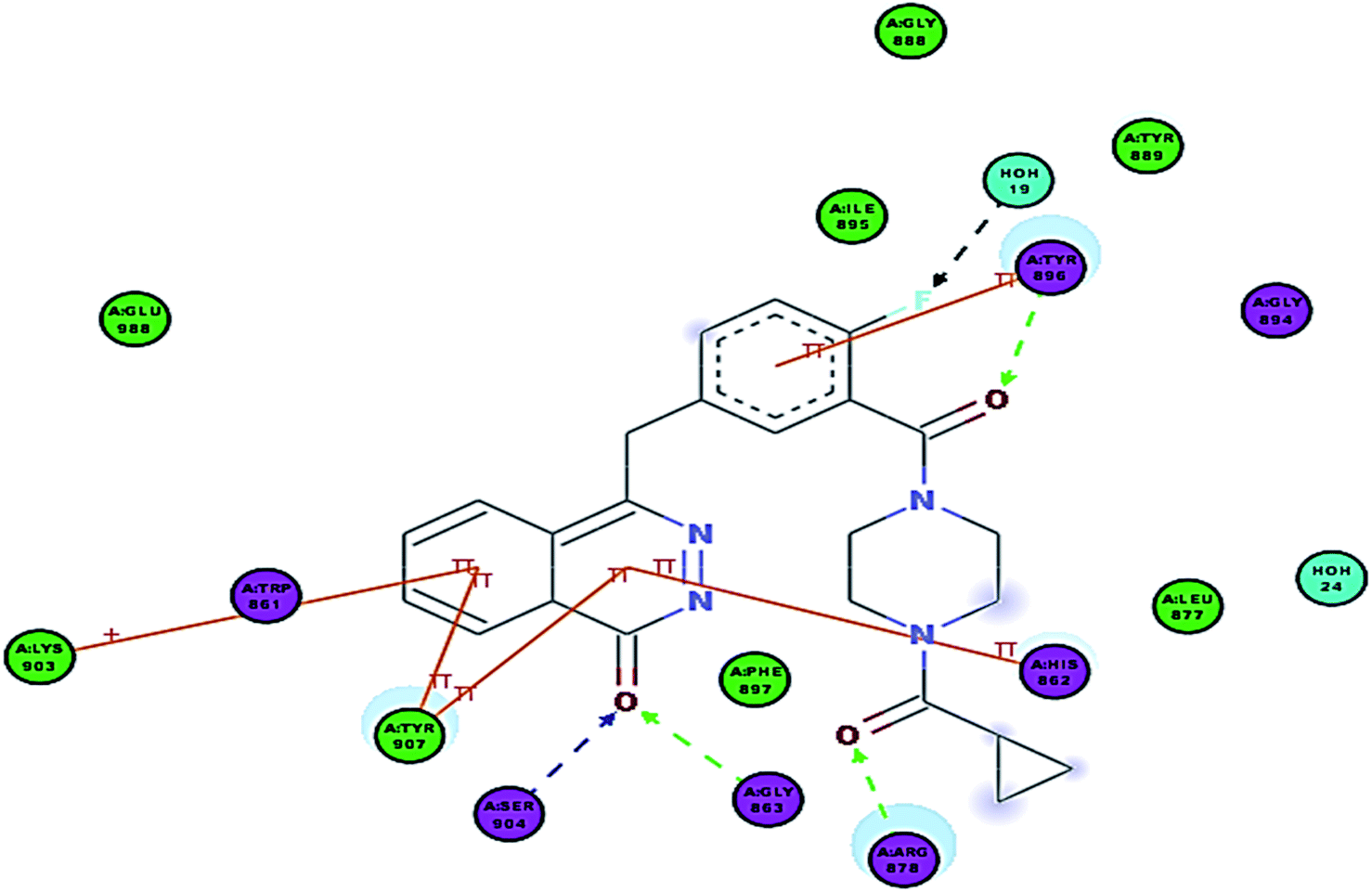 | ||
| Fig. 6 2D interaction diagram showing Olaparib (lead compound) docking pose interactions with the key amino acids in the PARP-1 active site. | ||
3.3. In silico studies
| Compd | ΔG kcal mol−1 | No. of hydrogen bonding | No. of cationic-π interactions |
|---|---|---|---|
| 5a | −51.47 | 4 (TYR907, GLY863, TYR896, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 5b | −53.18 | 4 (TYR907, HIS862, TYR896, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 5c | −51.30 | 6 (TYR907, GLY863, HIS862, TYR896, ARG878, TYR907) | 3 (TYR907, HIS862, LYS903) |
| 6a | −57.87 | 5 (GLY863, HIS862, TYR896, ARG878, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 6b | −55.23 | 5 (TYR907, GLY863, HIS862, TYR896, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 7a | −58.20 | 7 (TYR907, GLY863, HIS862, GLY888, ALA880, ARG878, ARG878) | 3 (TYR907, HIS862, ARG878) |
| 7b | −58.43 | 6 (TYR907, GLY863, HIS862, GLY888, ALA880, ARG878, ARG878) | 3 (TYR907, HIS862, ARG878) |
| 8a | −62.52 | 6 (TYR907, GLY863, HIS862, TYR896, ARG878, ASN868) | 3 (TYR907, HIS862, HIS862) |
| 8b | −57.54 | 5 (TYR907, GLY863, HIS862, TYR896, ARG878) | 3 (TYR907, HIS862, HIS862) |
| 8c | −54.18 | 5 (TYR907, GLY863, HIS862, TYR896, ARG878) | 3 (TYR907, HIS862, HIS862) |
| 9a | −61.58 | 6 (TYR907, GLY863, HIS862, GLY894, ARG878, ASN868) | 3 (TYR907, HIS862, LYS903) |
| 9b | −61.10 | 4 (TYR907, GLY863, GLY894, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 9c | −58.25 | 5 (TYR907, GLY863, HIS862, GLY894, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 12a | −59.61 | 9 (TYR907, SER904, GLY863, HIS862, GLY888, TYR889, TYR896, ARG878, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 12b | −58.01 | 8 (TYR907, SER904, GLY863, HIS862, GLY888, TYR889, TYR896, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 12c | −59.56 | 8 (TYR907, SER904, GLY863, HIS862, GLY888, TYR889, TYR896, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 13a | −53.14 | 5 (TYR907, TYR907, GLY863, HIS862, GLY888) | 3 (TYR907, HIS862, LYS903) |
| 13b | −55.98 | 6 (TYR907, TYR907, GLY863, HIS862, ALA880, ARG878) | 3 (TYR907, HIS862, LYS903) |
| 13c | −55.01 | 5 (TYR907, TYR907, GLY863, HIS862, TYR896) | 3 (TYR907, HIS862, LYS903) |
| 13d | −50.56 | 5 (TYR907, TYR907, GLY863, HIS862, TYR889) | 3 (TYR907, HIS862, LYS903) |
| Olaparib | −60.60 | 4 (GLY863, SER904, TYR896, ARG878) | 3 (TYR907, HIS862, LYS903, TYR896) |
In Olaparib, the phthalazinone scaffold occupies the NI-site and the carbonyl group of the ring interacts with Gly863 and Ser904 through characteristic hydrogen bonds, and it also shows π–π stacking interactions with Tyr907 and His862. Interestingly, the 2-fluorobenzamide linker formed a π–π interaction with Tyr896 and a hydrogen bond with the backbone of Tyr896. Also, the carbonyl on the piperazine ring formed a H-bond with Arg878. The cyclopropyl group was nicely inserted into a deep hydrophobic pocket.
Interpretation of the docking results showed that the majority of the designed compounds fit into the active site of PARP-1; most of them displayed comparable docking scores and binding modes similar to that of Olaparib and were able to reproduce the main interactions observed in Olaparib.
As expected, the quinazoline scaffold occupied the NI site, where the carbonyl group formed two hydrogen bonds with His862 and Gly863. Also, the N of the ring formed a hydrogen bond with Tyr907 in all of the synthesized compounds, except for 6a and 6b which formed a hydrogen bond with Tyr907 but via the ring sulfur. The quinazoline scaffold also showed π–π stacking interactions with Tyr907 and His862 previously observed in Olaparib. Additionally, it formed a π-cation interaction with Lys903 (Fig. 7).
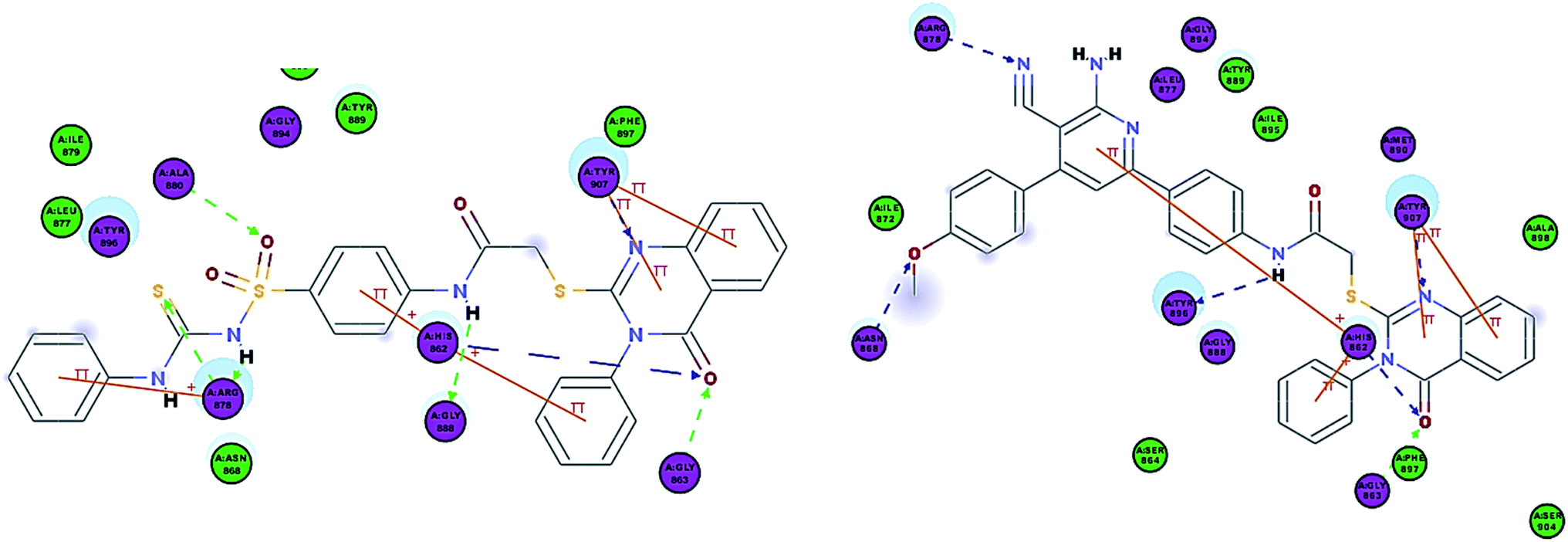 | ||
| Fig. 7 2D interaction diagram of 7b in the active site of PARP-1 and 2D interaction diagram of 8a in the active site of PARP-1. | ||
In compounds 5a–c and 6a–b, the carbonyl group of the amide formed a hydrogen bond with Tyr896 similar to that of Olaparib and their terminal groups formed a hydrogen bond with Arg878 similar to that of Olaparib. When comparing this group of compounds, 6a and 6b showed better docking energies in comparison to 5a and 5b, indicating that the substitution on the nitrogen is better than that on the sulfur of the quinazolinone ring and this was also consistent with their enzyme assay results.
Interestingly, compounds 7a and 7b showed extra interactions; the amino group forms a hydrogen bond with Gly888 and the SO group forms a hydrogen bond with Ala880 (Fig. 8), and this explained the increase in potency in comparison to 5c.
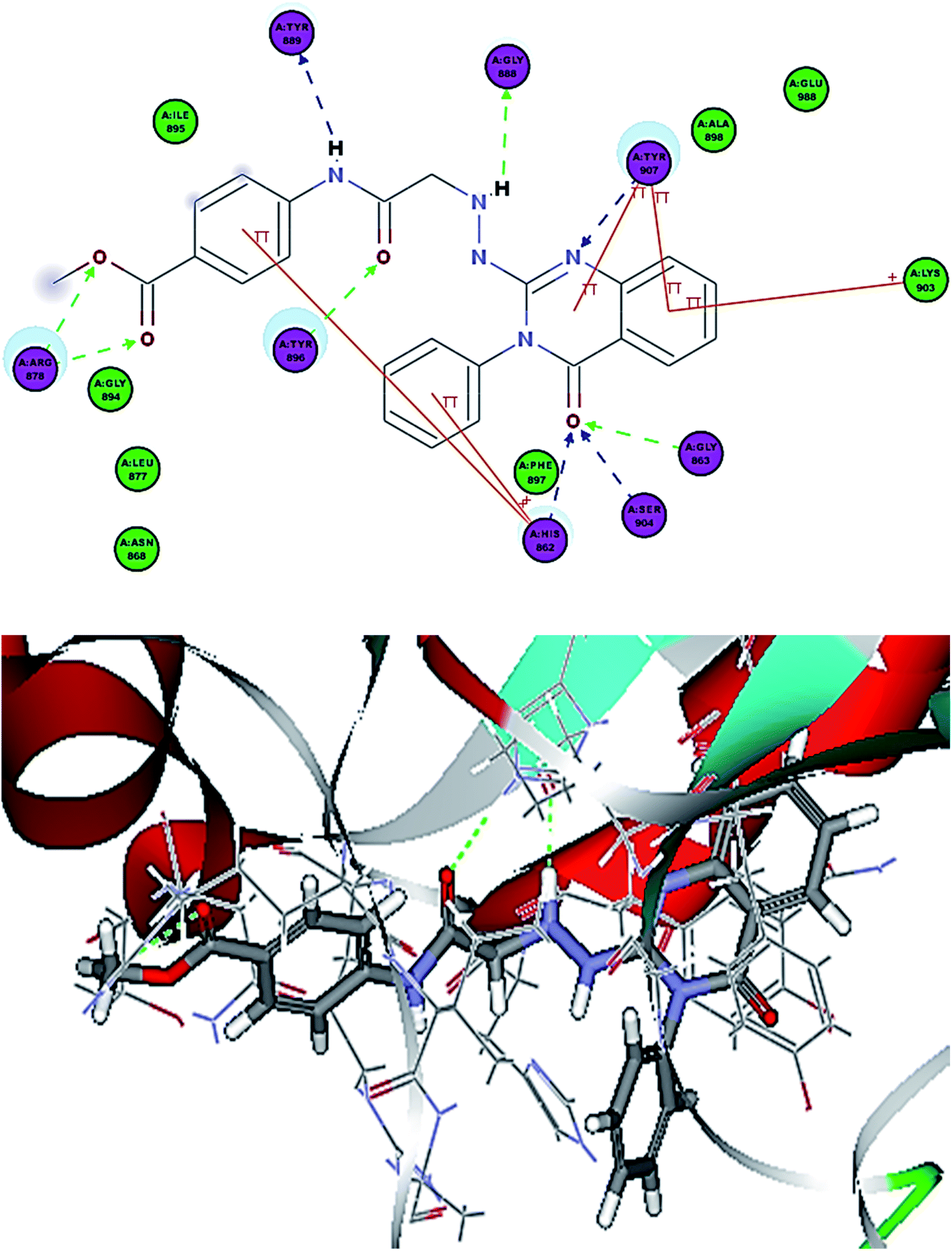 | ||
| Fig. 8 2D interaction diagram of 12a in the active site of PARP-1 and 3D interaction diagram of 12a in the active site of PARP-1. | ||
Compounds 8a–c showed the same interactions as 5a–b but Arg878 formed a hydrogen bond with the cyano group instead of the terminal substitution (Fig. 8). The methoxy group in 8a and 9a shows an extra hydrogen bond with Asn868, which might explain their higher potency than 8b–c and 9b–c, respectively, suggesting that methoxy substitution was better than chloro and nitro groups.
Compounds 12a–c showed additional interactions through hydrogen bonds between the NH of the hydrazine formed and Gly888, and between the amide NH and Tyr889. Additionally, the ester function or sulfonamide moieties form two H bonds with Arg878, which could explain the high potency of this series (Fig. 9). Compounds 13a–c showed the essential interactions with the NI site, but they lacked the hydrogen bond with the linker. Moreover, different substitutions on the terminal phenyl ring formed different interactions with various amino acids in the AD site of the enzyme. The presence of a hydroxyl group in position-4 gave the best docking score and the hydroxyl group was involved in two hydrogen bond interactions, which explains why 13b showed the best potency among this group.
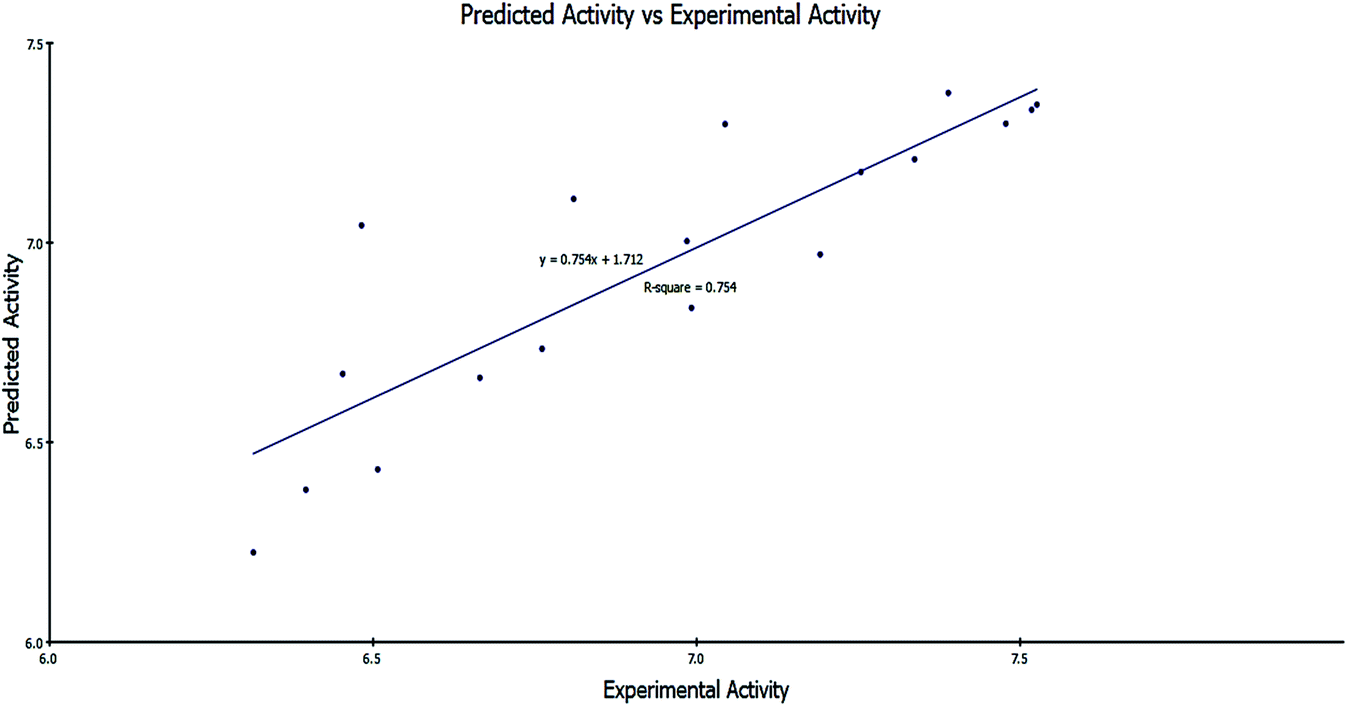 | ||
Fig. 9 Predicted activity versus experimental activity (−log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50) values of the training set according to eqn (1) (r2 = 0.754). IC50) values of the training set according to eqn (1) (r2 = 0.754). | ||
|
−logIC50 = 2.98977 + 1.06691 × CHI_V_3_C − 0.141044 × Dipole_mag + 6.03536 × Jurs_FPSA_1
| (1) |
IC50: the negative logarithmic value of the concentration required to produce 50% inhibition of MCF-7 cells. CHI_V_3_C: Kier & Hall valence-modified connectivity index. This index is a refinement of the molecular connectivity index that takes into account the electron configuration of the atoms.37 Dipole_mag: 3D electronic descriptor that indicates the strength and orientation behavior of a molecule in an electrostatic field.38 Jurs_FPSA_1: fractional charged partial surface area, which combines shape and electronic information to characterize molecules. The descriptors are calculated by mapping atomic partial charges on solvent-accessible surface areas of individual atoms.39| Compd | Experimental activity (−logIC50) |
Predicted activity (−logIC50) |
Residual |
|---|---|---|---|
| a 7a, 7b, and 13b were used for external validation through calculating their predicted activity from the QSAR model constructed using the training set. | |||
| 5a | 6.482 | 7.043 | −0.561 |
| 5b | 6.665 | 6.662 | 0.003 |
| 5c | 6.453 | 6.671 | −0.218 |
| 6a | 7.254 | 7.176 | 0.078 |
| 6b | 7.191 | 6.970 | 0.221 |
| 8a | 7.478 | 7.298 | 0.18 |
| 8b | 6.810 | 7.109 | −0.299 |
| 8c | 6.396 | 6.381 | 0.015 |
| 9a | 6.992 | 6.836 | 0.156 |
| 9b | 6.761 | 6.734 | 0.027 |
| 9c | 6.507 | 6.432 | 0.075 |
| 12a | 7.389 | 7.375 | 0.014 |
| 12b | 7.044 | 7.297 | −0.253 |
| 12c | 7.526 | 7.346 | 0.180 |
| 13a | 6.985 | 7.004 | −0.019 |
| 13c | 7.337 | 7.209 | 0.128 |
| 13d | 6.315 | 6.224 | 0.091 |
| Olaparib | 7.518 | 7.333 | 0.185 |
| 7aa | 6.886 | 6.947 | −0.061 |
| 7ba | 7.103 | 7.335 | −0.232 |
| 13ba | 7.348 | 7.196 | 0.152 |
The regression values were as follows: r2 = 0.754, q2 = 0.638 and least-squared error = 0.0403.
It was clearly illustrated that the IC50 values predicated by our QSAR model were very close to those experimentally observed, indicating that this model can be used productively for the predication of more effective hits having the same skeletal framework.
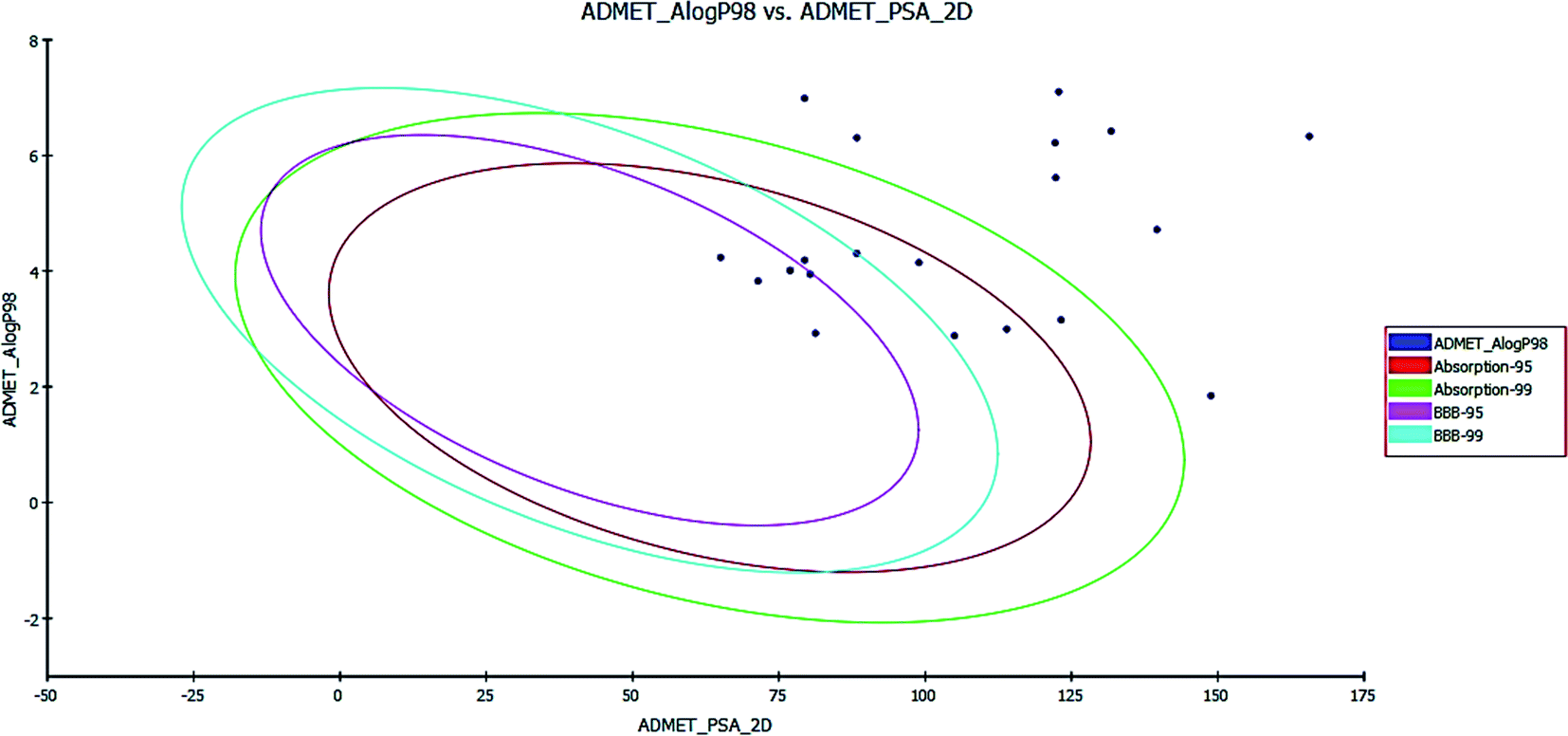 | ||
| Fig. 10 ADMET plot for the newly synthesized compounds. | ||
In the BBB plot, most of the compounds except for 5a, 5b, 6a, 6b, 13a, 13b and 13c fall outside of the 99% ellipse. Hence, these compounds may not be able to penetrate the blood brain barrier; hence, the chances of CNS side effects are predicted to be low. Meanwhile, for compounds 5a, 5b, 6a, 6b, 13a, 13b and 13c there is a probability of causing CNS side effects.
In the HIA plot, most of the compounds fell inside the 99% ellipse, and are thus estimated to have good human intestinal absorption, except for 7a, 7b, 8a, 8b, 8c, 9a, 9b, 9c and 12c, which show poor absorption. The ADME aqueous solubility level of most of the compounds was found to be 2 or 1, which indicates low aqueous solubility. The hepatotoxicity level of all compounds was 1. Hence, the compounds are predicted to possess hepatotoxicity. Further experimental studies are required to determine the hepatotoxic dose levels.
Most of the compounds are predicted as non-inhibitors of CYP2D6, and hence side effects (i.e. liver dysfunction) are not expected upon administration of these compounds, except for compounds 5a, 5b, 6b, 12a, 12b, 13a, 13b, 13c and 13d.
The plasma protein-binding model predicts whether a compound is likely to be highly bound to carrier proteins in the blood. There is a high probability that the synthesized compounds bind to plasma proteins.
PSA is a key property that has been linked to drug bioavailability. Thus, passively absorbed molecules with PSA > 140 are thought to have low bioavailability. Most of the synthesized compounds have PSA values ranging from 65.04–139.61, and thus, they are predicted to present good passive oral absorption, except for compounds 8c and 12c which had a PSA of more than 140. The calculated parameters from the ADMET study are tabulated in Table 7 (cf. ESI†).
| Cpd ID | BBB_Leva | Absorp_Levb | AQ SOlLEVc | Hepatoxd | Hepatox probe | CYP2D6f | CYP2D6Probg | PPB_Levh | AlogP98i | ADEM_PSA_2Dj |
|---|---|---|---|---|---|---|---|---|---|---|
| a Blood brain barrier level; 4 = undefined, 2 = medium penetration, 1 = high penetration.b Absorption level; 3 = very low absorption, 2 = low absorption, 1 = moderate absorption, 0 = good absorption.c Aqueous solubility level; 4 = optimal, 3 = good, 2 = low solubility, 1 = very low but soluble, 0 = extremely low.d Hepatotoxicity level; 1 = toxic, 0 = nontoxic.e Hepatotoxicity probability.f CYP2D6 inhibition; 1 = likely to inhibit, 0 = non inhibitor.g Cyp2D6 inhibition probability.h Plasma protein binding; 2 = more than 95%, 1 = more than 90%, 0 = less than 90%.i Lipophilicity descriptor; compounds must have logp value not greater than 5.0 to attain a reasonable probability of being well absorbed.j Polar surface area. |
||||||||||
| 5a | 2 | 0 | 2 | 1 | 0.841 | 1 | 0.732 | 2 | 4.305 | 88.318 |
| 5b | 2 | 0 | 2 | 1 | 0.821 | 1 | 0.712 | 2 | 4.189 | 79.388 |
| 5c | 4 | 1 | 2 | 1 | 0.827 | 0 | 0.485 | 2 | 3.154 | 123.22 |
| 6a | 2 | 0 | 2 | 1 | 0.94 | 0 | 0.495 | 2 | 3.944 | 80.347 |
| 6b | 2 | 0 | 2 | 1 | 0.927 | 1 | 0.633 | 2 | 3.828 | 71.417 |
| 7a | 4 | 2 | 2 | 1 | 0.88 | 0 | 0.297 | 2 | 4.717 | 139.61 |
| 7b | 4 | 2 | 1 | 1 | 0.874 | 0 | 0.297 | 2 | 5.616 | 122.31 |
| 8a | 4 | 2 | 1 | 1 | 0.952 | 0 | 0.346 | 2 | 6.419 | 131.75 |
| 8b | 4 | 3 | 1 | 1 | 0.953 | 0 | 0.316 | 2 | 7.1 | 122.83 |
| 8c | 4 | 3 | 1 | 1 | 0.951 | 0 | 0.316 | 2 | 6.33 | 165.64 |
| 9a | 4 | 2 | 1 | 1 | 0.821 | 0 | 0.366 | 2 | 6.305 | 88.318 |
| 9b | 4 | 2 | 1 | 1 | 0.814 | 0 | 0.475 | 2 | 6.986 | 79.388 |
| 9c | 4 | 2 | 1 | 1 | 0.834 | 0 | 0.425 | 2 | 6.216 | 122.21 |
| 12a | 4 | 0 | 2 | 1 | 0.814 | 1 | 0.613 | 2 | 2.995 | 113.93 |
| 12b | 4 | 0 | 2 | 1 | 0.834 | 1 | 0.623 | 2 | 2.88 | 105.01 |
| 12c | 4 | 2 | 2 | 1 | 0.827 | 0 | 0.475 | 2 | 1.845 | 148.84 |
| 13a | 2 | 0 | 2 | 1 | 0.953 | 1 | 0.603 | 2 | 4.011 | 76.925 |
| 13b | 2 | 0 | 2 | 1 | 0.973 | 1 | 0.742 | 2 | 4.011 | 76.925 |
| 13c | 1 | 0 | 2 | 1 | 0.933 | 1 | 0.871 | 2 | 4.236 | 65.04 |
| 13d | 4 | 0 | 2 | 1 | 0.953 | 1 | 0.524 | 2 | 4.147 | 98.933 |
4. Conclusion
In this study, a new series of quinazolinone derivatives were designed and synthesized in an eco-friendly manner using a microwave-assisted method. All of the newly synthesized compounds were evaluated for their inhibitory activity towards PARP-1 and they showed inhibitory activity with IC50 values ranging from 29.78 nM to 483.75 nM. The most active compound 12c (IC50 = 29.78 nM) showed the highest activity compared with Olaparib (IC50 = 30.27 nM).Cell cycle analysis showed that 12a and 12c caused cell growth arrest at the G2/M phase in MCF-7 cells. Also, both compounds were shown to increase the programmed apoptosis compared with the control. Moreover, molecular docking of the most active compounds into the PARP-1 active site displayed a similar binding mode to Olaparib. A computational QSAR study was performed for the synthesized compounds. Collectively, these results revealed that some of the newly synthesized compounds could serve as hit potent PARP-1 inhibitors and anticancer agents.
5. Experimental section
5.1. Chemistry
3-Phenyl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one (3). A mixture of anthranilic acid 1 (1.37 g, 10 mmol), phenyl isothiocyanate 2 (1.79 mL, 15 mmol) and triethylamine (2 mL) in ethanol (30 mL) was heated under reflux for 4 h. The obtained white precipitate, while hot, was collected by filtration and recrystallized from an ethanol/dioxane mixture (1
:1) to give quinazoline 3 as colorless crystals, mp 308–310 °C [lit.23 305–307 °C], IR (ν, cm−1): 3264, 3135 (NH), 3070 (CH aromatic), 1663 (CO). 1H NMR (DMSO-d6-D2O) δ (ppm): 7.24–7.26 (m, 1H, Ar–H), 7.31–7.47 (m, 6H, Ar–H), 7.74–7.78 (m, 1H, Ar–H), 7.92–7.94 (m, 1H, Ar–H), 13.01 (br.s, 1H, NH, exchangeable). Anal. calcd for C14H10N2OS (254.3): C, 66.12; H, 3.96; N, 11.02. Found: C, 65.91; H, 3.90; N, 11.11%.
Synthesis of the S- and N-alkylated quinazoline derivatives (5a–c and 6a–c).
(a) Conventional method. Method I. A mixture of compound 3 (5 mmol) and 2-chloro-N-(substituted phenyl)acetamide 4a–c (5 mmol), namely methyl 4-(2-chloroacetamido)benzoate, N-(4-acetylphenyl)-2-chloroacetamide and 2-chloro-N-(4-sulfamoylphenyl)acetamide, in absolute ethanol (20 mL) containing freshly prepared anhydrous sodium acetate (5 mmol) was heated under reflux for 4 h. The precipitated solid, while hot, was filtered off and recrystallized from dioxane to afford the corresponding N-alkylated products 6a–c, while the precipitate obtained after cooling the filtrate was collected and recrystallized from ethanol to give the S-alkylated products 5a–c, respectively.
Method II. An equimolar mixture of compound 3 (5 mmol) and 2-chloro-N-(substituted phenyl)acetamide 4a–c (5 mmol), namely methyl 4-(2-chloroacetamido)benzoate, N-(4-acetylphenyl)-2-chloroacetamide and 2-chloro-N-(4-sulfamoylphenyl)acetamide, in DMF (10 mL) containing two drops of triethylamine was stirred at room temperature for 10 h. The reaction mixture was poured onto ice-cooled water and acidified by dil. HCl (10%). The formed precipitate was filtered off and dried. This solid was a mixture of compounds 5a–c and 6a–c, which was separated by fractional recrystallization from ethanol to give compounds 5a–c. The insoluble part in ethanol was recrystallized from dioxane to afford compounds 6a–c.
(b) Microwave method. A mixture of compound 3 (5 mmol), the previously mentioned N-acetamido derivatives 4a–c (5 mmol), namely methyl 4-(2-chloroacetamido)benzoate, N-(4-acetylphenyl)-2-chloroacetamide and 2-chloro-N-(4-sulfamoylphenyl)acetamide, 2 mL of DMF and two drops of triethylamine as a catalyst were added to the reaction vessel of the monomodal Emrys™ Creator microwave synthesizer and allowed to react under microwave irradiation at 200–400 W power for 2–4 min. The automatic mode stirring helped with mixing and uniform heating of the reactants. The reaction vessel was cooled to room temperature. The solid mixture of compounds 5a–c and 6a–c was collected by filtration, washed with water, and separated by fractional crystallization from ethanol to give compounds 5a–c. The insoluble part in ethanol was recrystallized from dioxane to afford compounds 6a–c.
Methyl 4-(2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamido)benzoate (5a). White crystals, mp 190–192 °C, IR (ν, cm−1): 3304 (NH), 3064 (CH aromatic), 2846 (CH aliphatic), 1699 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 3.79 (s, 3H, CH3), 4.09 (s, 2H, CH2), 7.42–7.50 (m, 4H, Ar–H), 7.56–7.61 (m, 3H, Ar–H), 7.70–7.73 (dd, 2H, Ar–H, Jo = 10.8 Hz, Jm = 2 Hz), 7.76–7.80 (m, 1H, Ar–H), 7.88–7.91 (dd, 2H, Ar–H, Jo = 10.8 Hz, Jm = 2 Hz), 8.04–8.06 (dd, 1H, Ar–H, Jo = 8 Hz, Jm = 1.2 Hz), 10.68 (br.s, 1H, NH, exchangeable). MS, m/z, (%): 447 (M+ + 2), (9.03%), 246 (M+ + 1, 100%), 445 (33%). Anal. calcd for C24H19N3O4S (445.49): C, 64.71; H, 4.30; N, 9.43. Found: C, 64.55; H, 4.21; N, 9.54%.
N-(4-Acetylphenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (5b). White crystals, mp 160–162 °C, IR (ν, cm−1): 3245 (NH), 3040 (CH aromatic), 1686, 1671 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 2.47 (s, 3H, CH3), 4.10 (s, 2H, CH2), 7.42–7.50 (d, 4H, Ar–H), 7.56–7.60 (m, 3H, Ar–H), 7.72 (d, 2H, Ar–H, J = 8.8 Hz), 7.76–7.80 (m, 1H, Ar–H), 7.91 (d, 2H, Ar–H, J = 8.8 Hz), 8.04–8.06 (dd, 1H, Ar–H, Jo = 9.2 Hz, Jm = 1.2 Hz), 10.69 (br.s, 1H, NH, exchangeable). MS, m/z, (%): 429 (M+, 7.04%), 386 (27.28%), 295 (100%). Anal. calcd for C24H19N3O3S (429.49): C, 67.12; H, 4.46; N, 9.78. Found: C, 66.87; H, 4.38; N, 9.81%.
2-((4-Oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)-N-(4-sulfamoylphenyl)acetamide (5c). White crystals, mp 250–252 °C, IR (ν, cm−1): 3311, 3245 (NH), 3070 (CH aromatic), 1669 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.11 (s, 2H, CH2), 7.21–7.75 (m, 9H, Ar–H), 7.76–7.80 (m, 1H, Ar–H), 7.93 (d, 2H, Ar–H, J = 7.2 Hz), 8.04–8.06 (dd, 1H, Ar–H, Jo = 8.8 Hz, Jm = 0.8 Hz), (br.s, 2H, NH2, exchangeable), 13.02 (br.s, 1H, NH, exchangeable). Anal. calcd for C22H18N4O4S2 (466.53): C, 56.64; H, 3.89; N, 12.01. Found: C, 56.44; H, 3.80; N, 11.93%.
Methyl 4-(2-(4-oxo-3-phenyl-2-thioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamido)benzoate (6a). White crystals, mp >360 °C, yield: 25%. IR (ν, cm−1): 3414 (NH), 3054 (CH aromatic), 2926 (CH aliphatic), 1664 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 3.81 (s, 3H, CH3), 4.19 (s, 2H, CH2), 7.41–7.54 (m, 5H, Ar–H), 7.56–7.61 (m, 3H, Ar–H), 7.76–7.85 (m, 3H, Ar–H), 7.91 (d, 1H, Ar–H J = 8 Hz), 8.06 (d, 1H, Ar–H, J = 8 Hz), 11.21 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 37.9, 52.3, 119.0 (3), 119.7, 124.6, 126.3 (2), 126.8, 130.0 (3), 130.3, 130.9 (3), 135.5, 136.0, 144.1, 147.4, 157.4, 161.2, 166.6. Anal. calcd for C24H19N3O4S (445.49): C, 64.71; H, 4.30; N, 9.43. Found: C, 64.54; H, 4.22; N, 9.33%.
N-(4-Acetylphenyl)-2-(4-oxo-3-phenyl-2-thioxo-3,4-dihydroquinazolin-1(2H)-yl)acetamide (6b). White crystals, mp 220–222 °C, IR (ν, cm−1): 3238 (NH), 3040 (CH aromatic), 1687, 1671 (C
O). 1H-NMR (DMSO-d6-D2O) δ (ppm): 2.53 (s, 3H, CH3), 4.14 (s, 2H, CH2), 7.44–7.55 (m, 4H, Ar–H), 7.59–7.63 (m, 2H, Ar–H), 7.75–7.83 (m, 5H, Ar–H), 7.93 (d, 1H, Ar–H, J = 8 Hz), 8.08 (d, 1H, Ar–H, J = 8 Hz), 10.79 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 26.7, 37.4, 118.7 (3), 120.0, 125.7, 126.3 (2), 127.1, 130.0 (4), 130.3, 131.9, 135.2, 136.0, 143.6, 147.1, 156.9, 160.7, 166.9, 196.7. Anal. calcd for C24H19N3O3S (429.49): C, 67.12; H, 4.46; N, 9.78. Found: C, 66.89; H, 4.39; N, 9.67%.
2-(4-Oxo-3-phenyl-2-thioxo-3,4-dihydroquinazolin-1(2H)-yl)-N-(4-sulfamoylphenyl) acetamide (6c). White crystals, mp 264–266 °C, IR (ν, cm−1): 3317, 3215 (NH, NH2), 3074 (CH aromatic), 1671 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.33 (s, 2H, CH2), 7.26 (br.s, 2H, 2NH, exchangeable), 7.42–7.81 (m, 12H, Ar–H), 8.04–8.06 (dd, 1H, Ar–H, Jo = 8.8 Hz, Jm = 0.8 Hz), 10.85 (br.s, 1H, NH, exchangeable). Anal. calcd for C22H18N4O4S2 (466.53): C, 56.64; H, 3.89; N, 12.01. Found: C, 56.44; H, 3.80; N, 11.93%.
Reaction of 5c with phenyl isocyanate/phenyl isothiocyanate: general procedure for the synthesis of compounds 7a and b.
(a) Conventional method. A mixture of the sulfonamide derivative 5c (2 mmol) and phenyl isocyanate or phenyl isothiocyanate (2 mmol) in dioxane (20 mL) was heated under reflux for 8 h. The reaction mixture was then cooled and poured onto ice-cold water. The precipitated solid was collected and recrystallized from ethanol to produce urea/thiourea derivatives 7a and b, respectively.
(b) Microwave method. A mixture of the sulfonamide derivative 5c (2 mmol) and phenyl isocyanate or phenyl isothiocyanate (2 mmol) in DMF (2 mL) was added to the reaction vessel of the monomodal Emrys™ Creator microwave synthesizer and allowed to react under microwave irradiation at 200–400 W power for 2–4 min. The automatic mode stirring helped with mixing and uniform heating of the reactants. The reaction vessel was cooled to room temperature. The solid compound was collected by filtration, washed with water, and recrystallized from ethanol to give products 7a and b, respectively.
2-((4-Oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)-N-(4-(N-(phenylcarbamoyl) sulfamoyl)phenyl)acetamide (7a). Beige crystals, mp 220–222 °C, IR (ν, cm−1): 3330, 3286, 3194 (3NH), 3062 (CH aromatic), 1684, 1651 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.12 (s, 2H, CH2), 6.94–7.20 (m, 5H, Ar–Hphenyl ring), 7.29–7.48 (m, 6H, Ar–H), 7.57–7.80 (m, 5H, Ar–H), 7.82–8.09 (m, 2H, Ar–H quinazolinone), 9.23 (br.s, 2H, NH, exchangeable), 10.38 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 37.6, 114.4, 118.5 (3), 119.6, 119.9, 122.0 (2), 123.7, 126.3, 126.4, 127.1, 129.1, 129.2, 129.9 (3), 130.0, 130.9, 135.3, 136.0, 139.3, 140.4, 147.7, 153.2 (2), 161.2, 166.1. MS, m/z (%): 584 (M+ − 1, 60.25%), 570 (43%), 532 (52.03), 474 (43.45), 233 (69.90), 103 (100). Anal. calcd for C29H23N5O5S2 (585.65): C, 59.48; H, 3.96; N, 11.96. Found C, 59.31; H, 3.90; N, 11.88%.
2-((4-Oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)-N-(4-(N-(phenylcarbamothioyl) sulfamoyl)phenyl)acetamide (7b). Yellow crystals, mp 244–246 °C, IR (ν, cm−1): 3225, 3289, 3194 (3NH), 3062 (CH aromatic), 1649 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.19 (s, 2H, CH2), 7.43–7.59 (m, 11H, Ar–H), 7.78–7.83 (m, 3H, Ar–H), 7.90–7.92 (m, 3H, Ar–H), 8.06 (d, 1H, Ar–H, J = 8 Hz), 10.53 (br.s, 2H, 2NH, exchangeable), 11.04 (br.s, 1H, 1NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 52.3, 118.9 (3), 119.9, 124.5, 126.4 (2), 127.02 (2), 129.9 (3), 130.0 (3), 130.4, 130.7 (3), 135.3, 136.2 (2), 143.9, 147.5, 157.27, 161.1, 166.2, 167.7. Anal. calcd for C29H23N5O4S3 (601.7): C, 57.89; H, 3.85; N, 11.64. Found: C, 57.72; H, 3.77; N, 11.55%.
Synthesis of N-(4-(6-amino-5-cyano-4-(4-aryl)pyridin-2-yl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8a–c).
(a) Conventional method. A mixture of the acetyl derivative 5b (2 mmol), malononitrile (2 mmol) and the appropriate benzaldehyde derivative, namely 4-methoxy-, 4-chloro- or 4-nitrobenzaldehyde (2 mmol), and ammonium acetate (4 mmol) in absolute ethanol (20 mL) was heated under reflux for 8 h. The solid obtained upon cooling was collected and recrystallized from ethanol to produce the enaminonitrile derivatives 8a–c, respectively.
(b) Microwave method. A mixture of the acetyl derivative 5b (2 mmol), malononitrile (2 mmol) and the appropriate benzaldehyde derivative, namely 4-methoxy-, 4-chloro- or 4-nitrobenzaldehyde (2 mmol), in dioxane (4 mL) containing ammonium acetate (4 mmol) was added to the reaction vessel of the monomodal Emrys™ Creator microwave synthesizer and allowed to react under microwave irradiation at 200–400 W power for 2–4 min. The automatic mode stirring helped with mixing and uniformly heating the reactants. The reaction vessel was cooled to room temperature. The solid compound was collected by filtration, washed with water, and recrystallized from the ethanol mixture to afforded products 8a–c, respectively.
N-(4-(6-Amino-5-cyano-4-(4-methoxyphenyl)pyridin-2-yl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8a). Yellow crystals, mp 164–166 °C, IR (ν, cm−1): 3318 (NH), 3062 (CH aromatic), 2836 (CH aliphatic), 2216 (CN), 1687 (C
O); 1H NMR (DMSO-d6-D2O) δ (ppm): 3.85 (s, 3H, OCH3), 4.13 (s, 2H, CH2), 6.89–7.22 (m, 4H, Ar–H), 7.48 (br.s, 2H, NH2, exchangeable), 7.50–7.74 (m, 6H, Ar–H), 7.81–7.83 (m, 4H, Ar–H), 7.93–7.95 (d, 2H, Ar–H, J = 8 Hz), 8.08–8.12 (d, 2H, Ar–H, J = 8 Hz), 10.71 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 27.0, 55.8, 114.0 (3), 114.3, 115.1 (2), 118.7, 119.0 (2), 119.7, 112.2 (2), 126.3 (2), 126.8 (2), 130.0 (6), 130.6 (2), 131.4 (2), 136.0, 144.1 (2), 147.1 (2), 160.7, 166.4. Anal. calcd for C35H26N6O3S (615.69): C, 68.84; H, 4.29; N, 13.76. Found: C, 68.66; H, 4.20; N, 13.65%.
N-(4-(6-Amino-4-(4-chlorophenyl)-5-cyanopyridin-2-yl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8b). White crystals, mp 132–134 °C, IR (ν, cm−1): 3242, 3179 (NH), 3037 (CH aromatic), 2855 (CH aliphatic), 2226 (CN), 1686, 1671 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.12 (s, 2H, CH2), 7.45–7.53 (m, 5H, Ar–H of phenyl ring), 7.60–7.62 (m, 2H, Ar–H), 7.72–7.75 (m, 4H, Ar–H), 7.79–7.83 (m, 1H, Ar–H), 7.93–7.97 (m, 5H, Ar–H), 8.07–8.09 (d, 1H, Ar–H, J = 8 Hz), 8.55 (br.s, 2H, NH2, exchangeable), 10.70 (br.s, 1H, NH, exchangeable). MS, m/z, (%): 616 (M+ + 1, 19.83%), 614 (17.20), 605 (42.65), 156 (100%). Anal. calcd for C34H23ClN6O2S (615.11): C, 66.39; H, 3.77; N, 13.66. Found: C, 66.18; H, 3.61; N, 13.54%.
N-(4-(6-Amino-4-(4-nitrophenyl)-5-cyanopyridin-2-yl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (8c). Yellow crystals, mp 152–154 °C, IR (ν, cm−1): 3285 (NH, NH2), 2855 (CH aliphatic), 3037 (CH aromatic), 2226 (CN), 1686 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.12 (s, 2H, CH2), 7.47–7.53 (m, 5H, Ar–H of phenyl ring), 7.51–7.73 (m, 6H, Ar–H), 7.75–8.35 (m, 7H, Ar–H), 9.04 (br.s, 2H, NH2, exchangeable), 10.71 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 27.7, 89.7, 117.0, 118.7, 119.0, 119.7 (5), 124.1, 126.3 (2), 126.8 (2), 129.9, 130.6 (4), 130.3 (2), 132.5 (2), 135.2, 136.0, 143.4, 147.4, 157.4, 161.2, 166.6, 170.2, 166.95, 197.0. Anal. calcd for C34H23N7O4S (625.66): C, 65.27; H, 3.71; N, 15.67. Found: C, 65.14; H, 3.60; N, 15.70%.
Synthesis of chalcones, N-(4-(3-(4-aryl)acryloyl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio) acetamide (9a–c).
(a) Conventional method. To a solution of the acetyl derivative 5b (2 mmol) and the appropriate benzaldehyde derivatives, namely 4-methoxy-, 4-chloro- or 4-nitro-benzaldehyde (2 mmol) in ethanol (20 mL), sodium hydroxide (40%) was added under stirring. The reaction mixture was further stirred at room temperature for 8 h. The precipitated solid was collected, washed with water, dried, and recrystallized from dioxane to produce the chalcone derivatives 9a–c, respectively.
(b) Microwave method. A mixture of 5b (2 mmol) and the appropriate benzaldehyde derivatives, namely 4-methoxybenzaldehyde, 4-chlorobenzaldehyde or 4-nitrobenzaldehyde (2 mmol) in dioxane (3 mL) containing two drops of piperidine, were added to the reaction vessel of the monomodal Emrys™ Creator microwave synthesizer and allowed to react under microwave irradiation at 200–400 W power for (2–3) min. The automatic mode stirring helped with mixing and uniformly heating the reactants. The reaction vessel was cooled to room temperature. The solid compound was collected by filtration, washed with water, and recrystallized from dioxane to give products 9a–c, respectively.
N-(4-(3-(4-Methoxyphenyl)acryloyl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (9a). Yellow crystals, mp 338–340 °C, IR (ν, cm−1): 3266 (NH), 3037 (CH aromatic), 2836 (CH aliphatic), 1652 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 3.79 (s, 3H, CH3), 3.84 (s, 2H, CH2) 6.88–6.92 (d, 1H, CHC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) , J = 18 Hz), 6.99–7.12 (m, 5H, Ar–H of phenyl ring), 7.57–7.86 (m, 6H, Ar–H + 1H, CHC), 7.89–8.26 (m, 6H, Ar–H), 9.85 (br.s, 1H, NH, exchangeable). Anal. calcd for C32H25N3O4S (547.63): C, 70.18; H, 4.60; N, 7.67. Found: C, 70.02; H, 4.65; N, 7.73%.
, J = 18 Hz), 6.99–7.12 (m, 5H, Ar–H of phenyl ring), 7.57–7.86 (m, 6H, Ar–H + 1H, CHC), 7.89–8.26 (m, 6H, Ar–H), 9.85 (br.s, 1H, NH, exchangeable). Anal. calcd for C32H25N3O4S (547.63): C, 70.18; H, 4.60; N, 7.67. Found: C, 70.02; H, 4.65; N, 7.73%.
N-(4-(3-(4-Chlorophenyl)acryloyl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (9b). Yellow crystals, mp 186–187 °C (decomp.), IR (ν, cm−1): 3318 (NH), 3062 (CH aromatic), 2855 (CH aliphatic), 1677 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.11 (s, 2H, CH2), 7.23–7.35 (m, 2H, Ar–H), 7.43–7.60 (m, 5H, Ar–H + 2H, CC), 7.69–7.82 (m, 5H, Ar–H), 7.90–7.99 (m, 3H, Ar–H), 8.08 (d, 1H, Ar–H, J = 8 Hz), 8.19 (d, 1H, Ar–H, J = 8 Hz), 10.78 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 26.7, 116.3, 119.0, 119.2, 119.5, 126.5, 127.1 (2), 129.2 (4), 129.5 (2), 130.3, 130.6 (3), 131.4, 134.1, 134.4 (2), 134.9, 136.3, 136.6, 161.2, 167.1, 169.1, 169.7, 174.4, 184.2. MS, m/z, (%): 554 (M+ + 2, 48), 552 (M+, 100), 533 (89), (479, 40). Anal. calcd for C31H22ClN3O3S (552.05): C, 67.45; H, 4.02; N, 7.61. Found: C, 67.28; H, 3.89; N, 7.64%.
N-(4-(3-(4-Nitrophenyl)acryloyl)phenyl)-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamide (9c). Yellow crystals, mp 244–246 °C, IR (ν, cm−1): 3362 (NH), 2855 (CH aliphatic), 1645 (C
O). 1H-NMR (DMSO-d6-D2O) δ (ppm): 4.10 (s, 2H, CH2), 6.58–6.65 (m, 2H, Ar–H of phenyl), 7.01–8.29 (m, 15H, Ar–H + 2H, CC), 10.67 (br.s, 1H, NH, exchangeable). Anal. calcd for C31H22N4O5S (562.60): C, 66.18; H, 3.94; N, 9.96. Found: C, 65.88; H, 3.83; N, 9.84%.
Synthesis of 2-hydrazinyl-3-phenylquinazolin-4(3H)-one (11). A mixture of quinazoline derivative 5a (2mmol) and hydrazine hydrate (2 mmol, 80%) in n-butanol (20 mL) was heated under reflux for 6 h. The obtained solid after cooling the reaction mixture was collected by filtration and recrystallized from ethanol to furnish hydrazinoquinazoline 11 as colorless crystals, mp 207–209 °C, which was identical in all respects (melting point, mixed melting point, TLC, and IR) to an authentic sample prepared from refluxing a solution of quinazoline 3 and hydrazine hydrate in absolute ethanol for ∼4–5 h (until all H2S evolved).23
Synthesis of dihydroquinazoline derivatives 12a–c.
(a) Conventional method. A mixture of compound 11 (2 mmol), 2-chloro-N-acetamide derivatives, namely methyl 4-(2-chloroacetamido)benzoate, N-(4-acetylphenyl)-2-chloroacetamide or 2-chloro-N-(4-sulfamoylphenyl)acetamide (4a–c) (2 mmol), and anhydrous sodium acetate (2 mmol) in dioxane (15 mL) was heated under reflux for 6 h. The obtained product after cooling was collected and recrystallized from the appropriate solvent to acquire compounds 12a–c, respectively.
(b) Microwave method. An equimolar mixture of compound 11 and 2-chloro-N-acetamide derivatives, namely methyl 4-(2-chloroacetamido)benzoate, N-(4-acetylphenyl)-2-chloroacetamide or 2-chloro-N-(4-sulfamoyl-phenyl)acetamide (4a–c) (2 mmol) in DMF (2 mL) containing two drops of triethylamine, was added to the reaction vessel of the monomodal Emrys™ Creator microwave synthesizer and allowed to react under microwave irradiation at 200–400 W power for (2–3) min. The automatic mode stirring helped with mixing and uniformly heating the reactants. The reaction vessel was cooled to room temperature. The solid compound was collected by filtration, washed with water, and recrystallized from a suitable solvent to afford quinazoline derivatives 12a–c.
Methyl 4-(2-(2-(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)hydrazinyl)acetamido)benzoate (12a). White crystals, mp >360 °C (ethanol/dioxane, 1
:1), IR (ν, cm−1): 3420, 3285 (NH), 2855 (CH aliphatic), 1690, 1663 (CO). 1H NMR (DMSO-d6-D2O) δ (ppm): 3.79 (s, 3H, CH3), 4.14 (s, 2H, CH2), 4.67 (br.s, 1H, 1NH, exchangeable), 7.42–7.47 (m, 2H, Ar–H + 1H, NH, exchangeable), 7.51–7.58 (m, 3H, Ar–H), 7.76–7.79 (m, 5H, Ar–H), 7.87–7.89 (m, 2H, Ar–H), 8.04 (d, 1H, Ar–H, J = 8 Hz), 11.20 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 23.5, 40.3, 117.12, 118.3, 123.9 (2), 127.5 (2), 128.1 (2), 129.2 (4), 129.5 (4), 135.4 (2), 140.5, 142.7, 160.9, 176.3. Anal. calcd for C24H21N5O4 (443.46): C, 65.00; H, 4.77; N, 15.79. Found: C, 64.78; H, 4.69; N, 15.66%.
N-(4-Acetylphenyl)-2-(2-(4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)hydrazinyl)acetamide (12b). Beige crystals, mp 183–185 °C (decomp.) (ethanol), IR (ν, cm−1): 3336 (NH), 3056 (CH aromatic), 1678 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 2.55 (s, 3H, COC3), 3.44 (s, 2H, CH2), 4.32 (br.s, 1H, NH, exchangeable), 7.14–7.32 (m, 2H, Ar–H), 7.36–7.51 (m, 5H, Ar–H + 1H, NH exchangeable), 7.56–7.77 (m, 3H, Ar–H), 7.81–7.99 (m, 3H, Ar–H), 10.56 (br.s, 1H, NH, exchangeable). Anal. calcd for C24H21N5O3 (427.46): C, 67.44; H, 4.95; N, 16.38. Found: C, 67.31; H, 4.86; N, 16.40%.
2-(2-(4-Oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)hydrazinyl)-N-(4-sulfamoylphenyl)-acetamide (12c). Beige crystals, mp 274–276 °C (ethanol/dioxane, 1
:1), IR (ν, cm−1): 3317, 3217 (NH, NH2), 3068 (CH aromatic), 1672 (CO). 1H NMR (DMSO-d6-D2O) δ (ppm): 4.13 (s, 2H, CH2), 7.27 (br.s, 2H, NH2, exchangeable), 7.31 (br.s, 1H, NH, exchangeable), 7.45–7.53 (m, 5H, Ar–H of phenyl ring), 7.60–7.62 (m, 2H, Ar–H + 1H, NH, exchangeable), 7.78–7.84 (m, 5H, Ar–H), 8.09 (d, 1H, Ar–H, J = 8 Hz), 10.71 (br.s, 1H, NH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 37.9, 119.0 (2), 120.0, 126.1, 126.3, 126.5, 127.2 (3), 129.8 (2), 130.2, 130.5, 135.3, 136.0, 138.9, 142.2, 147.7, 157.0, 160.7, 166.7. Anal. calcd for C22H20N6O4S (464.50): C, 56.89; H, 4.34; N, 18.09. Found: C, 56.76; H, 4.26; N, 17.98%.
Condensation of 11 with different aromatic aldehydes.
(a) Conventional method. A solution of hydrazine derivative 11 (2 mmol) and the appropriate aromatic aldehyde, namely 2-hydroxybenzaldehyde, 4-hydroxybenzaldehyde, 2-methoxybenzaldeyde, and 4-nitrobenzaldehyde (2 mmol) in glacial acetic acid (10 mL), was heated under reflux for 30 min. The precipitated solid while heating was collected and recrystallized from dioxane to afford the corresponding hydrazone derivatives 13a–d, respectively.
(b) Microwave method. A mixture of hydrazine derivative 11 (2 mmol) and the appropriate aromatic aldehyde, namely 2-hydroxybenzaldehyde, 4-hydroxybenzaldehyde, 2-methoxybenzaldeyde, or 4-nitrobenzaldehyde (2 mmol) in glacial acetic acid (3 mL), was added to the reaction vessel of the monomodal Emrys™ Creator microwave synthesizer and allowed to react under microwave irradiation at 200–400 W power for 1–2 min. The automatic mode stirring helped with mixing and uniformly heating the reactants. The reaction vessel was cooled to room temperature. The solid compound was collected by filtration, washed with water, and recrystallized from dioxane to achieve products 13a–d, respectively.
2-(2-(2-Hydroxybenzylidene)hydrazinyl)-3-phenylquinazolin-4(3H)-one (13a). Yellow crystals, mp 341–343 °C, IR (ν, cm−1): 3248, 3221 (NH), 3036 (CH aromatic), 2872 (CH aliphatic), 1662 (C
O). 1H NMR (DMSO-d6-D2O) δ (ppm): 6.83–7.90 (m, 2H, Ar–H), 7.15–7.34 (m, 3H, Ar–H), 7.49–7.56 (m, 4H, Ar–H), 7.64–7.67 (m, 2H, Ar–H), 7.82 (d, 1H, Ar–H, J = 8 Hz), 7.91–7.97 (m, 1H, Ar–H), 8.22 (s, 1H, NC), 10.25 (br.s, 1H, NH, exchangeable), 10.67 (br.s, 1H, OH, exchangeable). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 114.8, 116.2, 116.4, 119.6, 120.7, 122.3, 124.9, 127.8, 128.3, 129.3 (2), 129.4, 129.7, 131.46, 135.5, 137.0, 140.3, 150.7, 153.7, 157.2, 161.0. MS, m/z, (%): 357 (M+ + 1, 44%), 356 (M+, 100%), 355, (76). Anal. calcd for C21H16N4O2 (356.39): C, 70.77; H, 4.53; N, 15.72. Found: C, 70.45; H, 4.42; N, 15.61%.
2-(2-(4-Hydroxybenzylidene)hydrazinyl)-3-phenylquinazolin-4(3H)-one (13b). White crystals, mp 320–322 °C, IR (ν, cm−1): 3322 (NH&OH), 3059 (CH aromatic), 2925 (CH aliphatic), 1700 (C
O), 1620 (CN). 1H NMR (DMSO-d6-D2O) δ (ppm): 6.79 (d, 2H, Ar–H C6H4OH, J = 8.4 Hz), 7.13–7.17 (m, 1H, Ar–H of phenyl group), 7.32 (d, 2H, Ar–H, C6H4OH, J = 8.4 Hz), 7.40–7.76 (m, 7H, Ar–H), 7.90 (d, 1H, Ar–H, J = 8.4 Hz), 7.95 (s, 1H, NC), 9.83 (br.s, 1H, NH, exchangeable), 10.47 (br.s, 1H, OH, exchangeable). Anal. calcd for C21H16N4O2 (356.39): C, 70.77; H, 4.53; N, 15.72; Found: C, 70.59; H, 4.60; N, 15.60%.
2-(2-(2-Methoxybenzylidene)hydrazinyl)-3-phenylquinazolin-4(3H)-one (13c). White crystals, mp 274–276 °C, IR (ν, cm−1): 3339 (NH), 3054 (CH aromatic), 2834 (CH aliphatic), 1686 (C
O), 1613 (CN). 1H NMR (DMSO-d6-D2O) δ (ppm): 3.79 (s, 3H, OCH3), 7.00–7.05 (m, 2H, Ar–H), 7.16–7.18 (m, 1H, Ar–H), 7.32–7.52 (m, 4H, Ar–H), 7.68 (m, 2H, Ar–H), 7.91 (d, 2H, Ar–H, J = 8 Hz), 8.28 (s, 1H, NC), 8.29 (d, 2H, Ar–H, J = 8 Hz), 10.59 (br.s, 1H, NH, exchangeable). Anal. calcd for C22H18N4O2 (370.41): C, 71.34; H, 4.90; N, 15.13; Found: C, 71.21; H, 4.82; N, 15.06%.
2-(2-(4-Nitrobenzylidene)hydrazinyl)-3-phenylquinazolin-4(3H)-one (13d). Orange crystals, mp 330–332 °C, IR (ν, cm−1): 3202 (NH), 3061 (CH aromatic), 2925 (CH aliphatic), 1665 (C
O), 1620 (CN). 1H NMR (DMSO-d6-D2O) δ (ppm): 7.20–7.24 (m, 1H, Ar–H), 7.36 (d, 2H, Ar–H, C6H4–NO2, J = 8 Hz), 7.41–7.53 (m, 5H, Ar–H), 7.70–7.76 (m, 2H, Ar–H), 7.95 (d, 1H, Ar–H, J = 8 Hz), 8.18–8.27 (m, 2H, Ar–H + 1H, NC), 10.59 (br.s, 1H, NH, exchangeable). Anal. calcd for C21H15N5O3 (385.38): C, 65.45; H, 3.92; N, 18.17. Found: C, 65.22; H, 3.83; N, 18.10%.
5.2. Bioassay
Assay procedure. PARP-1 enzyme inhibition activity was measured using a colorimetric 96-well PARP-1 assay kit (catalog no. 80580) (BPS Bioscience), according to the manufacturer's protocol. Briefly, the histone mixture was diluted 1
:5 with 1× PBS, and 50 μL of histone solution was added to each well and incubated at 4 °C overnight. The plate was washed three times using 200 μL PBST buffer (1× PBS containing 0.05% Tween-20) per well. Liquid was removed from the wells by tapping the strip wells on clean paper towels. To each well, 200 μL of blocking buffer was added, followed by 60–90 min incubation at room temperature. Then, 25 μL of PARP master mixture (consisting of 2.5 μL 10× PARP buffer + 2.5 μL 10× PARP assay mixture + 5 μL activated DNA + 15 μL distilled water) was added to each well. Olaparib was used as a positive control. 5 μL of inhibitor solution was added to each well labeled as “Test Inhibitor”. For the “Positive Control” and “Blank”, 5 μL of the same solution without inhibitor was added. 1× PARP buffer was prepared by adding 1 part of 10× PARP buffer to 9 parts H2O (v/v), and 20 μL of 1× PARP buffer was added to the wells designated as “Blank”. The amount of PARP-1 required for the assay was then calculated. The reaction was initiated by adding 20 μL of diluted PARP-1 enzyme to the wells designated “Positive Control” and “Test Inhibitor Control”. The strip wells were incubated at room temperature for 1 hour. The strip wells were then washed three times with 200 μL of PBST buffer. Then, 50 μL of 50 times diluted Streptavidin-HRP with blocking buffer was added to each well, and the strips were further incubated at room temperature for 30 min. After washing the wells three times with 200 μL of PBST buffer, HRP colorimetric substrate was added to each well and the plate was incubated at room temperature until a blue color was developed in the positive control well. Then, the reaction was quenched with 100 mL per well of 2 M sulfuric acid. Carrier solvent (DMSO 5%, v/v) was used as a negative control. All compounds including Olaparib were tested at 5 concentrations (100, 10, 1, 0.1, and 0.01 μM). The optical density (OD) of each well was measured spectrophotometrically at 450 nm with an ELISA microplate reader (ChroMate-4300, FL, USA). The IC50 values were calculated according to the equation for Boltzmann sigmoidal concentration–response curves using the nonlinear regression fitting model (GraphPad Prism Version 8). All assays were performed in triplicate in 3 repeated experiments. The obtained data are presented means ± standard error of the means from 3 independent repeats (n = 3).
5.3. In silico studies
The genetic function approximation model was employed to search for optimal QSAR models that combine high quality binding pharmacophores with other molecular descriptors and are capable of correlating bioactivity variation across the used training set collection. The trials were held while changing the independent properties until the best model with the least variables was obtained. The QSAR model was validated employing leave one-out cross-validation, r2 (squared correlation coefficient value), and external validation using compounds 7a, 7b and 13b, as well as residuals between the predicted and experimental activity of the training and test set.
Conflicts of interest
The authors declare no conflict of interest, financial or otherwise.Acknowledgements
The authors would like to extend their sincere appreciation to Dr Ahmed Esmat from the Department of Pharmacology, Faculty of Pharmacy, ASU for his help in revising the biology. We also thank the center for drug discovery and development research at ASU for performing NMR spectral analysis. In addition, we gratefully acknowledge the scientific research team of VASERA CO. Giza, Egypt, for providing laboratory facilities for biological activity.References
- M. Rouleau, A. Patel, M. J. Hendzel, S. H. Kaufmann and G. G. Poirier, Nat. Rev. Canc., 2010, 10, 293–301 CrossRef CAS PubMed.
- E. A. Comen and M. Robson. Oncology, Williston Park, N.Y., 2010, vol. 24, pp. 55–62 Search PubMed.
- F. J. Bock and P. Chang, New directions in poly(ADP-ribose) polymerase biology, FEBS J., 2016, 83, 4017–4031 CrossRef PubMed.
- H. E. Bryant, N. Schultz, H. D. Thomas, K. M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, N. J. Curtin and T. Helleday, Nature, 2005, 434, 913–917 CrossRef CAS PubMed.
- T. A. Yap, S. K. Sandhu, C. P. Carden and J. S. de Bono, Ca-Cancer J. Clin., 2011, 61, 31–49 CrossRef PubMed.
- R. Scully, Breast Cancer Res., 2000, 2, 324–330 CrossRef CAS PubMed.
- N. Curtin, PARP inhibitors for anticancer therapy, Biochem. Soc. Trans., 2014, 42, 82–88 CrossRef CAS PubMed.
- Y. Nonomiya, K. Noguchi, K. Katayama and Y. Sugimoto, Biochem. Biophys. Res. Commun., 2019, 510, 501–507 CrossRef CAS PubMed.
- B. Sahu, A. Narota and A. S. Naura, Eur. J. Pharmacol., 2020, 87715, 173091 CrossRef PubMed.
- M. M. AlHilli, M. A. Becker and P. Haluska, J. Gynecol. Oncol., 2016, 143, 379–388 CrossRef CAS PubMed.
- A. C. Houts, T. Olufade, R. Shenolikar, M. S. Walker and L. S. Schwartzberg, Cancer Treat. Res. Commun., 2019, 19, 100121 CrossRef PubMed.
- L. Lehtio, A. S. Jemth, R. Collins, O. Loseva, A. Johansson, N. Markova, M. Hammarstrom, A. Flores, L. Holmberg-Schiavone, J. Weigelt, T. Helleday, H. Schuler and T. Karlberg, J. Med. Chem., 2009, 52, 3108–3111 CrossRef CAS PubMed.
- Y. Q. Wang, P. Y. Wang, Y. T. Wang, G. F. Yang, A. Zhang and Z. H. Miao, J. Med. Chem., 2016, 59, 9575–9598 CrossRef CAS PubMed.
- P. G. Jain and B. D. Patel, Eur. J. Med. Chem., 2019, 165, 198–215 CrossRef CAS PubMed.
- X. Song, X. Han, F. Yu, X. Zhang, L. Chen and C. Lv, Theranostics, 2018, 8(8), 2217–2228 CrossRef CAS PubMed.
- I. A. Shagufta, RSC Med. Chem., 2017, 8, 871 RSC.
- J. Zhou, M. Ji, H. Yao, R. Cao, H. Zhao, X. Wang, X. Chen and B. Xu, Org. Biomol. Chem., 2018, 16, 3189 RSC.
- H. Yao, M. Ji, Z. Zhu, J. Zhou, R. Cao, X. Chen and B. Xu, Bioorg. Med. Chem., 2015, 23, 681–693 CrossRef CAS PubMed.
- I. H. Eissa, A. M. El-Naggar and M. A. El-Hashash, Bioorg. Chem., 2016, 67, 43–56 CrossRef CAS PubMed.
- S. K. Ramadan and H. A. Sallam, J. Heterocycl. Chem., 2018, 55, 1942–1954 CrossRef CAS.
- (a) H. Yan, X.-Q. Xiao, R. C. Hider and Y. Ma, Front. Chem., 2019, 7, 584 CrossRef CAS PubMed; (b) K. A. M. Abouzid, G. H. Al-Ansary and A. M. El-Naggar, Eur. J. Med. Chem., 2017, 134, 357–365 CrossRef CAS PubMed.
- (a) M. Haghighat, F. Shirini and M. Golshekan, J. Mol. Struct., 2018, 1171, 168–178 CrossRef CAS; (b) A. M. El-Naggar, A. K. Khalil, H. M. Zeidan and W. M. El-Sayed, Anti-Cancer Agents Med. Chem., 2017, 17, 1644–1651 CAS.
- (a) A. M. Alafeefy, M. Ceruso, A. S. Al-Tamimi, S. D. Prete, C. Capasso and C. T. Supuran, Bioorg. Med. Chem., 2014, 22(19), 5133–5140 CrossRef CAS PubMed; (b) S. K. Pandey, A. Singh and A. Singh, Eur. J. Med. Chem., 2009, 44, 1188–1197 CrossRef CAS PubMed.
- T. S. Straub, Tetrahedron Lett., 1995, 36, 663–664 CrossRef CAS.
- S. Sandler and W. Karo, in Organic Functional Group Preparations, 1972, vol. 3, p. 372 Search PubMed.
- E. D. Bergman, L. Ginsibm and R. Pappo, Org. React., 1959, 10, 179 Search PubMed.
- J. F. Ballesteros, M. J. Sanz, A. Ubeda, M. A. Miranda, S. Iborra, M. Paya and M. Alcaraz, J. Med. Chem., 1995, 38, 2794–2797 CrossRef CAS PubMed.
- M. L. Go, X. Wu and X. L. Liu, Curr. Med. Chem., 2005, 12, 483–499 CrossRef CAS PubMed.
- V. K. Mukerjee, A. K. Prased, A. G. Raj, M. E. Brakhe, C. E. Olsen, S. C. Jain and V. S. Parmar, Bioorg. Med. Chem., 2001, 9, 337–345 CrossRef.
- U. M. Liu, P. Wilairat, S. L. Croft, A. L. Tan and M. Go, Bioorg. Med. Chem., 2003, 11, 2729–2738 CrossRef.
- P. M. Sivakumar, S. K. GeethaBabu and D. Mukesh, Chem. Pharm. Bull., 2007, 55, 44 CrossRef CAS PubMed.
- N. Tiwari, B. Dwivedi, K. F. Nizamuddin, Y. Nakanshi and K. H. Lee, Bioorg. Med. Chem., 2000, 10, 699–701 CrossRef.
- S. Ducki, R. Forrest, J. A. Hadfield, A. Kendall, N. J. Lawrence, A. T. Mc-Gown and D. Rennison, Bioorg. Med. Chem., 1998, 8, 1051 CrossRef CAS.
- J. Wang and M. J. Lenardo, J. Cell Sci., 2000, 113, 753–757 CAS.
- J. M. Brown and B. G. Wouters, Cancer Res., 1999, 59, 1391–1399 CAS.
- (a) Y. Huang, F. Yu, J. Wang and L. Chen, Anal. Chem., 2016, 88(7), 4122–4129 CrossRef CAS PubMed; (b) K. K.-W. Lo, T. K.-M. Lee, J. S.-Y. Lau, W.-L. Poon and S.-H. Cheng, Inorg. Chem., 2008, 47, 200–208 CrossRef CAS PubMed; (c) W. Zhang, Y. Wang, X. Sun, W. Wang and L. Chen, Nanoscale, 2014, 6, 14514–14522 RSC.
- J. L. Faulon and A. Bender, Handbook of Chemoinformatics Algorithms, CRC Press, 2010 Search PubMed.
- R. F. George, N. S. M. Ismail, J. Stawinski and A. S. Girgis, Eur. J. Med. Chem., 2013, 68, 339–351 CrossRef CAS PubMed.
- A. S. Girgis, J. Stawinski, N. S. M. Ismail and H. Farag, Eur. J. Med. Chem., 2012, 47, 312–322 CrossRef CAS.
- L. B. Kier and L. H. Hall, in Molecular Connectivity in Structure-Activity Analysis, Chemometrics Series, ed. D. D. Bawden, Research Studies Press Ltd, New York, 1985, vol. 9 Search PubMed.
- C. J. F. Bottcher, Theory of Electric Polarization, Elsevier Press: Amsterdam, 1973 Search PubMed.
- D. T. Stanton and P. C. Jurs, Anal. Chem., 1990, 62, 2323–2329 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra05943a |
| This journal is © The Royal Society of Chemistry 2020 |