
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Translational incorporation of modified phenylalanines and tyrosines during cell-free protein synthesis†
Zhongqiang Wang *abc and
Hayden Matthews*d
*abc and
Hayden Matthews*d
aKey Laboratory of Biocatalysis & Chiral Drug Synthesis of Guizhou Province, Generic Drug Research Center of Guizhou Province, School of Pharmacy, Zunyi Medical University, Zunyi, 563000, China. E-mail: zqwang@zmu.edu.cn
bKey Laboratory of Basic Pharmacology of Ministry of Education and Joint International Research Laboratory of Ethnomedicine of Ministry of Education, Zunyi Medical University, Zunyi, 563000, China
cResearch School of Chemistry, Australian National University, 137 Sullivans Creek Road, Acton ACT 2601, Australia
dAustralian National University Medical School, 54 Mills Road, Acton ACT 2601, Australia. E-mail: Hayden.Matthews@anu.edu.au
First published on 18th March 2020
Abstract
Inherent promiscuity of bacterial translation is demonstrated by mass spectrometric quantification of the translational incorporation of ring-substituted phenylalanines and tyrosines bearing fluoro-, hydroxyl-, methyl-, chloro- and nitro-groups in an E. coli-derived cell-free system. Competitive studies using the cell-free system show that the aminoacyl-tRNA synthetases (aaRS) have at least two orders of magnitude higher specificity for the native substrate over these structural analogues, which correlates with studies on the purified synthetase.
Introduction
Protein synthesis is a strictly regulated biosynthetic pathway. The remarkable substrate specificity exhibited by aminoacyl-tRNA synthetases (aaRSs),1a a family of enzymes that catalyze the aminoacylation of amino acids with the cognate tRNA, is important for the high degree of accuracy.1a However, examples of non-canonical amino acids mistakenly incorporated into proteins via a natural biosynthetic pathway have been reported,2 indicating that there is a limit to aaRS with regard to distinguishing substrates with structures slightly varied from the canonical amino acid. Here, studies probing the substrate range of wild-type E. coli phenylalanyl-tRNA synthetase (PheRS) and tyrosyl-tRNA synthetase (TyrRS) through the expression of protein with structural analogues of L-phenylalanine (Phe) and L-tyrosine (Tyr) (Fig. 1), respectively, are described. The two aaRSs are known for their ability to recognize cognate substrates that differ by only a hydroxyl group at the para position of the substrate phenyl ring.1 Some non-canonical amino acids display quite diverse physiological and pharmaceutical activity,3,4 whereas others are involved in many regards, such as the use for biological markers,4 altering protein properties5 and the production of new biological materials.6 Further, some hydroxylated derivatives are found to be the most abundant modified amino acids bound to proteins that are commonly detected in pathological tissues, and their misincorporation has been associated with a wide variety of pathological conditions such as aging,7 atherosclerosis,4,8 cataractogenesis,4,9 myocardial ischaemia and reperfusion10,11 and neurodegenerative disorders such as Parkinson's diseases.12,26a | ||
| Fig. 1 Amino acids used in this study. | ||
The aminoacylation reaction catalyzed by aaRSs is the first step in protein synthesis and is accomplished via two stages.1 The amino acid first uses adenosine triphosphate (ATP) to form an aminoacyl-adenylate along with the release of pyrophosphate, followed by transfer of the amino acid to the terminal adenosine of the cognate tRNA.1 This has been identified as a rate determining step for both phenylalanyl-tRNA synthetase and tyrosyl-tRNA synthetase, with initial rates of transfer of the amino acid from aminoacyladenylate to cognate tRNA similar to the overall turnover number for the steady state aminoacylation of tRNA.14c The aminoacylation is a key point in translational quality control as it provides the link between an amino acid, the tRNA anticodon and mRNA codon to enable accurate translation. Under physiological conditions, the estimated average error rate of amino acid misaminoacylation by aaRSs is 10−4.13 For some aaRSs, the accurate amino acid selection is accomplished mainly at their active sites, discriminating non-cognate amino acids on steric grounds. In other cases, there is an additional proofreading mechanism assisting the selection,14 which occurs at a site referred to as an “editing site”.14b The latter is known as “double-sieve amino acid selection”.14 While protein synthesis using non-canonical amino acids is an indirect measure of the aaRS activity, work presented here was aimed to investigate the enzyme in a more direct manner, by evaluating the interactions between the non-canonical amino acids and the aaRS through an aminoacylation reaction.
Cell-free expression systems use a cell extract, which is partially purified and supplemented with the target DNA, amino acids and other ingredients, to enable in vitro protein synthesis in a controlled manner. The cell-free systems allow substitution of a standard amino acid with an unusual one, therefore can bias the competition in favor of the unusual amino acids. By this technique, incorporation of chloro,15a,b dehydro15c and hydroxyl17a amino acids has been reported. Here, an E. coli-derived cell-free system was used to study the modified phenylalanines 1–14 and tyrosines 15–19 (Fig. 1). In these amino acid analogue systems the volume of the substituent on the phenyl ring of non-canonical amino acids is varying, and so is the hydrophobicity. We investigated how the size and hydrophobicity of substituent affect the incorporation of non-canonical amino acids. In earlier reports, translational incorporation of 5a–c into proteins as substitutes for Phe,16 and 15a, 16a, 17a and 19a for Tyr17 have been described. However, the incorporation levels of many of the amino acids and the extent to which they compete with the natural substrate have not been determined.
Results and discussion
E. coli cell-free protein synthesis with Phe analogues
It is known that PheRS is highly selective for Phe over Tyr.1 However, Tyr isomers m-Tyr (1b) and o-Tyr (1c), and well as 3,4-dihydroxyphenylalanine (3,4-DOPA, 16a) with two OH groups on the ring of the substrate, were reportedly activated18a–c and incorporated18d–f into proteins by PheRSs from various organisms. These suggest that, although PheRS has evolved to distinguish between Phe and Tyr primarily through recognition of the OH group at para position, it imparts far lower selectivity against the OH at other positions on the substrate ring.E. coli peptidyl-Pro cis–trans isomerase B, which has been extensively studied,19 with a hexahistidine tag attached at the N-terminus (His6-PpiB) was expressed as a test protein. It contains 12 and 3 sites for Phe and Tyr, respectively.19 SDS-PAGE analysis of the synthesized His6-PpiB shows that protein was produced when all components required for protein synthesis were supplied (Fig. 2, with DNA and Phe). Mass spectral analysis (Fig. 2d) confirmed that it is wild-type protein, and corresponds to the pair of the predominant peaks. The presence of two signals in mass spectra is due to incomplete deformylation of PpiB by deformylase in the E. coli S30 extract.17a By comparison, only a trace of protein was produced when Phe was excluded from the reaction mixture (Fig. 2, with DNA but no Phe). Mass spectral analysis (data not shown) showed that it is wild-type His6-PpiB, suggesting that the production of background protein was due to the presence of traces of Phe during cell-free reaction. This experiment also confirms that Tyr is not incorporated into His6-PpiB in place of Phe, as the reaction mixture contained a typical amount of Tyr (1.0 mM) for cell-free synthesis. No protein band corresponding to His6-PpiB was observed when the DNA plasmid was not added.
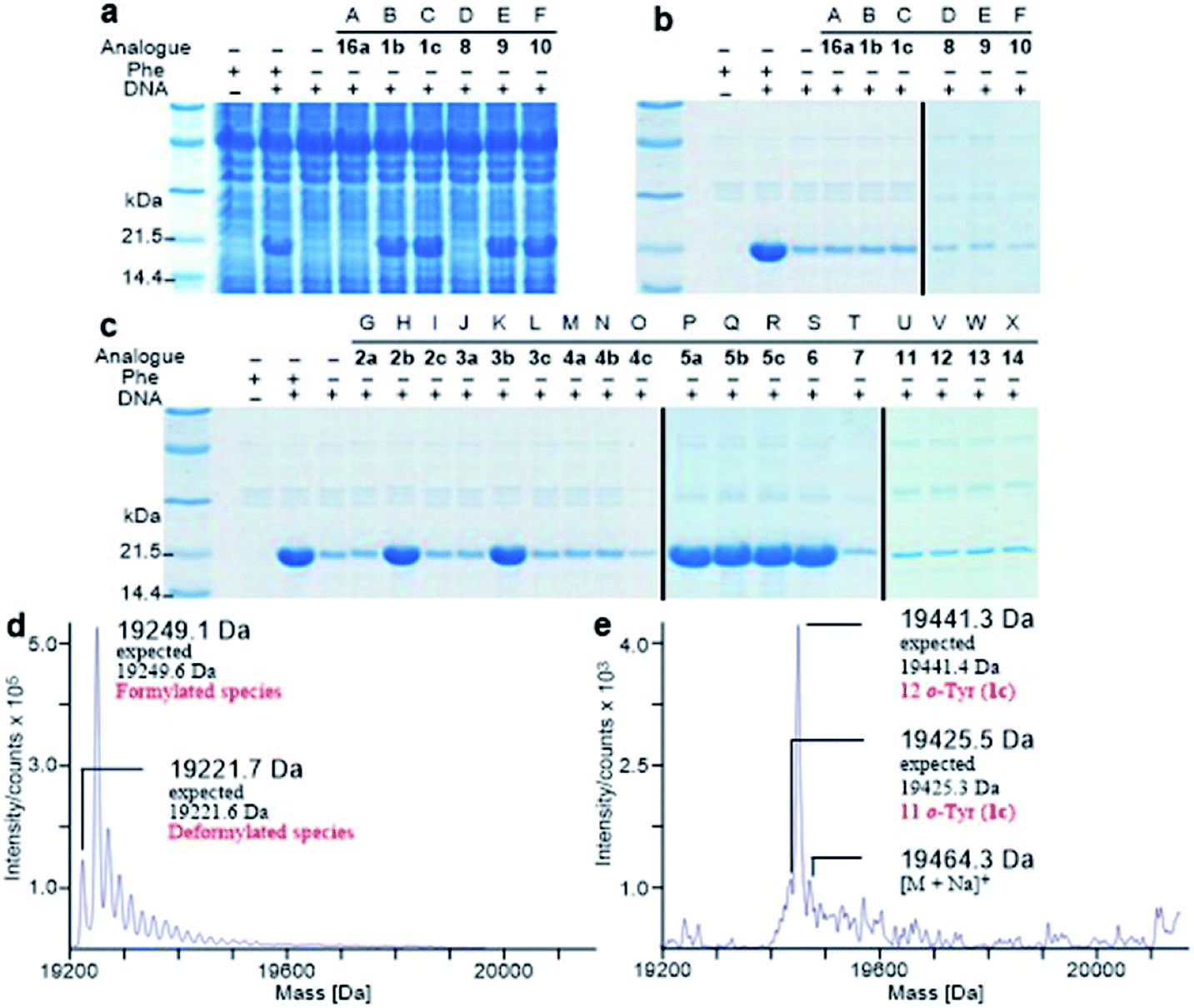 | ||
| Fig. 2 Analysis of synthesized His6-PpiB. (a) SDS-PAGE analysis before purification; (b and c) SDS-PAGE analysis after purification. Lanes A–X: no Phe but with 16a, 1b, 1c, 8, 9, 10, 2a, 2b, 2c, 3a, 3b, 3c, 4a, 4b, 4c, 5a, 5b, 5c, 6, 7, 11, 12, 13 or 14. (d and e) Mass spectral analysis of wild-type His6-PpiB and His6-PpiB synthesized in the presence of 1c. | ||
Protein expression was efficient when Phe was substituted with 1b,c (Fig. 2a, lanes B and C), reaching almost wild-type levels. Interestingly, the expressed proteins precipitated during purification (Fig. 2b, lanes B and C). This hinted at the possible effects the non-standard amino acid can have on protein structural stability, in particular given the abundance in the protein (12 Phe sites). To ensure that the tested amino acids were indeed incorporated into the protein, the small amounts of isolated protein were analyzed by mass spectrometry (Fig. 2e). Given the presence of peaks mainly corresponding to completely substituted protein species, the average level of substitution of 1c for Phe was concluded to be greater than 95%, whereas a 90% was given for 1b (ESI Fig. S1†). This incorporation level was assessed semiquantitatively, by following a method described in an earlier report,17a through a comparison of the height of the peaks corresponding to 12, 11 and 10 substitutions in the mass spectrum (Fig. 2e). The amount of His6-PpiB produced with 16a (Fig. 2a and b, lane A) is comparable to that produced in the absence of Phe, suggesting that 16a does not incorporate instead of Phe. The mass spectra of this protein (data not supplied) showed wild-type His6-PpiB.
Subsequently, Phe derivatives containing Me, Cl and NO2 substituents including mono-substituted 2a–c, 3a–c, 4a–c and di-substituted 8–10, as well as para-substituted derivatives 11–14, were studied in an analogous manner. Of these, protein was only produced with the meta isoforms, including mono-derivatized 2b and 3b (Fig. 2c, lanes H and K), and di-derivatized 9 and 10 (Fig. 2a and b, lanes E and F), suggesting greater flexibility around that position at the synthetase active site. Interestingly, the proteins synthesized with 2b and 3b remained soluble and were successfully isolated without apparent loss of yield, whereas in the cases of 9 and 10, white precipitates were observed after the completion of reaction. Consequently, samples of 9 and 10 for mass spectral analysis were prepared with two methods, by chromatographic purification of the supernatant, which resulted in neither protein bands in SDS-PAGE nor signals in the expecting region in mass spectra, and by solubilizing the insoluble part with buffer containing a high concentration of urea, which showed a high ratio of signal-to-noise in the mass spectra (ESI Fig. S8 and S9†). This suggests that the insoluble part was mainly His6-PpiB containing 9 and 10. The expression was then repeated with derivatives bearing one or more fluorines on the aromatic ring, of which all but the pentafluorophenylalanine (7) were incorporated into the expressed His6-PpiB (Fig. 2c, lanes P–S). In all successful cases soluble proteins were obtained and the incorporation of intact amino acid was verified by mass spectrometry (ESI Fig. S4–S7†). Incorporation of the fluorides 5a–c as substitutes for Phe using whole cell-based methods has been reported previously.16 The results obtained here are consistent with previous studies and confirm that the incorporation will occur in a cell-free system.
The reported crystal structure of the E. coli PheRS–Phe complex (Fig. 3a) indicates that the active site of E. coli PheRS is a mainly hydrophobic pocket with stringent steric requirements for the phenyl ring.27 The Me group of alanine at position 294 (Ala294) provides a steric constraint at the para position, which has been proposed as the primary factor assuring the high degree of selection of Phe over Tyr (Fig. 3a1). Previous mutagenesis studies have shown that substituting alanine with glycine (Ala294Gly) expands the volume of the active site and thus relaxes the substrate specificity of E. coli PheRS. As a consequence, Tyr and derivatives of Phe with para-substituents larger than an OH group, including Cl, Br, I, and ethynyl-, cyano- and azido-groups, are accepted and activated in aminoacylation reaction.28 In comparison, substitution with serine (Ala294Ser) decreases the volume and leads to a narrower substrate range for the engineered enzyme, rejecting even 5a.28a,b The results of the present work correlate with these previous studies and support the role of synthetic pocket size as a determinant of specificity. The incorporation of 5a, and the lack of incorporation of the para-substituted derivatives 1a, 2a, 3a, 4a, 11–14, indicate slight flexibility at the active site of the wild-type E. coli PheRS near the para position, being able to accommodate only an F group. The requirements for the ortho and meta positions, on the other hand, are not so rigid and the synthetase has been shown to accept larger ring substituents. On one side of the ring the positions are observed projecting toward a small hydrophobic space (Fig. 3a2), surrounded by the side chains of Phe248 and Phe250 arranged in a “edge-to-face” manner for each aromatic pair, whereas the corresponding positions on the opposite side are enclosed with the side chains of Ser171, Gln174, Gln208, Glu210 and Gly296, pointing to a hydrophilic area (Fig. 3a).27 Incorporation of 5b,c in place of Phe suggests acceptance of an F at each position, which might have been expected given the flexibility of the para position. In addition to an F, an OH was also accepted at the ortho position, as suggested by the results with 1c, and an OH, Me and Cl at the meta position by the results with 1b, 2b and 3b. Further evidence by di-substituted amino acids 6, 9 and 10 reveals difference of the meta positions on the two ring sides, with one being able to accept a group as large as an Me and the other an OH.
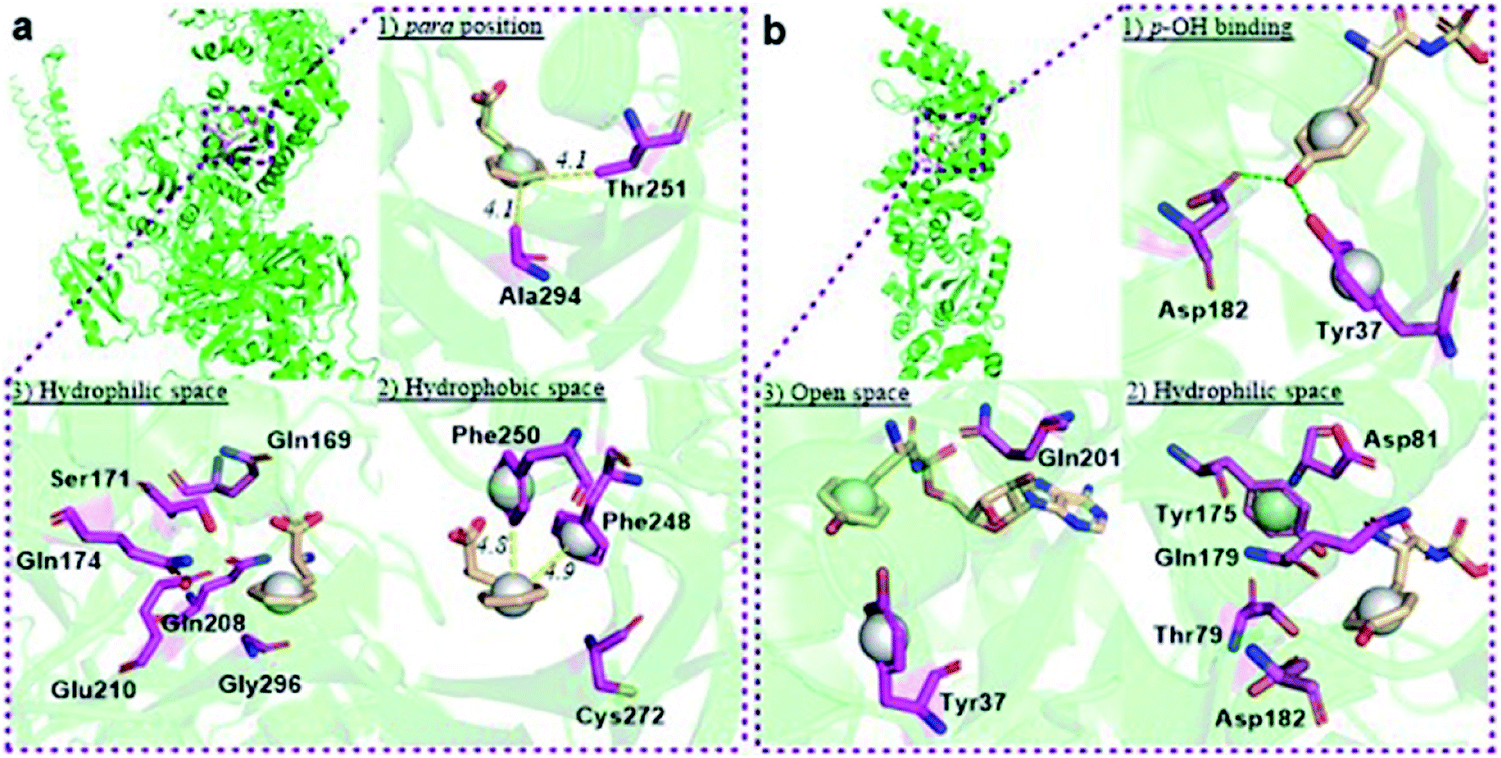 | ||
| Fig. 3 Structural analyses of E. coli PheRS and TyrRS. (a) E. coli PheRS synthetic active site with bound Phe;27 (b) E. coli TyrRS synthetic active site with bound tyrosyl-adenylate.23 The substrates are colored tan and amino acid residues purple. Yellow dashes measure distance between atoms. Green dashes represent hydrogen bond interactions. Round balls are placed in substrate rings to enhance views. Diagrams reproduced with the permission of Elsevier. | ||
To assist the selection for Phe, there is also an editing site located in the B3/4 domain of the β-subunit, about 35 Å from the synthetic active site, providing a post-transfer proofreading mechanism through hydrolysis of the tRNAPhe mistakenly charged with non-cognate amino acid.27 However, this proofreading activity appears to require a phenol OH for hydrogen bond formation with the side chain of Glu334 and the main chain amide of Gly315.27 Previous reports have shown that E. coli PheRS hydrolyzes the tRNAPhe misacylated with Tyr1b and 1b,22 but does not cleave the tRNAPhe charged with para-substituted (fluoro-, chloro-, bromo- and amino-) Phe derivatives,28 further evidence comes from incorporation of 5a via wild-type machinery,16,28a,b as discussed above, and of p-Cl-Phe (3a) using a Ala294GlyPheRS mutant-containing E. coli strain.28b Consistent with these, results of the current work support the requirement for hydrogen bond interactions and showed translational incorporation of a series of amino acid derivatives. Of particular interest was a mutant with significantly reduced editing activity, by which formation of the m-Tyr-tRNAPhe was successful in aminoacylation.22 This indicates that 1b is a substrate of both the active and editing sites of E. coli PheRS, but the rate of hydrolysis is far lower than the rate of activation. This may account for the incorporation of 1b, and that of the 1c, 9 and 10 with at least one OH group on the substrate ring.
E. coli cell-free protein synthesis with Tyr analogues
Analogously, the substrate scope of E. coli TyrRS was studied through protein expression with Tyr analogues. To assess the requirement for the ring location of the OH, Tyr isomers 1b,c were studied. To assess the importance of the p-OH group, derivatives bearing a range of para-substituents, including 2a, 3a, 4a, 5a, 11–14 were studied. None of these amino acids were able to support protein expression (Fig. 4a).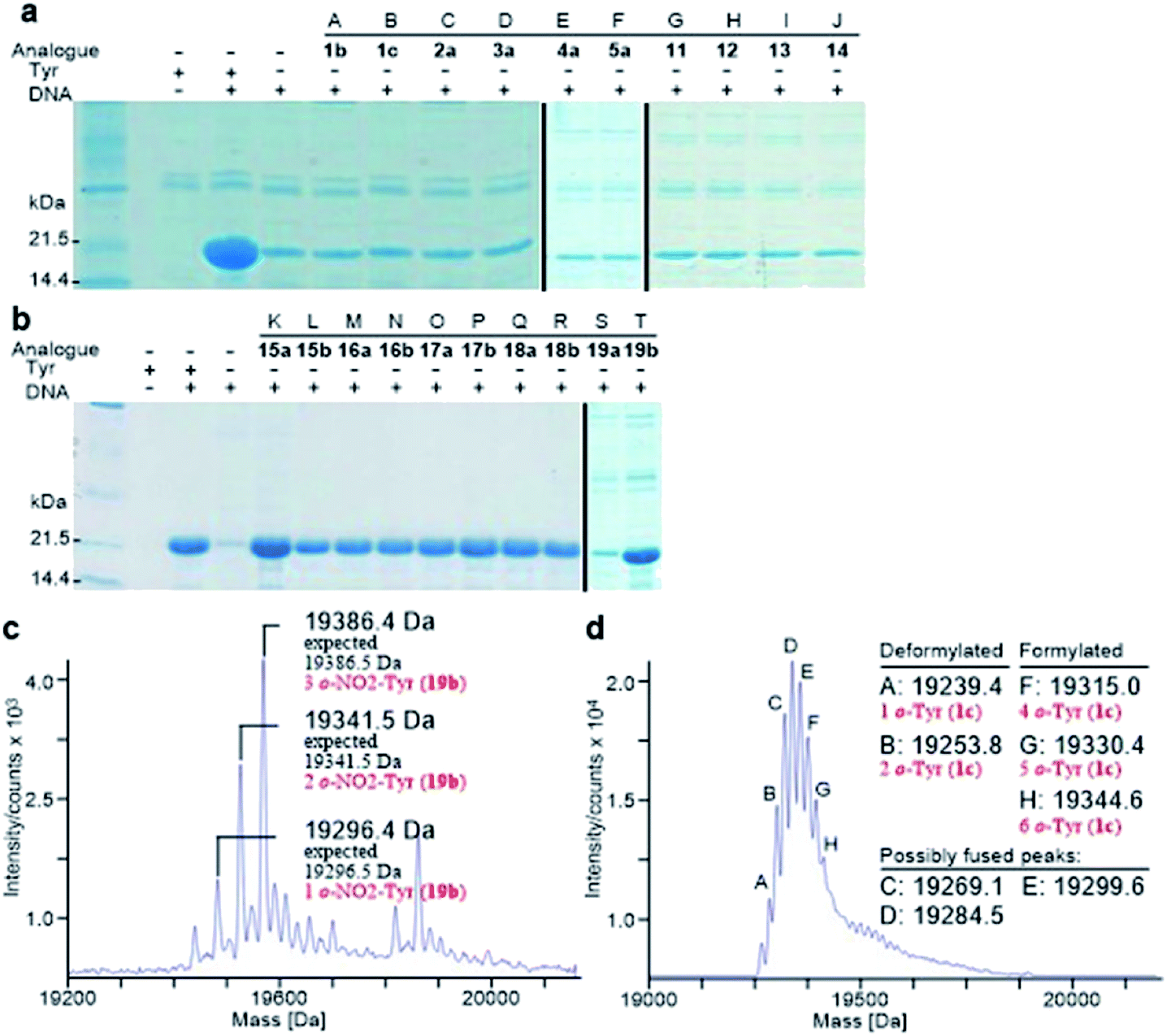 | ||
Fig. 4 Analysis of synthesized His6-PpiB. (a and b) SDS-PAGE analysis of synthesized His6-PpiB. Lanes A–T: no Tyr but with 1b, 1c, 2a, 3a, 4a, 5a, 11, 12, 13, 14, 15a, 15b, 16a, 16b, 17a, 17b, 18a, 18b, 19a or 19b. (c) Mass spectral analysis of His6-PpiB synthesized in the presence of 19b; (d) mass spectral analysis of His6-PpiB synthesized in the presence of 0.2 mM Phe and 2.0 mM 1c. In (d), the peaks at 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 239.4 Da (A) and 19253.8 Da (B) correspond to the respective 1 and 2 replacements of Phe residues for deformylated His6-PpiB, and peaks at 19315.0 Da (F), 19330.4 Da (G) and 19344.6 Da (H) to the respective 4, 5 and 6 replacements of Phe residues for formylated His6-PpiB. The peaks at 19269.1 Da (C), 19284.5 Da (D) and 19299.6 Da (E) are probably a result of fused peaks of the substituted deformylated and formylated His6-PpiB. 239.4 Da (A) and 19253.8 Da (B) correspond to the respective 1 and 2 replacements of Phe residues for deformylated His6-PpiB, and peaks at 19315.0 Da (F), 19330.4 Da (G) and 19344.6 Da (H) to the respective 4, 5 and 6 replacements of Phe residues for formylated His6-PpiB. The peaks at 19269.1 Da (C), 19284.5 Da (D) and 19299.6 Da (E) are probably a result of fused peaks of the substituted deformylated and formylated His6-PpiB. | ||
Having established that p-OH is not replaceable for substrate recognition, Tyr derivatives with substituents at other ring positions, including both ortho and meta positions, were investigated. Derivatives studied involve the fluorides 15a,b, and hydroxyl-, chloro-, methyl- and nitro-substituted 16a,b, 17a,b, 18a,b and 19a,b. Apart from 19a, soluble proteins were obtained and successfully isolated with all derivatives (Fig. 4b), even with 16a,b that bear two OH groups. Compared to protein loss observed for 1b,c, successful isolation with 16a,b is probably attributable to a low level of abundance in the protein (3 Tyr sites). With a similar cell-free system, incorporation of 19a in place of Tyr was reported.17d It was accomplished by considerably enhancing the concentrations of E. coli TyrRS, which was prepared separately and supplemented to the system exogenously, and the substrate 19a.17d The necessity for this suggests a poor activation by E. coli TyrRS, which is most probably the cause of the lack of incorporation in the present study.
The reported crystal structure of the E. coli TyrRS–tyrosyl-adenylate complex (Fig. 3b) shows that the enzyme forms hydrogen bonds with the p-OH group of Tyr by the side chains of Tyr37 and Asp182 (Fig. 3b).23 The p-OH group is a key element for the substrate specificity of E. coli TyrRS as it offers complementary hydrogen bonds for the recognition of the amino acid,29 that of the twenty normal amino acids only Tyr can form. Studies of the Tyr isomers and para-substituted derivatives show that this OH group must locate at the para position and it is not replaceable. In analogy to the E. coli PheRS active site, spaces near ring positions are observed for E. coli TyrRS.23 The ortho and meta positions on one side of the substrate ring, enclosed by the side chains of Tyr175, Thr76 and Gln179, project to a small hydrophilic space (Fig. 3b), whereas the corresponding positions on the other side point to an open area (Fig. 3b), in particular the ortho position. Consistent with earlier findings,17a–c results obtained with 15a, 16a and 17a indicate the space near the meta position at the E. coli TyrRS active site can accommodate an F, OH or Cl. In addition, this space has been shown to accommodate a Me (18a). The ortho position displayed the most flexibility, being able to accept not only an F, OH, Cl and Me but also a NO2.
Competition between the cognate and derivatized amino acids
A major advantage of the cell-free expression system over whole cell-based techniques is to precisely control the concentration of substrate. This allows to determine the extent to which the non-canonical amino acids compete with the natural counterpart. Competitive experiments between Phe and Tyr and their related derivatives were therefore performed. Although many amino acid analogues were found to be efficient substrates of E. coli PheRS or TyrRS, the natural amino acids were highly favoured against all analogues investigated (Table 1). Inclusion of a small amount of Phe in the reaction mixture precluded the incorporation of most Phe analogues. The inefficient competition of only 1c (2.1%) and the fluorides 5a–c (3.6%, 2.1% and 1.4%, respectively, Table 1) shows that the selectivity for Phe is at least two orders of magnitude higher than for all surrogates. The KM value of E. coli PheRS for Phe (0.05 mM) is reported to be approximately 100-, 30- and 10-fold lower than that for 5c (5.0 mM), 5b (1.4 mM) and 5a (0.56 mM),20 respectively (Table 1). These ratios correlate with the competition efficiencies obtained. In an analogous manner, preclusion of most Tyr analogues was also observed with a small amount of Tyr in the reaction mixture. The inefficient competition of 15a,b (2.9% and 2.3%, respectively, Table 1) suggests that the selectivity for Tyr is at least two orders of magnitude higher. The ratio of the KM value of E. coli TyrRS for Tyr (0.0061 mM)21 relative to that of 15a (0.13 mM)21 (4.7%) also correlates with the competition efficiencies obtained for 15a.| Derivative (2.0 mM) | Incorporation levela (%) | Competition efficiency (%) | kcata (s−1) | KM (mM) | kcat/KM (s−1 M−1) | |
|---|---|---|---|---|---|---|
| No Phe | 0.2 mM Phe | |||||
| Phe | — | — | — | — | 0.05(ref. 20) | — |
| 1b | 90 | N.D. | N.D.b | 2.1 (ref. 22) | 0.25 (ref. 22) | 0.8 × 104 |
| 1c | >95 | 21 ± 0 | 2.1 | — | — | — |
| 2b | >95 | N.D. | N.D. | — | — | — |
| 3b | >95 | N.D. | N.D. | — | — | — |
| 5a | >95 | 36 ± 0.9 | 3.6 | — | 0.56 (ref. 20) | — |
| 5b | >95 | 21 ± 1.7 | 2.1 | — | 1.4 (ref. 20) | — |
| 5c | >95 | 14 ± 2 | 1.4 | — | 5.0 (ref. 20) | — |
| 6 | >95 | N.D. | N.D. | — | — | — |
| 9 | >95 | N.D. | N.D. | — | — | — |
| 10 | >95 | N.D. | N.D. | — | — | — |
| Derivative (2.0 mM) | Incorporation levela (%) | Competition efficiency (%) | kcata (s−1) | KM (mM) | kcat/KM (s−1 M−1) | |
|---|---|---|---|---|---|---|
| No Tyr | 0.2 mM Ty | |||||
| a Incorporation levels and rates of reaction determined in duplicate experiments varied by less than 20%. Data are the mean of duplicate experiments.b N.D. = not determined. | ||||||
| 1a (Tyr) | — | — | — | 11.3 ± 0.15 | 0.006 (ref. 21) | 1.85 × 106 |
| 15a | >95 | 29 ± 0.6 | 2.9 | 6.5 ± 0.14 | 0.13 (ref. 21) | 5.02 × 104 |
| 15b | >95 | 23 ± 1.2 | 2.3 | 4.8 ± 0.12 | — | — |
| 16a | >95 | N.D. | N.D. | 3.3 ± 0.01 | 1.4 (ref. 21) | 2.33 × 103 |
| 16b | >95 | N.D. | N.D. | 0.89 ± 0.01 | — | — |
| 17a | >95 | N.D. | N.D. | 0.34 ± 0.07 | — | — |
| 17b | >95 | N.D. | N.D. | 0.17 ± 0.08 | — | — |
| 18a | ∼90 | N.D. | N.D. | 0.09 ± 0.07 | — | — |
| 18b | >95 | N.D. | N.D. | 0.13 ± 0.06 | — | — |
| 19a | <10 | N.D. | N.D. | N.D. | — | — |
| 19b | ∼80 | N.D. | N.D. | 0.06 ± 0.01 | — | — |
Monitoring activity of E. coli TyrRS
Apart from the protein expression experiments, the interactions of the tested analogues with the E. coli TyrRS were assessed directly through adenosine monophosphate (AMP) production assays. Of the two synthetases indirectly investigated by protein expression, E. coli TyrRS was selected as it only has one active site.23 From Table 1 it is evident that E. coli TyrRS catalyzes the reactions of Tyr analogues at different rates, suggesting varying degrees of binding of the synthetase toward each analogue. Among all substrates, a highest kcat value was obtained for Tyr, whereas the reaction with Phe was not detected. This is expected as Tyr is the natural substrate while Phe is not. No reaction with 19a was observed either, despite that an earlier report has described the detection of aminoacylation of tRNATyr with 19a by E. coli TyrRS using a mass spectrometry-based technique.25 As shown in control experiment (Fig. 4c), 0.5 μM AMP is in the limit of quantification by HPLC. This suggests that the AMP produced with 19a was below detection limit. As mentioned above, it is probably a result of poor activation by the enzyme. Attempts to enhance the concentrations of components in the present study to measure the reaction rate were unsuccessful, due to the poor solubility of 19a.In view of the kcat values of the meta-substituted tyrosines 15a, 16a, 17a and 18a, it appears that the relative reaction rate decreases in relation to an increase of van der Waals radius of the substituent on the aromatic ring of each analogue. The same pattern was also observed for the ortho-substituted species 15b, 16b, 17b, 18b and 19b. This shows that E. coli TyrRS distinguishes small structural changes and catalyzes reaction of each analogue at a varying rate. A combination of the kcat for Tyr and its KM value affords a catalytic efficiency (kcat/KM) being the greatest among the three ligands with reported KM values (Tyr itself, 15a and 16a) (Table 1). A comparison of the catalytic efficiency of Tyr with 15a shows that 15a is activated only 2.7% as efficiently as Tyr by E. coli TyrRS. This ratio correlates with the 2.9% competition efficiency observed in competitive experiment (Table 1), indicating that competing for incorporation is essentially a competition for the activation by E. coli TyrRS. These show a direct relationship between the ability of the synthetase to bind different substrates and catalyze the aminoacylation reactions at varying rates with the incorporation of non-canonical amino acids.
Incorporation of hydroxylated amino acids during protein synthesis has been associated with deposition and accumulation of oxidized proteins,18e as in biological systems the aromatic amino acids Phe and Tyr are prone to modification by cellular oxidants such as reactive oxygen species to give the corresponding products 1b,c and 16a.4,18e,26 Elevated levels of the three amino acids have been found bound to oxidatively damaged proteins that are commonly detected in pathological tissues.12,18e Although the involvement of aaRSs in this process has been hypothesized, the specific pathway by which oxidized amino acids are incorporated remains poorly understood. While both whole-cell and cell-free,18d–f as well as aminoacylation18a–c studies have shown that 1b,c could be incorporated via PheRS and 16a via TyrRS,6,17a the incorporation may occur by the other way around, with 16a via PheRS and 1b,c via TyrRS, since activation of 16a by both bacterial and eukaryotic PheRSs has been observed,18b,c as mentioned above and, as isomers of Tyr, 1b,c could in principle replace Tyr. In this work, we have clarified that, in an E. coli system, 1b,c can only be incorporated at the Phe location, not at the Tyr location, and that 16a would only be incorporated at the Tyr location (Fig. 5). This may aid the study of age-related pathologies associated with oxidized protein accumulation.
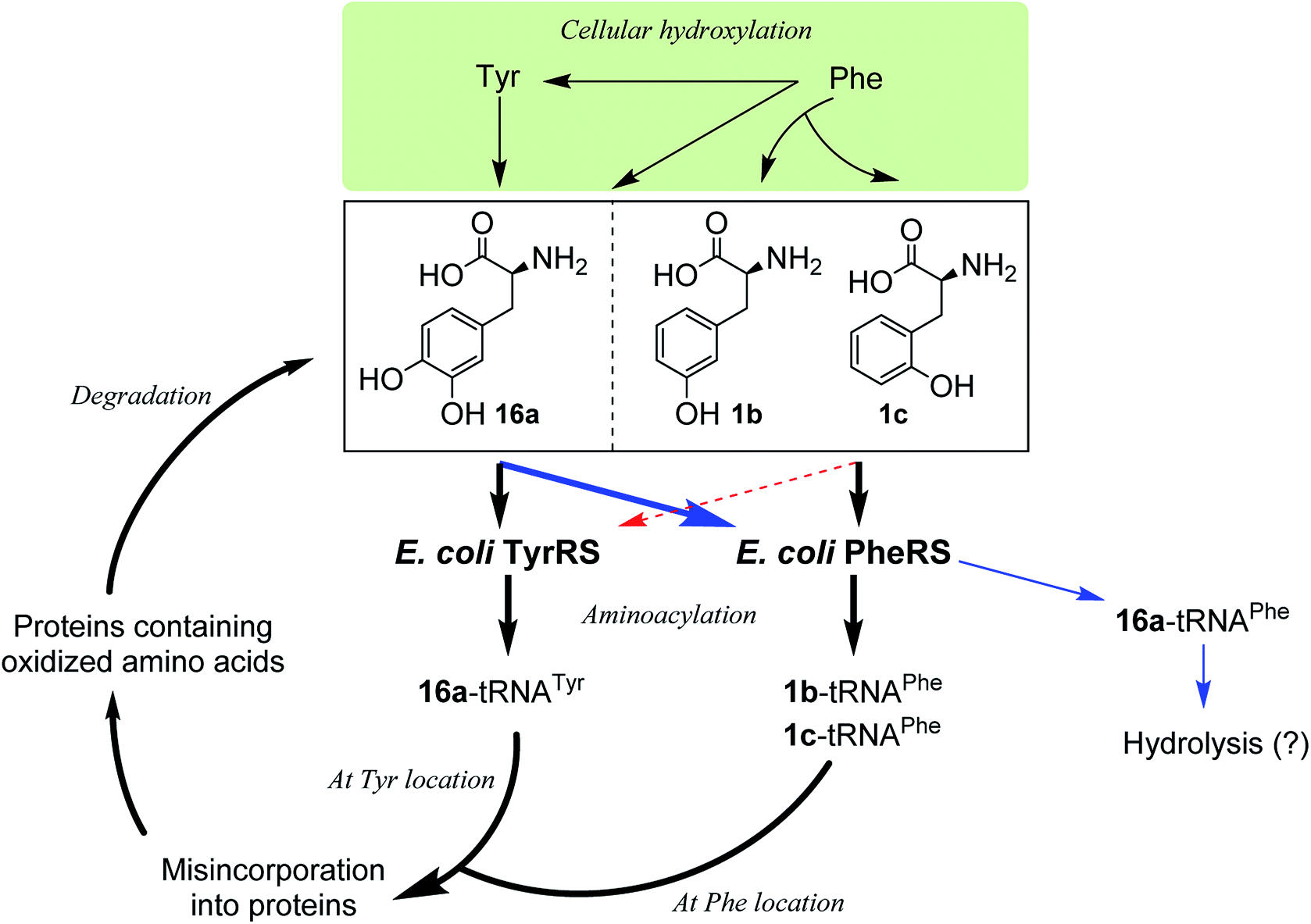 | ||
| Fig. 5 Proposed oxidized protein synthetic pathways by incorporation of oxidized amino acid via E. coli PheRS and TyrRS. Intracellular oxidation of Phe and Tyr produces noncanonical hydroxylated derivatives that are substrates of E. coli PheRS and TyrRS. 3,4-DOPA (16a) is a substrate for both PheRS and TyrRS, while p- and m-tyrosine (1b,c) are both substrates of PheRS. The proofreading domain of PheRS is likely to prevent incorporation of 16a into protein in place of Phe, however misincorporation of 16a in place of Tyr may occur along with misincorporation of 1b or 1c in place of Phe. | ||
Conclusions
The work described here shows inherent substrate promiscuity of the E. coli PheRS and TyrRS. They can activate a range of structural analogues and subsequently charge them onto the cognate tRNA for protein expression. To the best of our knowledge, it is the first time incorporation of the amino acid analogues 2b, 3b, 6, 9, 10, and 15b, 16b, 17b, 18 and 19b into protein via wild-type translational machinery has been observed.The inherent substrate promiscuity of aaRS may prove useful as new tools for biotechnology, as it allows the production of proteins engineered with nonproteinogenic amino acids in a direct manner, without the need for aaRSs with relaxed substrate specificity. Unlike the case of expanded genetic codes, this system does not accommodate an additional noncanonical amino acid, instead replacing a Phe or Tyr partially or completely. There is no site specificity with regard to partial replacement and hence a plasmid cannot be generated to direct modifications in a predetermined position of the protein it encodes. In this work, the incorporation levels of most amino acids are greater than 95% and thus enabled a high degree of amino acid modification, including fluorination, oxidation, methylation, chlorination and nitration, which can not only be used for applications such as isotopic labeling for use in spectroscopic studies, but also for investigation of general rather than specific effects of amino acid derivatives on protein structure and function. In addition, this study also identifies the oxidized protein synthetic pathways by incorporation of oxidized amino acids involving aaRSs. Further, the substrate promiscuity of bacterial PheRS and TyrRS may be different from that of eukaryotic enzymes, making these two synthetases potential targets for the development of antibacterial agents.
Experimental
Syntheses
Most of the amino acid derivatives used in this study were commercially available, except for 8–10, 15b, 16b, 17b, 18 and 19b, which were synthesized in the laboratory. Apart from 3-hydroxy-5-methylphenylalanine (10), the preparation of the rest of the derivatives was described previously.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH3), 1.28 (t, J = 7.2 Hz, 6H; (CH2CH3)2). 13C NMR (100 MHz, CDCl3): δ 169.03 (NHCO), 167.69 (2C, (CO)OEt), 156.97 (Ar C), 132.26 (Ar C), 128.07 (Ar C), 126.67 (Ar C), 126.29 (Ar C), 109.74 (Ar C), 67.43 (quaternary C), 62.60 (2C, CH2CH3), 55.27 (OCH3), 37.03 (Ar-CH2), 23.09 ((CO)CH3), 16.25 (Ar-CH3), 14.10 (2C, CH2CH3). HRMS (ESI) calcd for C18H25NO6Na [M + Na]+ m/z 374.1580; found 374.1580. Calcd for C18H26NO6 [M + H]+ m/z 352.1760; found 352.1763. Anal. calcd for C18H25NO6: C 61.53, H 7.17, N 3.99; found: C 61.31, H 7.17, N 4.12.O), 153.17 (Ar-C), 131.93 (Ar-C), 127.87 (Ar-C), 125.60 (Ar-C), 125.51 (Ar-C), 115.47 (Ar-C), 53.99 (CH), 34.60 (CH2), 15.01 (CH3). HRMS (ESI) calcd for C10H13NO3Na [M + Na]+ m/z 218.0793; found 218.0793. Anal. calcd for C10H13NO3: C 61.53, H 6.71, N 7.17; found: C 61.87, H 6.96, N 7.26.
O)CH3), 1.28 (t, J = 7.2 Hz, 6H; (CH2CH3)2). 13C NMR (100 MHz, CDCl3): δ 169.03 (NHCO), 167.69 (2C, (CO)OEt), 156.97 (Ar C), 132.26 (Ar C), 128.07 (Ar C), 126.67 (Ar C), 126.29 (Ar C), 109.74 (Ar C), 67.43 (quaternary C), 62.60 (2C, CH2CH3), 55.27 (OCH3), 37.03 (Ar-CH2), 23.09 ((CO)CH3), 16.25 (Ar-CH3), 14.10 (2C, CH2CH3). HRMS (ESI) calcd for C18H25NO6Na [M + Na]+ m/z 374.1580; found 374.1580. Calcd for C18H26NO6 [M + H]+ m/z 352.1760; found 352.1763. Anal. calcd for C18H25NO6: C 61.53, H 7.17, N 3.99; found: C 61.31, H 7.17, N 4.12.O), 153.17 (Ar-C), 131.93 (Ar-C), 127.87 (Ar-C), 125.60 (Ar-C), 125.51 (Ar-C), 115.47 (Ar-C), 53.99 (CH), 34.60 (CH2), 15.01 (CH3). HRMS (ESI) calcd for C10H13NO3Na [M + Na]+ m/z 218.0793; found 218.0793. Anal. calcd for C10H13NO3: C 61.53, H 6.71, N 7.17; found: C 61.87, H 6.96, N 7.26.Construction of plasmids
The gene in pND706 vector encoding the E. coli His6-TyrRS was kindly provided by the Dixon group at the University of Wollongong (Australia). The genes in pND1098 vector encoding E. coli His6-PpiB and in pND706 vector encoding the E. coli His6-TyrRS were transformed into the respective E. coli DH5α and E. coli AN1459 for amplification and were isolated using Qiagen® Mini kit following procedures recommended by the manufacturer. The gene encoding the E. coli TyrRS was then cloned in pETMCSIII vector within the Ndel-HindIII sites to generate the plasmid pET-TyrRS-His. The sequence of pET-TyrRS-His was confirmed using ABI 3730 Genetic Analyzer. The concentration of the DNA was measured using a NanoDrop spectrophotometer.Preparation of E. coli S30 extract
The E. coli S30 extract was prepared from E. coli star BL21(DE3) by following a protocol established earlier.15,17aCell-free protein synthesis
The E. coli cell-free protein synthesis was conducted by following a procedure, as described in earlier reports.15,17a The natural amino acids were used at 1.0 mM. The unnatural amino acids were used at 2.0 mM and the corresponding natural amino acid was not added. The reactions were proceeded for 6 h at 30 °C, with shaking at 200 rpm, unless otherwise stated. The competitive experiments between unnatural amino acids and their corresponding natural counterparts were conducted in a system identical to that used for usual cell-free protein synthesis, with the cognate amino acid supplied to the expression at 0.2 mM concentration to compete with the foreign substrate at 2 mM. In cases where racemic mixtures were used, this concentration refers to the L-enantiomer content. The levels of unnatural amino acid incorporation in competitive experiments were concluded from the mass spectra. The competition efficiencies are given as percentages and were calculated based on the ratio of the concentrations between the natural amino acid and its corresponding competitor, and the level of unnatural amino acid incorporation (incorporation level (%) × [natural amino acid]/[derivative]). The amino acid derivatives that did not incorporate were excluded from this assay, except for 19a, which was studied for the purpose of comparison.Protein analysis
The crude contents of the inner reaction was analyzed by SDS-PAGE, to confirm the production of soluble protein. The inner reaction contents were then purified using the HisGraviTrap® Kit following the native conditions recommended by the manufacturer, and the resulting elution fraction was concentrated using Amicon Ultra-4 (YM-3, 000) centrifugal filter devices. Protein concentrations were measured using a NanoDrop. The mass of the protein produced was confirmed by the Agilent 1100 series LC/MSD TOF instrument.E. coli His6-TyrRS preparation and activity assay
The E. coli TyrRS with a polyhistidine tag at the C-terminus (E. coli His6-TyrRS) was overproduced from pET-TyrRS-His in E. coli BL21 (DE3). The resulting enzyme was purified using HisGraviTrap® Kit following the conditions recommended by the manufacturer, and was concentrated using Amicon Ultra-4 (YM-3, 000) centrifugal filter devices. The prepared enzyme was analyzed by SDS-PAGE (20% acrylamide) with mass confirmed by Agilent 1100 series LC/MSD TOF instrument (Fig. 6a and b). The enzyme concentration was measured using a NanoDrop spectrophotometer. The method used to evaluate the activity of the prepared enzyme determines the catalytic rate constant (kcat) through HPLC monitoring the formation of AMP by analyzing aliquots taken from the reaction mixture and quenched with 0.1% SDS every 15 seconds (Fig. 6c). It gave Tyr a kcat of 11.3 s−1 (Fig. 6c and d), which is in excellent agreement with reported value of 12 s−1.24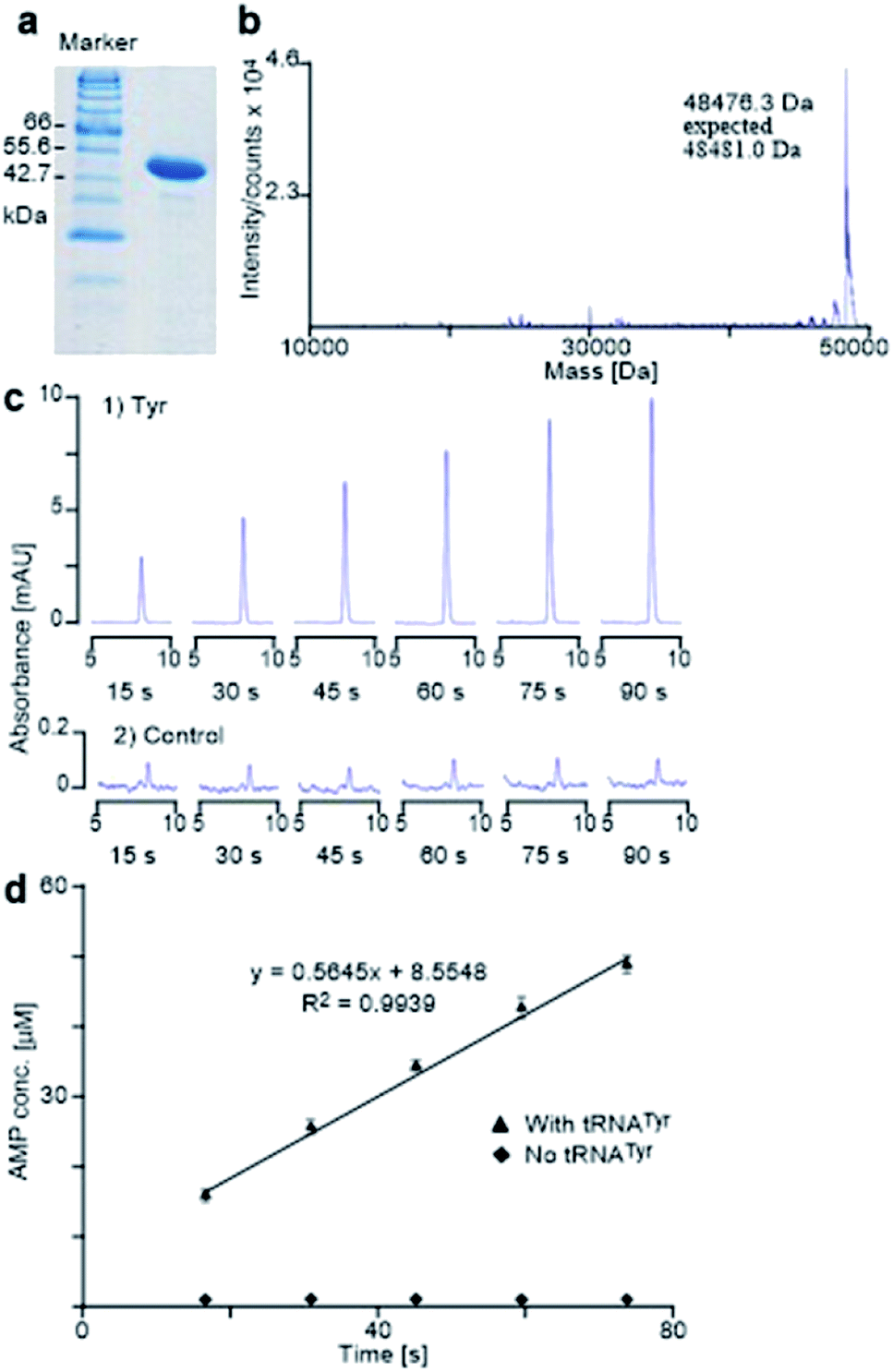 | ||
| Fig. 6 E. coli His6-TyrRS activity assay. (a and b) SDS-PAGE and mass spectral analyses of prepared E. coli TyrRS. (c) Partial HPLC traces of aliquots collected every 15 seconds from the reactions of Tyr (1) and the control experiment (2) that contained no tRNATyr. (d) The plot of the concentration of AMP against time for the aminoacylation of Tyr. Data points are the mean of duplicate experiments, with errors bars representing ± SD. | ||
Aminoacylation assay
The aminoacylation of amino acid derivative was conducted in a manner identical to Tyr. Stock solutions were prepared in Tris–HCl buffer containing 100.0 mM Tris, 15.0 mM MgCl2, 40.0 mM KCl and 1.0 mM DTT, at pH 7.6. The reaction was performed at 37 °C in a 0.5 mL microcentrifuge, with a total volume of 60 μL containing 0.05 or 0.15 μM E. coli His6-TyrRS, 1.0 mM ATP, 16.7 or 60.0 μM tRNATyr, and 0.1 mg mL−1 bovine serum albumin (BSA). After pre-incubation for one minute, the reaction was initiated by adding in the E. coli His6-TyrRS. A 10 μL sample was taken out by hand from the proceeding reaction every 15 or 40 seconds and quenched into 5.0 μL 0.3% (w/v) aqueous sodium dodecylsulfate (SDS). The SDS-treated samples were either immediately analyzed by HPLC or stored at −20 °C for later analysis. In the latter case, the storage was never longer than 12 hours. The solvent system used for HPLC analysis was as follows:| Time (min) | Buffer A (%) | Buffer B (%) |
|---|---|---|
| 0 | 87 | 13 |
| 18 | 87 | 13 |
| 19 | 70 | 30 |
| 23 | 70 | 30 |
| 24 | 87 | 13 |
In this solvent system, Buffer A contains 60.0 mM ammonium phosphate and 5.0 mM tetrabutylammonium phosphate, at pH 5.0, while Buffer B contains 5.0 mM tetrabutylammonium phosphate in methanol. Both buffers were filtered and degased before use.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support by the Doctoral Research Startup Fund Project of the Zunyi Medical University (F-946). We express our sincere thanks to M. Liutkus and D. Fankhauser for helpful discussions regarding the preparation of the manuscript. We are also indebted to J. E. Hennessy and A. Alissandratos for assistance in recording protein mass spectra.References
- (a) M. Ibba and D. Söll, Aminoacyl-tRNA synthesis, Annu. Rev. Biochem., 2000, 69, 617–650 CrossRef CAS PubMed; (b) O. Kotik-Kogan, N. Moor, D. Tworowski and M. Safro, Structural basis for discrimination of L-phenylalanine from L-tyrosine by phenylalanyl-tRNA synthetase, Structure, 2005, 13, 1799–1807 CrossRef CAS PubMed.
- (a) S. A. Fraser and C. J. Easton, Biosynthetic incorporation of fluorinated amino acids into peptides and proteins, Aust. J. Chem., 2015, 68, 9–12 CrossRef CAS; (b) N. Budisa, Prolegomena to future experimental efforts on genetic code engineering by expanding its amino acid repertoire, Angew. Chem., Int. Ed., 2004, 43, 6426–6463 CrossRef CAS PubMed.
- (a) T. H. Johnston, A. M. Lacoste, N. P. Visanji, A. E. Lang, S. H. Fox and J. M. Brotchie, Repurposing drugs to treat L-DOPA-induced dyskinesia in Parkinson's disease, Neuropharmacology, 2019, 147, 11–27 CrossRef CAS PubMed; (b) R. A. Ruggiero, J. Bruzzo, P. Chiarella, P. Di Gianni, M. A. Isturiz, S. Linskens, N. Speziale, R. P. Meiss, O. D. Bustuoabad and C. D. Pasqualini, Tyrosine isomers mediate the classical phenomenon of concomitant tumor resistance, Cancer Res., 2011, 71, 7113–7124 CrossRef CAS PubMed.
- (a) B. R. Ipson and A. L. Fisher, Roles of the tyrosine isomers meta-tyrosine and ortho-tyrosine in oxidative stress, Ageing Res. Rev., 2016, 27, 93–107 CrossRef CAS PubMed; (b) G. Gueron, N. Anselmino, P. Chiarella, E. G. Ortiz, S. L. Vickers, A. V. Paez, J. Giudice, M. D. Contin, D. Leonardi, F. Jaworski, V. Manzano, A. Strazza, D. R. Montagna, E. Labanca, J. Cotignola, N. D'Accorso, A. Woloszynska-Read, N. Navone, R. P. Meiss, R. Ruggiero and E. Vazquez, Game-changing restraint of Ros-damaged phenylalanine, upon tumor metastasis, Cell Death Dis., 2018, 9, 140 CrossRef PubMed.
- F. Agostini, J. S. Völler, B. Koksch, C. G. Acevedo-Rocha, V. Kubyshkin and N. Budisa, Biocatalysis with unnatural amino acids: enzymology meets xenobiology, Angew. Chem., Int. Ed., 2017, 56, 9680–9703 CrossRef CAS PubMed.
- P. J. Nowatzki, C. Franck, S. A. Maskarinec, G. Ravichandran and D. A. Tirrell, Mechanically tunable thin films of photosensitive artificial proteins: preparation and characterization by nanoindentation, Macromolecules, 2008, 41, 1839–1845 CrossRef CAS.
- E. R. Stadtman, Protein oxidation and aging, Science, 1992, 257, 1220–1224 CrossRef CAS PubMed.
- (a) C. Amatore, S. Arbault, M. Guille and F. Lemaitre, Electrochemical monitoring of single cell secretion: vesicular exocytosis and oxidative stress, Chem. Rev., 2008, 108, 2585–2621 CrossRef CAS PubMed; (b) H. Ou, Z. Huang, Z. Mo and J. Xiao, The characteristics and roles of advanced oxidation protein products in atherosclerosis, Cardiovasc. Toxicol., 2017, 17, 1–12 CrossRef CAS PubMed.
- G. A. Molnár, V. Nemes, Z. Biró, A. Ludány, Z. Wagner and I. Wittmann, Accumulation of the hydroxyl free radical markers meta-, ortho-tyrosine and DOPA in cataractous lenses is accompanied by a lower protein and phenylalanine content of the water-soluble phase, Free Radical Res., 2005, 39, 1359–1366 CrossRef PubMed.
- J. Z. Sun, H. Kaur, B. Halliwell, X. Y. Li and R. Bolli, Use of aromatic hydroxylation of phenylalanine to measure production of hydroxyl radicals after myocardial ischemia in vivo. Direct evidence for a pathogenetic role of the hydroxyl radical in myocardial stunning, Circ. Res., 1993, 73, 534–549 CrossRef CAS PubMed.
- C. A. O'Neill, L. W. Fu, B. Halliwell and J. C. Longhurst, Hydroxyl radical production during myocardial ischemia and reperfusion in cats, Am. J. Physiol.: Heart Circ. Physiol., 1996, 271, H660–H667 CrossRef PubMed.
- S. W. Chan, R. A. Dunlop, A. Rowe, K. L. Double and K. J. Rodgers, L-DOPA is incorporated into brain proteins of patients treated for Parkinson's disease, inducing toxicity in human neuroblastoma cells in vitro, Exp. Neurol., 2012, 238, 29–37 CrossRef CAS PubMed.
- J. T. Ngo and D. A. Tirrell, Noncanonical amino acids in the interrogation of cellular protein synthesis, Acc. Chem. Res., 2011, 44, 677–685 CrossRef CAS PubMed.
- (a) A. R. Fersht and M. M. Kaethner, Enzyme hyperspecificity. Rejection of threonine by the valyl-tRNA synthetase by misacylation and hydrolytic editing, Biochemistry, 1976, 15, 3342–3346 CrossRef CAS PubMed; (b) A. R. Fersht, J. S. Shindler and W. C. Tsui, Probing the limits of protein-amino acid side chain recognition with the aminoacyl-tRNA synthetases. Discrimination against phenylalanine by tyrosyl-tRNA synthetases, Biochemistry, 1980, 19, 5520–5524 CrossRef CAS PubMed; (c) F. Fasiolo and A. R. Fersht, The aminoacyladenylate mechanism in the aminoacylation reaction of yeast phenylalanyl-tRNA synthetase, Eur. J. Biochem., 1978, 85, 85–88 CrossRef CAS PubMed.
- (a) D. J. Stigers, Z. I. Watts, J. E. Hennessy, H. K. Kim, R. Martini, M. C. Taylor, K. Ozawa, J. W. Keillor, N. E. Dixon and C. J. Easton, Incorporation of chlorinated analogues of aliphatic amino acids during cell-free protein synthesis, Chem. Commun., 2011, 47, 1839–1841 RSC; (b) I. N. Arthur, J. E. Hennessy, D. Padmakshan, D. J. Stigers, S. Lesturgez, S. A. Fraser, M. Liutkus, G. Otting, J. G. Oakeshott and C. J. Easton, In situ deprotection and incorporation of unnatural amino acids during cell-free protein synthesis, Chem.–Eur. J., 2013, 19, 6824–6830 CrossRef CAS PubMed; (c) M. Liutkus, S. A. Fraser, K. Caron, D. J. Stigers and C. J. Easton, Peptide synthesis through cell-free expression of fusion proteins incorporating modified amino acids as latent cleavage sites for peptide release, ChemBioChem, 2016, 17, 908–912 CrossRef CAS PubMed.
- (a) N. Voloshchuk, A. Y. Zhu, D. Snydacker and J. K. Montclare, Positional effects of monofluorinated phenylalanines on histone acetyltransferase stability and activity, Bioorg. Med. Chem. Lett., 2009, 19, 5449–5451 CrossRef CAS PubMed; (b) K. R. Mehta, C. Y. Yang and J. K. Montclare, Modulating substrate specificity of histone acetyltransferase with unnatural amino acids, Mol. BioSyst., 2011, 7, 3050–3055 RSC.
- (a) K. Ozawa, M. J. Headlam, D. Mouradov, S. J. Watt, J. L. Beck, K. J. Rodgers, R. T. Dean, T. Huber, G. Otting and N. E. Dixon, Translational incorporation of L-3,4-dihydroxyphenylalanine into proteins, FEBS J., 2005, 272, 3162–3171 CrossRef CAS PubMed; (b) N. Ayyadurai, N. S. Prabhu, K. Deepankumar, A. Kim, S. G. Lee and H. Yun, Biosynthetic substitution of tyrosine in green fluorescent protein with its surrogate fluorotyrosine in Escherichia coli, Biotechnol. Lett., 2011, 33, 2201 CrossRef CAS PubMed; (c) N. Ayyadurai, K. Deepankumar, N. S. Prabhu, N. Budisa and H. Yun, Evaluation and biosynthetic incorporation of chlorotyrosine into recombinant proteins, Biotechnol. Bioprocess Eng., 2012, 17, 679–686 CrossRef CAS; (d) S. J. Oh, K. H. Lee, H. C. Kim, C. Catherine, H. Yun and D. M. Kim, Translational incorporation of multiple unnatural amino acids in a cell-free protein synthesis system, Biotechnol. Bioprocess Eng., 2014, 19, 426–432 CrossRef CAS.
- (a) L. Klipcan, N. Moor, N. Kessler and M. G. Safro, Eukaryotic cytosolic and mitochondrial phenylalanyl-tRNA synthetases catalyze the charging of tRNA with the meta-tyrosine, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11045–11048 CrossRef CAS PubMed; (b) N. Moor, L. Klipcan and M. G. Safro, Bacterial and eukaryotic phenylalanyl-tRNA synthetases catalyze misaminoacylation of tRNAPhe with 3,4-dihydroxy-L-phenylalanine, Chem. Biol., 2011, 18, 1221–1229 CrossRef CAS PubMed; (c) E. A. First, L-DOPA ropes in tRNAPhe, Chem. Biol., 2011, 18, 1201–1202 CrossRef CAS PubMed; (d) O. Popp, V. Larraillet, H. Kettenberger, I. H. Gorr, M. Hilger, F. Lipsmeier, A. Zeck and N. Beaucamp, Molecular polygamy: the promiscuity of L-phenylalanyl-tRNA-synthetase triggers misincorporation of meta- and ortho-tyrosine in monoclonal antibodies expressed by Chinese hamster ovary cells, Biotechnol. Bioeng., 2015, 112, 1187–1199 CrossRef CAS PubMed; (e) K. J. Rodgers and N. Shiozawa, Misincorporation of amino acid analogues into proteins by biosynthesis, Int. J. Biochem. Cell Biol., 2008, 40, 1452–1466 CrossRef CAS PubMed; (f) H. Gurer-Orhan, N. Ercal, S. Mare, S. Pennathur, H. Orhan and J. W. Heinecke, Misincorporation of free m-tyrosine into cellular proteins: a potential cytotoxic mechanism for oxidized amino acids, Biochem. J., 2006, 395, 277–284 CrossRef CAS PubMed.
- K. J. Edwards, D. L. Ollis and N. E. Dixon, Crystal structure of cytoplasmic Escherichia coli peptidyl-prolyl isomerase: evidence for decreased mobility of loops upon complexation, J. Mol. Biol., 1997, 271, 258–265 CrossRef CAS PubMed.
- D. V. Santi and P. V. Danenberg, Phenylalanyl transfer ribonucleic acid synthetase from Escherichia coli. Analysis of the phenylalanine binding site, Biochemistry, 1971, 10, 4813–4820 CrossRef CAS PubMed.
- R. Calendar and P. Berg, The catalytic properties of tyrosyl ribonucleic acid synthetases from Escherichia coli and Bacillus subtilis, Biochemistry, 1966, 5, 1690–1695 CrossRef CAS PubMed.
- T. J. Bullwinkle, N. M. Reynolds, M. Raina, A. Moghal, E. Matsa, A. Rajkovic, H. Kayadibi, F. Fazlollahi, C. Ryan, N. Howitz, K. F. Faull, B. A. Lazazzera and M. Ibba, Oxidation of cellular amino acid pools leads to cytotoxic mistranslation of the genetic code, eLife, 2014, 3, e02501 CrossRef PubMed.
- T. Kobayashi, T. Takimura, R. Sekine, K. Vincent, K. Kamata, K. Sakamoto, S. Nishimura and S. Yokoyama, Structural snapshots of the KMSKS loop rearrangement for amino acid activation by bacterial tyrosyl-tRNA synthetase, J. Mol. Biol., 2005, 346, 105–117 CrossRef CAS PubMed.
- D. Kiga, K. Sakamoto, K. Kodama, T. Kigawa, T. Matsuda, T. Yabuki, M. Shirouzu, Y. Harada, H. Nakayama, K. Takio, Y. Hasegawa, Y. Endo, I. Hirao and S. Yokoyama, An engineered Escherichia coli tyrosyl-tRNA synthetase for site-specific incorporation of an unnatural amino acid into proteins in eukaryotic translation and its application in a wheat germ cell-free system, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9715–9720 CrossRef CAS PubMed.
- M. C. Hartman, K. Josephson and J. W. Szostak, Enzymatic aminoacylation of tRNA with unnatural amino acids, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4356–4361 CrossRef CAS PubMed.
- (a) K. J. Rodgers, Non-protein amino acids and neurodegeneration: the enemy within, Exp. Neurol., 2014, 253, 192–196 CrossRef CAS PubMed; (b) W. M. Garrison, Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins, Chem. Rev., 1987, 87, 381–398 CrossRef CAS.
- I. Mermershtain, I. Finarov, L. Klipcan, N. Kessler, H. Rozenberg and M. G. Safro, Idiosyncrasy and identity in the prokaryotic Phe-system: crystal structure of E. coli phenylalanyl-tRNA synthetase complexed with phenylalanine and AMP, Protein Sci., 2011, 20, 160–167 CrossRef CAS PubMed.
- (a) P. Kast and H. Hennecke, Amino acid substrate specificity of Escherichia coli phenylalanyl-tRNA synthetase altered by distinct mutations, J. Mol. Biol., 1991, 222, 99–124 CrossRef CAS PubMed; (b) M. Ibba, P. Kast and H. Hennecke, Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase, Biochemistry, 1994, 33, 7107–7112 CrossRef CAS PubMed; (c) N. Sharma, R. Furter, P. Kast and D. A. Tirrell, Efficient introduction of aryl bromide functionality into proteins in vivo, FEBS Lett., 2000, 467, 37–40 CrossRef CAS PubMed; (d) K. Kirshenbaum, I. S. Carrico and D. A. Tirrell, Biosynthesis of proteins incorporating a versatile set of phenylalanine analogues, ChemBioChem, 2002, 3, 235–237 CrossRef PubMed; (e) D. Datta, P. Wang, I. S. Carrico, S. L. Mayo and D. A. Tirrell, A designed phenylalanyl-tRNA synthetase variant allows efficient in vivo incorporation of aryl ketone functionality into proteins, J. Am. Chem. Soc., 2002, 124, 5652–5653 CrossRef CAS PubMed.
- A. R. Fersht, J. P. Shi, J. Knill-Jones, D. M. Lowe, A. J. Wilkinson, D. M. Blow, P. Brick, P. Carter, M. M. Y. Waye and G. Winter, Hydrogen bonding and biological specificity analyzed by protein engineering, Nature, 1985, 314, 235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of amino acid analogues; ESI mass spectrometry of His6-PpiB synthesized in the presence of Phe and Tyr analogues; representative HPLC of the aminoacylation of tyrosine. See DOI: 10.1039/d0ra00655f |
| This journal is © The Royal Society of Chemistry 2020 |