
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalization of C–H bonds in acetophenone oximes with arylacetic acids and elemental sulfur†
Phuc H. Pham‡
 ac,
Khang X. Nguyen‡ac,
Hoai T. B. Phamabc,
Thien T. Tranac,
Tung T. Nguyen*ac and
Nam T. S. Phan*ac
ac,
Khang X. Nguyen‡ac,
Hoai T. B. Phamabc,
Thien T. Tranac,
Tung T. Nguyen*ac and
Nam T. S. Phan*ac
aFaculty of Chemical Engineering, Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam. E-mail: tungtn@hcmut.edu.vn; ptsnam@hcmut.edu.vn
bDepartment of Chemistry, University of Colorado Denver, Denver, CO 80204, USA
cVietnam National University, Ho Chi Minh City, Vietnam
First published on 17th March 2020
Abstract
Fused thieno[3,2-d]thiazoles were synthesized via a coupling of acetophenone ketoximes, arylacetic acids, and elemental sulfur in the presence of Li2CO3 base. Functionalities including chloro, bromo, fluoro, trifluoromethyl, and pyridyl groups were compatible with reaction conditions. High yields and excellent regioselectivities were obtained even if meta-substituted ketoxime acetates were used. Ethyl esters of heteroarylacetic acids were competent substrates, which is very rare in the literature. Our method would offer a convenient protocol to afford polyheterocyclic structures from simple substrates.
Introduction
Although carbonyl groups are ubiquitously found in organic compounds, the use of the functionalities to assist the synthesis of complex molecules is hitherto challenging. Exploiting the presence of aldehyde and ketone groups for selective functionalization of C–H bonds appears to be one of the major targets in organic synthesis over the last decades.1 Among known reports, methods using N-oxime and isosteric groups to facilitate the functionalization of C–H bonds in carbonyl compounds have emerged.2 Oxime ester and oxime ether directed, metal-catalyzed transformations of arene carbon–hydrogen bonds into other carbon–carbon and carbon–heteroatom bonds are known. Cleavage of the N–O bond in oximes has been leveraged to stabilize high valent metal intermediates, thus promoting the activation of proximal C–H bonds.3 In 2008, Liebeskind reported a seminal method for copper-catalyzed coupling of vinyl ketoxime esters and vinylboronic acids to afford highly condensed pyridines.3a Functionalization of C–H bonds α to ketone in oximes was also feasible, furnishing N-heterocycles.3c–f Notably, most of the known examples often use transition metal catalysis.Functionalization of C–H bonds in acetophenones with elemental sulfur is a promising metal-free synthetic scheme to conveniently afford sulfur-containing aromatics. Given the importance of five-membered heterocycles such as thiazole and thiophene in organic synthesis and functional materials,4 developing new methods that allow for the use of simple, commercial substrates to obtain S-heterocycles is demanding. Notably, lack of general substrate scopes is still unsolved. Only a few methods for elemental sulfur-promoted, direct transformation of C–H bonds in acetophenones have been reported.5 Using prefunctionalized ketoximes have expanded the substrate scope.6 Deng recently described the annulation of acetophenone oxime acetates and aldehydes or methylazaarenes to afford 2-aryl thienothiazoles (Scheme 1).6a,b The successes leaned on the use of copper catalysis or a strong base, thus limiting the compatibility of functional groups. Moreover, the reactions often suffered from the mixture of regioisomeric products when oximes derived from meta-substituted acetophenones were used. Herein we report a method for annulation of acetophenone oxime acetates and arylacetic acids. Reactions used such a mild base Li2CO3 that many functionalities such as bromide or amine are tolerated. Notably, all of the substrates including those have meta-substituents afforded single regioisomers. We also attempted coupling of arylacetate esters, since elemental sulfur mediated decarboxylation is known.7
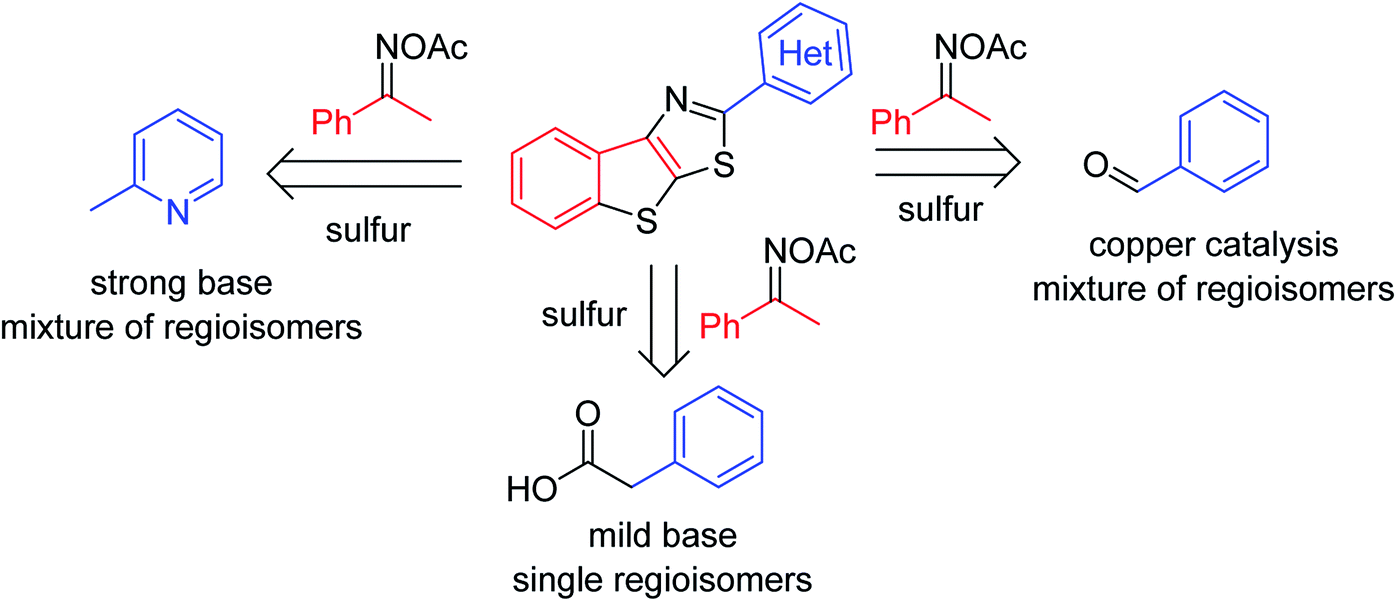 | ||
| Scheme 1 Retrosynthesis of 2-aryl benzothienothiazoles. | ||
Results and discussion
Our first study focused on the reaction of acetophenone oxime 1a and phenylacetic acid 2a to yield 2-phenyl benzothienothiazole 3aa. The reaction was optimized with respect to bases and solvents. The results are shown in Table 1. In general, inorganic bases were more active than organic bases in our conditions (entries 1–7). Both cesium carbonate and lithium carbonate afforded substantial conversion of reactants (entries 4 and 5). Dissimilar to previous studies,5a,7 DABCO base was inferior to carbonate bases (entry 7). Polar solvents such as DMF, DMAc, and NMP could not be used for the reaction (entries 8–10). Previous studies have often used DMSO solvent for sulfur-mediated, oxime-assisted annulation of C–H bonds,6b,6d presumably due to oxidative activity. One should be noted that changing the amount of base somewhat impacted the yield of 3aa (entries 11 and 12). Lastly, a 94% yield of the product was obtained if a diluted reaction mixture was run for 3 h (entry 13).
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yield of 3aa, % |
| a Oxime acetate 1a (0.15 mmol), phenylacetic acid 2a (0.1 mmol), elemental sulfur (0.3 mmol), base (0.1 mmol), solvent (0.5 mL), 120 °C for 6 h under air. Yields of 3aa are GC yields using diphenyl ether internal standard.b 0.05 mmol Li2CO3.c 0.15 mmol Li2CO3.d 1 mL DMSO, 3 h. Abbreviation: DABCO = 1,4-diazabicyclo[2.2.2]octane. | |||
| 1 | K2CO3 | DMSO | 52 |
| 2 | Na2CO3 | DMSO | 78 |
| 3 | tBuOK | DMSO | 29 |
| 4 | Cs2CO3 | DMSO | 81 |
| 5 | Li2CO3 | DMSO | 84 |
| 6 | N-Methylmorpholine | DMSO | 53 |
| 7 | DABCO | DMSO | 78 |
| 8 | Li2CO3 | DMF | 8 |
| 9 | Li2CO3 | DMAc | n.d. |
| 10 | Li2CO3 | NMP | n.d. |
| 11b | Li2CO3 | DMSO | 64 |
| 12c | Li2CO3 | DMSO | 93 |
| 13c,d | Li2CO3 | DMSO | 94c |

Scope of arylacetic acids was then investigated and presented in Scheme 2. Electron-rich (3ab, 3ae) and electron-poor (3ah, 3ai) substrates were all active. ortho-Substituted phenylacetic acids afforded the thienothiazoles in high yields (3ac). It should be noted that the transformation was scalable, up to 2 mmol run without a significant loss of the yield (3aa). Halogenated arylacetic acids were competent substrates (3ag, 3ah), thus are useful for further cross-coupling functionalization. Our first attempts to incorporate heteroaryl acetic acids failed to give the thienothiazoles. Fortunately, the uses of ethyl ester derivatives afforded the desired products in good yields. Such decarboxylation coupling of arylacetates has not been discussed in previous studies.7 In our conditions, ethyl esters of pyridyl acetates were found to be active regardless of the relative position (3aj, 3ak, 3al).
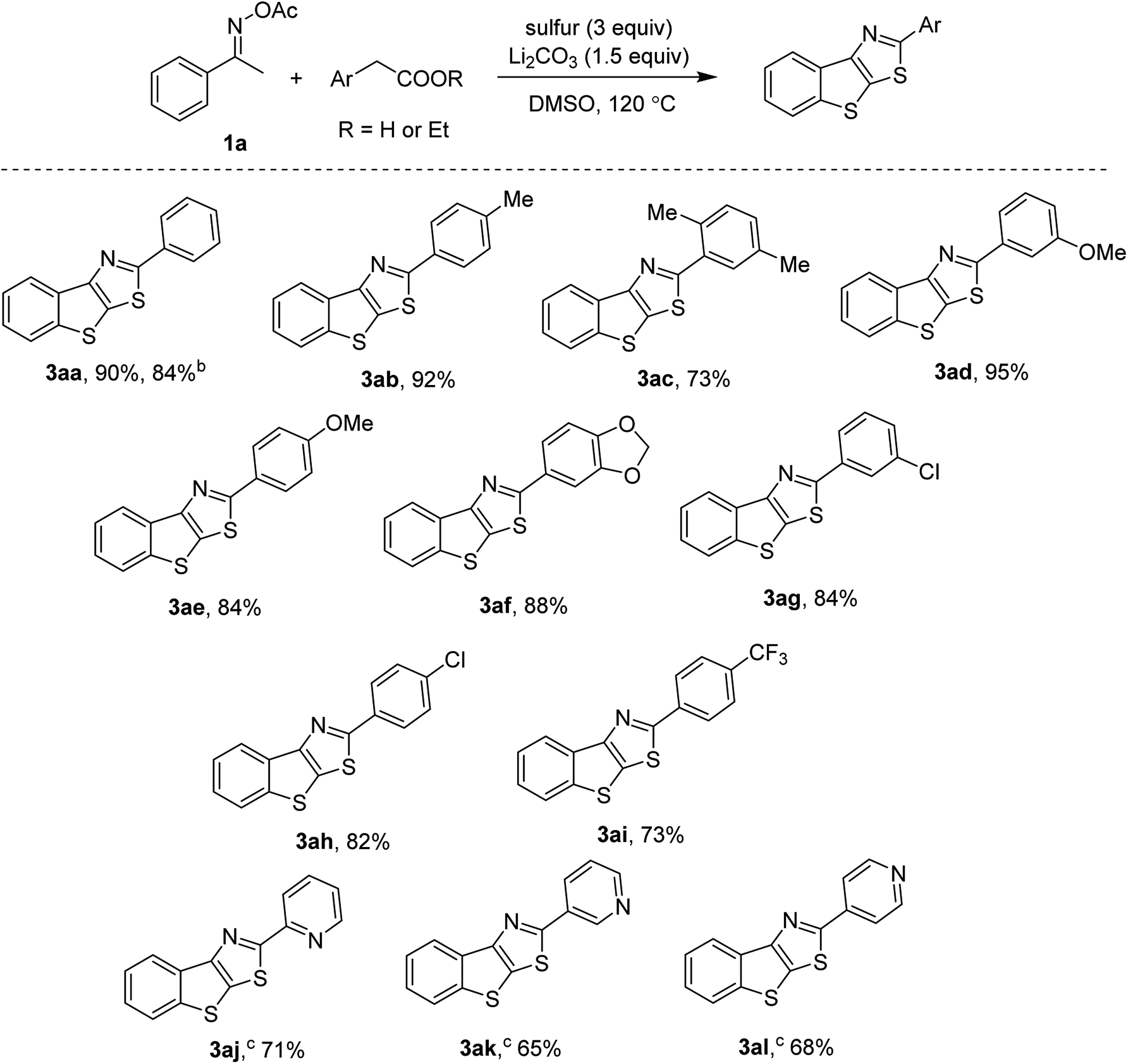 | ||
| Scheme 2 Annulation of arylacetic acid derivativesa. a1a (0.3 mmol), arylacetic acids (0.2 mmol), sulfur (0.6 mmol, 32 g mol−1), Li2CO3 (0.3 mmol), DMSO (1 mL), 120 °C, 3–12 h. Yields are isolated yields. b2 mmol scale. cEthyl esters of arylacetic acids (0.2 mmol) were used. | ||
Derivatives of ketoxime acetates could couple with phenylacetic acid 2a as well. Scope of the methyl oximes is presented in Scheme 3. Our conditions were compatible with many functionalities such as methoxy (3ca), chloro (3da), bromo (3ea, 3fa) and fluoro (3ga) groups. The most acidic C–H bonds were functionalized if meta-substituted ketoxime esters were used (3ca, 3ea). It should be noted that such substrates often suffered from the forging of regioisomeric mixtures.6a,b Since the bromo-substituted acetophenone oximes furnished the thienothiazoles in high yields (3ea, 3fa), our transformation is somewhat useful for further functionalization. Vinyl ketoxime esters were active, thus affording a trisubstituted thienothiazole in 81% yield (3ha).
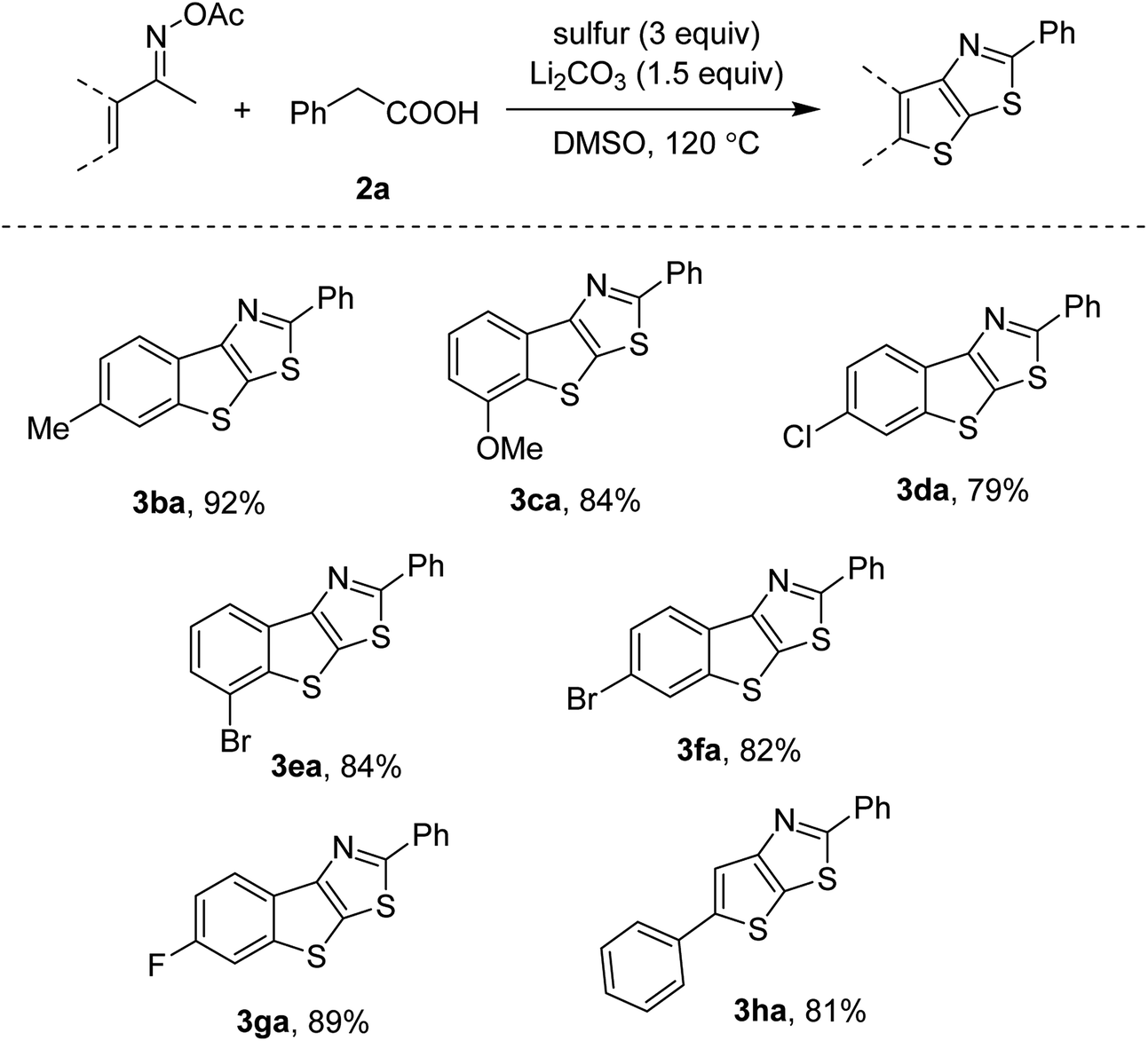 | ||
| Scheme 3 Scope of ketoxime acetatesa. aKetoxime acetates (0.3 mmol), phenylacetic acid 2a (0.2 mmol), sulfur (0.6 mmol, 32 g mol−1), Li2CO3 (0.3 mmol), DMSO (1 mL), 120 °C, 3–12 h. Yields are isolated yields. | ||
To explore the possible mechanism of the transformation, some control experiments were performed (Scheme 4). Running the reaction of acetophenone oxime ester 1a and phenylacetic acid 2a afforded some heterocyclic intermediates such as 4a and 5a after 30 min (detected by GC-MS). Since 2a unsuccessfully coupled with dithiazolethiones, such as 5g, under standard conditions, the observed species (i.e. 5a) was likely a resting state than an intermediate of the transformation. Meanwhile, reaction of 3-aminobenzothiophene hydrochloride (4a·HCl) afforded a quantitative yield of the desired thienothiazole 3aa. We speculated that the conversion of 1a to 4a likely proceeded through a single electron cyclization, since the reaction was slowed down in the presence of radical quenchers such as TEMPO or 1,1-diphenylethylene.5b Meanwhile, functionalization of phenylacetic acid was likely triggered by the formation of dibenzyl sulfide intermediate.7a The annulation of acetophenone oxime 1a with disulfide 6 furnished 3aa in 88% yield which was in comparison with the standard run.
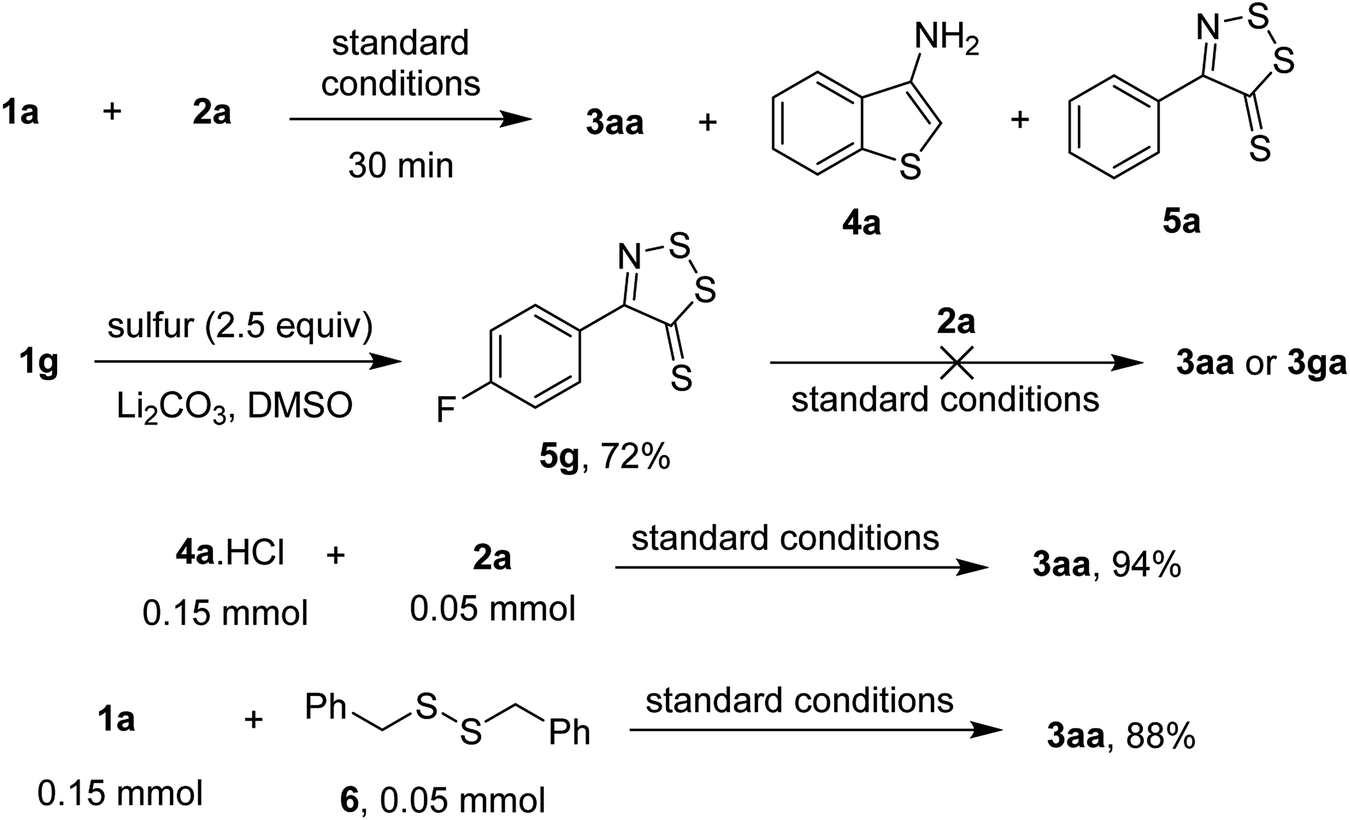 | ||
| Scheme 4 Mechanistic consideration. | ||
With the information in hands, we briefly proposed a possible mechanism (Scheme 5), commencing with a sulfur-mediated decarboxylative sulfuration of phenylacetic acid 2a to afford the benzylic polysulfide intermediate A. The interaction between S3˙−, formed by heating elemental sulfur in DMSO under basic conditions, and ketoxime acetate 1a delivered the iminosulfur radicals A or B, by which elimination of a disulfur moiety afforded radical C followed by a SET cyclization to furnish 3-aminobenzothiophene 4a. Such radical cyclization is supported by the results of intermolecular competition reactions and regioselectivity of meta-substituted ketoxime substrates Condensation of this species with the polysulfide A then afforded an iminobenzothiophene E. An addition of S3˙− to E would give the intermediates F or H. Subsequent cyclizations and cleavages of S–S bond afforded G followed by a further oxidation, in the presence of DMSO, to afford 3aa.
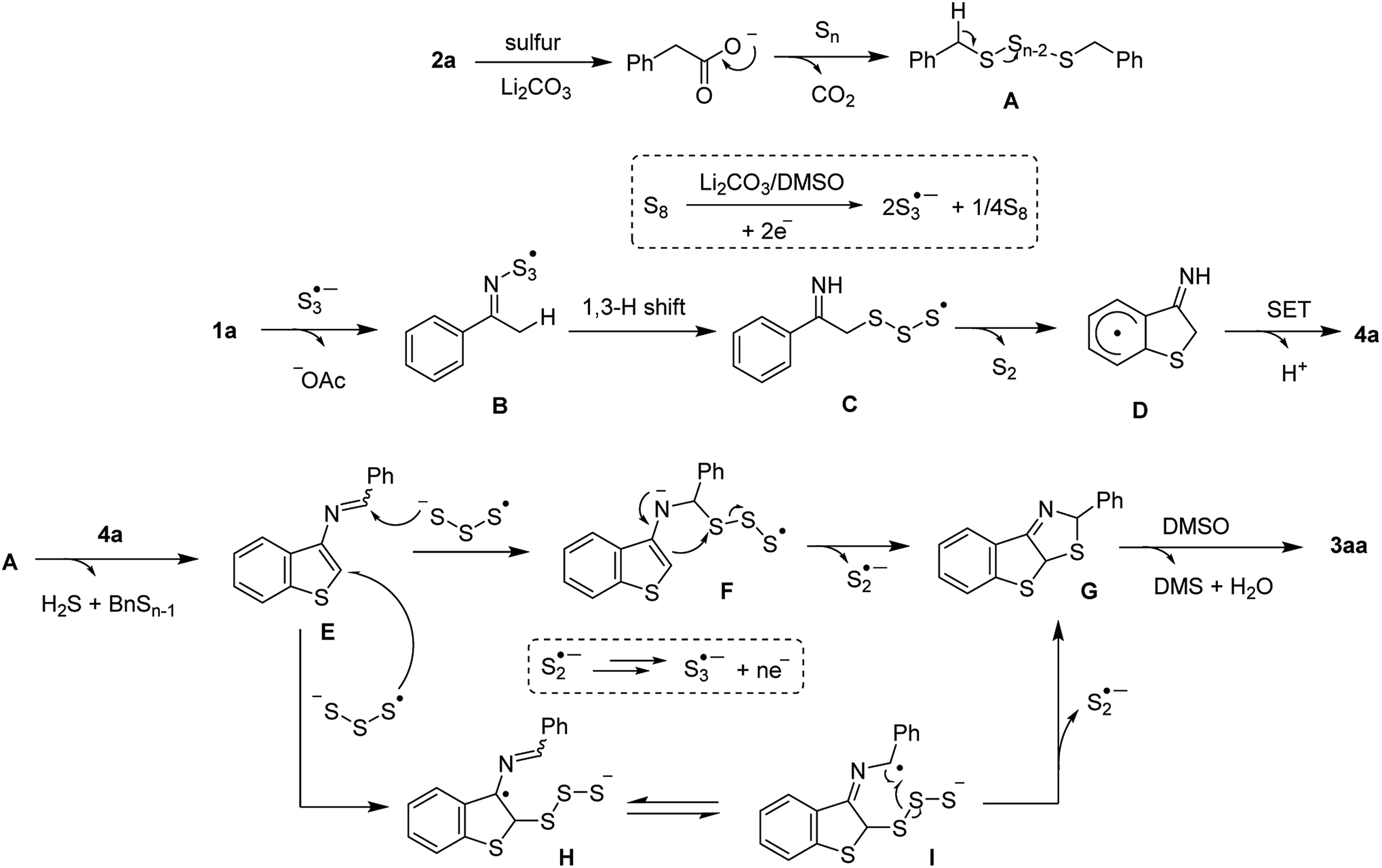 | ||
| Scheme 5 Plausible mechanism. | ||
It is perhaps more synthetically useful if late stage functionalization of 2-aryl benzothienothiazoles was possible. The presence of the chelating nitrogen in thiazole would help achieve the regioselective functionalization of C–H bonds (Scheme 6). Thus, thiazole-directed nitration of C–H bonds was achievable in the presence of CuCl2 catalyst and Fe(NO3)3·9H2O as a nitrating source, affording the product 7 in 77% yield.8
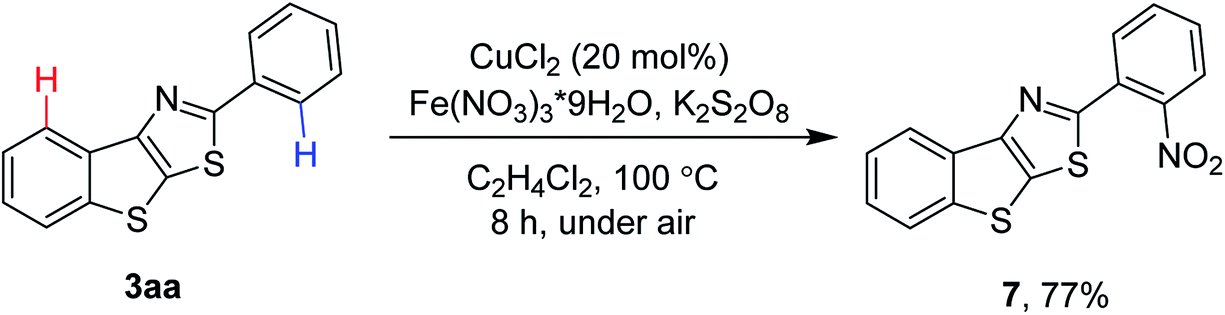 | ||
| Scheme 6 Directed C–H functionalization of 2-phenyl benzothienothiazolea. aCondition: 3aa (0.2 mmol), Fe(NO3)3·9H2O (0.4 mmol), anhydrous CuCl2 (20 mol%), K2S2O8 (0.4 mmol), 1,2-dichloroethane (3 mL), 100 °C, 8 h. Yield is isolated yield. | ||
Conclusions
In summary, we have developed a method for synthesis of fused thieno[3,2-d]thiazoles. The transformation only required Li2CO3 base to promote the coupling of C–H bonds in acetophenone oxime acetates with arylacetic acids and elemental sulfur. A number of functional groups were tolerant of reaction conditions. Reactions of ethyl arylacetates were also feasible, affording heteroaryl thienothiazoles in good yields. Ongoing projects will focus on selective C–H functionalization directed by the thieno-fused thiazole directing group.Experimental section
General information
All reagents and starting materials were obtained commercially and used as received without any further purification unless otherwise noted. Gas chromatographic (GC) analyses were performed using a Shimadzu GC 2010-Plus equipped with a flame ionization detector (FID) and an SPB-5 column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm). The GC yield was calculated using diphenyl ether as the internal standard. GC-MS analyses were analyzed on a Shimadzu GCMS-QP2010Ultra with a ZB-5MS column (length = 30 m, inner diameter = 0.25 mm, and film thickness = 0.25 μm). MS spectra were compared with the spectra gathered in the NIST library. The 1H NMR and 13C NMR were recorded on Bruker AV 500 spectrometers using residual solvent peak as a reference. HR-MS spectra were recorded by an Agilent HPLC 1200 Series coupled to Bruker micrOTOF-QII. Molecular mass of elemental sulfur is 32 g mol−1, unless otherwise noted. Compounds 4a·HCl6a and 6 (ref. 7a) were synthesized following the known procedures.2-Phenylbenzo[4,5]thieno[3,2-d]thiazole (3aa, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 48 mg (90%) of a white solid was obtained. This compound is known.6a Rf = 0.53 (hexane/dichloromethane = 2:1). Mp = 137–139 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.28 (d, J = 7.9 Hz, 1H), 8.08–8.03 (m, 2H), 7.85 (d, J = 8.1 Hz, 1H), 7.53–7.45 (m, 4H), 7.44–7.39 (m, 1H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.6, 156.1, 142.8, 134.0, 130.8, 130.5, 130.3, 129.1, 126.6, 125.1, 125.1, 123.3, 121.8.
:1), 48 mg (90%) of a white solid was obtained. This compound is known.6a Rf = 0.53 (hexane/dichloromethane = 2:1). Mp = 137–139 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.28 (d, J = 7.9 Hz, 1H), 8.08–8.03 (m, 2H), 7.85 (d, J = 8.1 Hz, 1H), 7.53–7.45 (m, 4H), 7.44–7.39 (m, 1H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.6, 156.1, 142.8, 134.0, 130.8, 130.5, 130.3, 129.1, 126.6, 125.1, 125.1, 123.3, 121.8.
Procedure for the synthesis of 3aa in 2 mmol scale. To a 50 mL Schlenk tube was added acetophenone ketoxime acetate 1a (531 mg, 3 mmol), phenylacetic acid 2a (272 mg, 2 mmol), elemental sulfur (192 mg, 6 mmol, 32 g mol−1), Li2CO3 (222 mg, 3 mmol), and DMSO (10 mL). The tube was sealed with septum and placed into a sand bath preheated to 120 °C. After 6 h, the mixture was cooled to room temperature and poured onto cold water. The precipitates were then extracted into ethyl acetate (3 × 30 mL). The organic phase was washed with saturated NaHCO3 solution (2 × 10 mL) and brine (2 × 10 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (gradient dichloromethane/hexane from 1
:8 to 1:2) to afford the desired product 449 mg of 3aa (84%) as a white solid. The spectrum information was consistent with that reported in the small scale.
2-(p-Tolyl)benzo[4,5]thieno[3,2-d]thiazole (3ab, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), p-tolylacetic acid 2b (30 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 3
:1), 52 mg (92%) of a white solid was obtained. This compound is known.6a Rf = 0.38 (hexane/dichloromethane = 3:1). Mp = 167–169 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.26 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 8.1 Hz, 2H), 7.83 (d, J = 8.1 Hz, 1H), 7.49 (t, J = 7.5 Hz, 1H), 7.39 (dd, J = 11.2, 4.1 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 2.42 (s, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.9, 156.0, 142.8, 140.6, 131.4, 130.6, 130.4, 129.8, 126.5, 125.08, 125.06, 123.4, 121.9, 21.5.
2-(2,5-Dimethylphenyl)benzo[4,5]thieno[3,2-d]thiazole (3ac, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 2,5-dimethylphenylacetic acid 2c (33 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 43 mg (73%) of a white solid was obtained. Rf = 0.44 (hexane/dichloromethane = 2:1). Mp = 174–176 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.23 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.51 (s, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.36–7.32 (m, 1H), 7.16 (d, J = 7.8 Hz, 1H), 7.11 (d, J = 7.8 Hz, 1H), 2.56 (s, 3H), 2.33 (s, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.5, 155.3, 142.7, 135.8, 133.8, 132.9, 131.5, 131.3, 130.65, 130.61, 129.5, 129.1, 125.1, 123.4, 121.9, 20.92, 20.91. HRMS (ESI+) m/z calculated for C17H14NS2+ (M + H)+ 296.0562, found 296.0564.
2-(3-Methoxyphenyl)benzo[4,5]thieno[3,2-d]thiazole (3ad, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 3-methoxyphenylacetic acid 2d (33 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 5
:1), 57 mg (95%) of a white solid was obtained. This compound is known.6a Mp = 123–125 °C Rf = 0.26 (hexane/dichloromethane = 5:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.28 (d, J = 7.9 Hz, 1H), 7.84 (d, J = 8.1 Hz, 1H), 7.65–7.62 (m, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.40 (ddd, J = 13.7, 10.4, 4.5 Hz, 2H), 7.01 (dd, J = 8.2, 2.5 Hz, 1H), 3.93 (s, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.4, 160.1, 156.1, 142.8, 135.3, 130.9, 130.6, 130.1, 125.17, 125.13, 123.4, 121.9, 119.2, 116.5, 111.4, 55.5.
2-(4-Methoxyphenyl)benzo[4,5]thieno[3,2-d]thiazole (3ae, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 4-methoxyphenylacetic acid 2e (33 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 50 mg (84%) of a white solid was obtained. This compound is known.6a Mp = 124–126 °C. Rf = 0.27 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.26 (d, J = 7.9 Hz, 1H), 8.03–7.95 (m, 2H), 7.84 (d, J = 8.1 Hz, 1H), 7.53–7.47 (m, 1H), 7.43–7.38 (m, 1H), 7.03–6.98 (m, 2H), 3.89 (s, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.6, 161.4, 155.9, 142.8, 130.6, 129.9, 128.1, 127.0, 125.05, 124.99, 123.4, 121.8, 114.4, 55.9.
2-(Benzo[d][1,3]dioxol-5-yl)benzo[4,5]thieno[3,2-d]thiazole (3af, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 3,4-(methylenedioxyphenyl)acetic acid 2f (36 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 1
:1), 55 mg (88%) of a white solid was obtained. Rf = 0.31 (hexane/dichloromethane = 1:1). Mp = 140–142 °C 1H NMR (500 MHz, CDCl3, ppm) δ 8.29 (d, J = 7.6 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.60 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.52 (t, J = 7.5 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 6.93 (d, J = 8.0 Hz, 1H), 6.08 (s, 2H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.3, 155.7, 149.6, 148.4, 142.8, 130.5, 130.1, 128.4, 125.11, 125.09, 123.4, 121.9, 121.2, 108.7, 106.9, 101.7. HRMS (ESI+) m/z calculated for C16H10NO2S2+ (M + H)+ 312.0147, found 312.0149.
2-(3-Chlorophenyl)benzo[4,5]thieno[3,2-d]thiazole (3ag, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 3-chlorophenylacetic acid 2g (34 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 51 mg (84%) of a white solid was obtained. This compound is known.6a Mp = 162–164 °C. Rf = 0.45 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.31 (d, J = 7.8 Hz, 1H), 8.11 (s, 1H), 7.96–7.92 (m, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.49–7.44 (m, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 168.7, 156.2, 142.9, 135.6, 135.2, 131.4, 130.4, 130.3, 130.2, 126.5, 125.3, 124.7, 123.4, 121.9. One carbon signal could not be located.
2-(4-Chlorophenyl)benzo[4,5]thieno[3,2-d]thiazole (3ah, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 4-chlorophenylacetic acid 2h (34 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 50 mg (82%) of a white solid was obtained. This compound is known.6a Mp = 160–161 °C. Rf = 0.48 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.19 (d, J = 7.8 Hz, 1H), 7.94–7.89 (m, 2H), 7.77 (d, J = 8.1 Hz, 1H), 7.46–7.41 (m, 1H), 7.41–7.32 (m, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 169.1, 156.2, 142.8, 136.2, 132.5, 131.1, 130.4, 129.3, 127.7, 125.3, 125.2, 123.4, 121.9.
2-(4-Trifluoromethylphenyl)benzo[4,5]thieno[3,2-d]thiazole (3ai, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), 4-(trifluoromethyl)phenylacetic acid 2i (41 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 3
:1), 49 mg (73%) of a white solid was obtained. This compound is known.6c Mp = 124–126 °C. Rf = 0.37 (hexane/dichloromethane = 3:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.28 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 8.5 Hz, 2H), 7.86 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.52 (t, J = 8.0 Hz, 1H), 7.44 (t, J = 8.0 Hz, 1H). 13C NMR (125 MHz, CDCl3, ppm) δ 168.4, 156.5, 142.9, 137.1, 131.9, 130.5 (q, J = 276.9 Hz), 128.1, 126.7, 126.1 (q, J = 3.7 Hz), 125.5, 125.3, 124.9, 123.4, 122.8, 121.9.
2-(Pyridin-2-yl)benzo[4,5]thieno[3,2-d]thiazole (3aj, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), ethyl 2-pyridylacetate 2j (33 mg, 0.2 mmol). After column chromatography (gradient hexane/ethyl acetate from 10
:1 to 2:1), 38 mg (71%) of a white solid was obtained. This compound is known.6a Mp = 194–196 °C. Rf = 0.63 (hexane/ethyl acetate = 2:1). 1H NMR (500 MHz, DMSO-d6, ppm) δ 8.70 (d, J = 3.8 Hz, 1.0 Hz), 8.27 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 7.6 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.04 (t, J = 7.0 Hz, 1H), 7.59–7.50 (m, 3H). 13C NMR (125 MHz, DMSO-d6, ppm) δ 171.9, 155.7, 151.1, 150.3, 143.2, 138.4, 134.6, 130.4, 125.9, 125.91, 125.87, 124.5, 121.4, 119.7.
2-(Pyridin-3-yl)benzo[4,5]thieno[3,2-d]thiazole (3ak, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), ethyl 3-pyridylacetate 2k (33 mg, 0.2 mmol). After column chromatography (gradient hexane/ethyl acetate from 10
:1 to 2:1), 35 mg (65%) of a white solid was obtained. This compound is known.6c Rf = 0.57 (hexane/ethyl acetate = 2:1). Mp = 122–124 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 9.21 (bs, 1H), 8.63 (bs, 1H), 8.29 (d, J = 7.8 Hz, 1H), 8.21 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 8.1 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.40–7.34 (m, 2H). 13C NMR (125 MHz, CDCl3, ppm) δ 166.5, 156.5, 150.4, 147.2, 142.9, 134.0, 131.7, 130.3, 125.5, 125.3, 124.0, 123.5, 122.0. One carbon signal could not be located.
2-(Pyridin-4-yl)benzo[4,5]thieno[3,2-d]thiazole (3al, Scheme 2): following the general procedure A. Acetophenone oxime acetate 1a (53 mg, 0.3 mmol), ethyl 4-pyridylacetate 2l (33 mg, 0.2 mmol). After column chromatography (gradient hexane/ethyl acetate from 4
:1 to 1:1), 36 mg (68%) of a yellow solid was obtained. This compound is known.6a Rf = 0.64 (hexane/ethyl acetate = 1:1). Mp = 170–172 °C. 1H NMR (500 MHz, DMSO-d6, ppm) δ 8.76 (dd, J = 4.5, 1.5 Hz, 2H), 8.20 (d, J = 7.7 Hz, 1H), 8.13 (d, J = 8.0 Hz, 1H), 8.00 (dd, J = 4.5, 1.5 Hz, 2H), 7.57 (td, J = 7.5, 1.0 Hz, 1H), 7.51 (td, J = 7.5 Hz, 1.0 Hz, 1H). 13C NMR (125 MHz, DMSO-d6, ppm) δ 167.8, 155.7, 151.3, 143.4, 140.4, 134.4, 130.1, 126.2, 125.9, 124.6, 121.6, 120.4.
6-Methyl-2-phenylbenzo[4,5]thieno[3,2-d]thiazole (3ba, Scheme 3): following the general procedure A. 4-Methylacetophenone oxime acetate 1b (57 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 52 mg (92%) of a white solid was obtained. This compound is known.6a Rf = 0.41 (hexane/dichloromethane = 2:1). Mp = 126–128 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.15 (d, J = 8.1 Hz, 1H), 8.04 (dd, J = 8.0, 1.4 Hz, 2H), 7.63 (s, 1H), 7.47 (tdd, J = 6.9, 4.6, 2.5 Hz, 3H), 7.32 (d, J = 8.1 Hz, 1H), 2.51 (s, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.4, 156.1, 143.1, 135.3, 134.1, 130.2, 129.8, 129.1, 128.3, 126.7, 126.6, 123.3, 121.4, 21.6.
5-Methoxy-2-phenylbenzo[4,5]thieno[3,2-d]thiazole (3ca, Scheme 3): following the general procedure A. 3-Methoxyacetophenone oxime acetate 1c (62 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 1
:1), 50 mg (84%) of a white solid was obtained. This compound is known.6a Rf = 0.42 (hexane/dichloromethane = 1:1). Mp = 132–134 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.00 (dd, J = 8.0 Hz, 1.5 Hz, 2H), 7.87 (d, J = 8.0 Hz, 1H), 7.44–7.38 (m, 4H), 6.83 (d, J = 8.0 Hz, 1H), 3.97 (s, 3H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.7, 154.3, 149.6, 133.8, 131.9, 131.4, 130.9, 130.4, 129.1, 126.6, 126.5, 114.6, 105.5, 55.7.
6-Chloro-2-phenylbenzo[4,5]thieno[3,2-d]thiazole (3da, Scheme 3): following the general procedure A. 4-Chloroacetophenone oxime acetate 1d (63 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 48 mg (79%) of a white solid was obtained. This compound is known.6a Mp = 186–188 °C. Rf = 0.52 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.12 (d, J = 8.4 Hz, 1H), 7.98 (dd, J = 7.8, 1.6 Hz, 2H), 7.76 (d, J = 1.7 Hz, 1H), 7.44–7.39 (m, 4H). 13C NMR (125 MHz, CDCl3, ppm) δ 171.2, 155.3, 143.7, 133.8, 131.1, 131.0, 130.5, 129.2, 129.0, 126.7, 125.9, 123.0, 122.5.
5-Bromo-2-phenylbenzo[4,5]thieno[3,2-d]thiazole (3ea, Scheme 3): following the general procedure A. 3-Bromoacetophenone oxime acetate 1e (77 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 58 mg (84%) of a white solid was obtained. This compound is known.6a Mp = 176–178 °C. Rf = 0.48 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.28 (d, J = 7.8 Hz, 1H), 8.08 (dd, J = 7.7, 1.5 Hz, 2H), 7.60 (d, J = 7.7 Hz, 1H), 7.56–7.50 (m, 3H), 7.44 (t, J = 7.8 Hz, 1H). 13C NMR (125 MHz, CDCl3, ppm) δ 171.0, 144.1, 133.8, 132.1, 131.5, 130.6, 129.2, 127.9, 126.7, 126.5, 120.7, 116.4. One carbon signal could not be located.
6-Bromo-2-phenylbenzo[4,5]thieno[3,2-d]thiazole (3fa, Scheme 3): following the general procedure A. 4-Bromoacetophenone oxime acetate 1f (77 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 57 mg (82%) of a white solid was obtained. This compound is known.6a Mp = 188–190 °C. Rf = 0.42 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.55 (d, J = 7.8 Hz, 2H), 8.15 (d, J = 8.4 Hz, 1H), 7.79 (s, 1H), 7.62 (t, J = 7.4 Hz, 1H), 7.51 (t, J = 7.7 Hz, 2H), 7.44 (dd, J = 8.4, 1.6 Hz, 1H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.1, 156.2, 144.1, 138.8, 134.8, 133.9, 132.2, 131.2, 129.0, 128.5, 126.3, 123.2, 122.8.
6-Fluoro-2-phenylbenzo[4,5]thieno[3,2-d]thiazole (3ga, Scheme 3): following the general procedure A. 4-Fluoroacetophenone oxime acetate 1g (59 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 2
:1), 51 mg (89%) of a white solid was obtained. This compound is known.6c Mp = 140–142 °C. Rf = 0.35 (hexane/dichloromethane = 2:1). 1H NMR (500 MHz, CDCl3, ppm) δ 8.21 (dd, J = 9.0 Hz, 5.0 Hz, 1H), 8.03 (d, J = 8.0 Hz, 1.5 Hz, 2H), 7.53–7.46 (m, 3H), 7.25 (d, J = 10 Hz, 2.5 Hz, 1H, overlapping with CHCl3 signal). 13C NMR (125 MHz, CDCl3, ppm) δ 171.0, 160.8 (d, 1JC–F = 245.6 Hz), 155.3, 143.6 (d, 3JC–F = 10.0 Hz), 133.9, 130.4, 129.8 (d, 4JC–F = 2.9 Hz), 129.1, 127.1, 126.6, 122.7 (d, 3JC–F = 9.2 Hz), 113.8 (d, 2JC–F = 23.9 Hz), 110.0 (d, 2JC–F = 25.8 Hz).
2,5-Diphenylthieno[3,2-d]thiazole (3ha, Scheme 3): following the general procedure A. (2E,3E)-4-Phenylbut-3-en-2-one O-acetyl oxime 1h (61 mg, 0.3 mmol), phenylacetic acid 2a (27 mg, 0.2 mmol). After column chromatography (hexane/dichloromethane = 4
:1), 47 mg (81%) of a white solid was obtained. This compound is known.6a Mp = 195–197 °C. Rf = 0.31 (hexane/dichloromethane = 4:1). 1H NMR (500 MHz, CDCl3, ppm) δ 7.92 (dd, J = 8.0 Hz, 1.0 Hz, 2H), 7.63 (s, 1H), 7.57 (d, J = 7.5 Hz, 2H), 7.42–7.34 (m, 5H), 7.27 (t, J = 7.5 Hz, 1H). 13C NMR (125 MHz, CDCl3, ppm) δ 170.7, 161.6, 147.8, 134.4, 133.8, 130.5, 129.5, 129.1, 128.2, 126.7, 125.8, 114.8. One carbon signal could not be located.
4-(4-Fluorophenyl)-5H-1,2,3-dithiazole-5-thione (5g, Scheme 4). To an 8 mL screw-cap vial was added 4-fluoroacetophenone ketoxime acetate 1g (71 mg, 0.4 mmol), Li2CO3 (15 mg, 0.2 mmol), elemental sulfur (32 mg, 1 mmol), and DMSO (0.5 mL). The vial was stirred at 120 °C for 12 h. Upon completion of the reaction, the mixture was cooled to room temperature, and NaHCO3 (5% aqueous solution, 2 mL) was added. The organic components were consequently extracted into ethyl acetate (3 × 2 mL), washed with brine (2 × 1 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (hexane/dichloromethane 10
:1) to afford 66 mg (72%) of a brown solid. This compound is known.9 Rf = 0.72 (hexane/dichloromethane 2:1). Mp = 150–152 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 7.98–7.95 (m, 2H), 7.16–7.12 (m, 2H). 13C NMR (125 MHz, CDCl3, ppm) δ 207.9, 166.7, 164.0 (d, 1JC–F = 251.9 Hz), 131.7 (d, 3JC–F = 8.5 Hz), 127.5 (d, 4JC–F = 3.5 Hz), 115.2 (d, 2JC–F = 21.7 Hz).
2-(2-Nitrophenyl)benzo[4,5]thieno[3,2-d]thiazole (7, Scheme 6). To a 25 mL Schlenk tube was added 2-phenylbenzo[4,5]thieno[3,2-d]thiazole (53 mg, 0.2 mmol), Fe(NO3)3·9H2O (162 mg, 0.4 mmol), anhydrous CuCl2 (5.4 mg, 0.04 mmol), K2S2O8 (108 mg, 0.4 mmol), and 1,2-dichloroethane (3 mL). The reaction mixture was stirred at 100 °C for 8 h in an oil bath. After cooling to room temperature, the mixture was diluted with chloroform (15 mL) and filtered through a short pad of silica gel. The organic layer was washed with brine (2 × 5 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. After column chromatography (hexane/dichloromethane 1
:1), 48 mg (77%) of a yellow solid was obtained. Rf = 0.2 (hexane/dichloromethane 1:1). Mp = 187–189 °C. 1H NMR (500 MHz, CDCl3, ppm) δ 8.21 (dd, J = 7.5, 1.5 Hz, 1H), 7.86 (dd, J = 8.0, 1.0 Hz, 1H), 7.77 (dd, J = 7.5 Hz, 1.5 Hz, 1H), 7.74 (dd, J = 8.0, 1.0 Hz, 1H), 7.49–7.42 (m, 4H). 13C NMR (125 MHz, CDCl3, ppm) δ 169.1, 155.2, 140.8, 135.4, 135.3, 132.9, 131.7, 131.5, 130.4, 126.5, 125.7, 124.7, 124.3, 121.8. HRMS (ESI+) m/z calculated for C15H9N2O2S2+ (M + H)+ 313.0100, found 313.0104. One carbon signal could not be located.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The Vietnam National University - Ho Chi Minh City (VNU-HCM) is acknowledged for financial support via project no. NCM2019-20-01.References
- For selected examples, see: (a) F. Kakiuchi, S. Kan, K. Igi, N. Chatani and S. Murai, J. Am. Chem. Soc., 2003, 125, 1698 CrossRef CAS PubMed; (b) N. Kimura, T. Kochi and F. Kakiuchi, J. Am. Chem. Soc., 2017, 139, 14849 CrossRef CAS; (c) R.-Y. Zhu, Z.-Q. Li, H. S. Park, C. H. Senanayake and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 3564 CrossRef CAS PubMed; (d) D. Xing, X. Qi, D. Marchant, P. Liu and G. Dong, Angew. Chem., Int. Ed., 2019, 58, 4366 CrossRef CAS; (e) S. St John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840 RSC; (f) Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617 CrossRef CAS PubMed; (g) B. Li, K. Seth, B. Niu, L. Pan, H. Yang and H. Ge, Angew. Chem., Int. Ed., 2018, 57, 3401 CrossRef CAS PubMed; (h) X.-Y. Chen and E. Sorensen, J. Chem. Sci., 2018, 9, 8951 RSC; (i) W. Xu and N. Yoshikai, Org. Lett., 2018, 20, 1392 CrossRef CAS PubMed; (j) Y. Hu, B. Zhou, H. Chen and C. Wang, Angew. Chem., Int. Ed., 2018, 57, 12071 CrossRef CAS PubMed.
- For review, see: (a) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155 RSCSelected examples: (b) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed; (c) J. M. Neely and T. Rovis, J. Am. Chem. Soc., 2013, 135, 66 CrossRef CAS PubMed; (d) B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896 CrossRef CAS PubMed; (e) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234 CrossRef CAS PubMed; (f) B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2015, 54, 12968 CrossRef CAS PubMed; (g) Y. Li, H. Chen, L.-B. Qu, K. N. Houk and Y. Lan, ACS Catal., 2019, 9, 7154 CrossRef CAS.
- (a) S. Liu and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 6918 CrossRef CAS PubMed; (b) Z.-H. Ren, Z.-Y. Zhang, B.-Q. Yang, Y. Y. Wang and Z.-H. Guan, Org. Lett., 2011, 13, 5394 CrossRef CAS PubMed; (c) Y. Wei and N. Yoshikai, J. Am. Chem. Soc., 2013, 135, 3756 CrossRef CAS PubMed; (d) H. Huang, J. Cai, X. Ji, F. Xiao, Y. Chen and G.-J. Deng, Angew. Chem., Int. Ed., 2016, 55, 307 CrossRef CAS PubMed; (e) C. Zhu, R. Zhu, H. Zeng, F. Chen, C. Liu, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2017, 56, 13324 CrossRef CAS PubMed; (f) Z. Zhu, X. Tang, J. Cen, J. Li, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 3767 RSC.
- Selected examples: (a) J. P. Lutz, M. D. Hannigan and A. J. McNeil, Coord. Chem. Rev., 2018, 376, 225 CrossRef CAS; (b) I. Nakanishi, S. Itoh, T. Suenobu and S. Fukuzumi, Angew. Chem., Int. Ed., 1998, 37, 992 CrossRef CAS; (c) C. Zhang and X. Zhu, Acc. Chem. Res., 2017, 50, 1342 CrossRef CAS PubMed; (d) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef CAS PubMed.
- (a) T. B. Nguyen and P. Retailleau, Green Chem., 2018, 20, 387 RSC; (b) P. H. Pham, K. X. Nguyen, H. T. B. Pham, T. T. Nguyen and N. T. S. Phan, Org. Lett., 2019, 21, 8795 CrossRef CAS PubMed.
- (a) H. Huang, Z. Xu, X. Ji, B. Li and G.-J. Deng, Org. Lett., 2018, 20, 4917 CrossRef CAS PubMed; (b) H. Huang, Q. Wang, Z. Xu and G.-J. Deng, Adv. Synth. Catal., 2019, 361, 591 CrossRef CAS; (c) P. Zhou, Y. Huang, W. Wu, W. Yu, J. Li, Z. Zhu and H. Jiang, Org. Biomol. Chem., 2019, 17, 3424 RSC; (d) P. Zhou, Y. Huang, W. Wu, J. Zhou, W. Yu and H. Jiang, Org. Lett., 2019, 21, 9976 CrossRef CAS PubMed.
- (a) T. B. Nguyen and P. Retailleau, Org. Lett., 2019, 21, 279 CrossRef CAS PubMed; (b) T. Guntreddi, R. Vanjari and K. N. Singh, Org. Lett., 2015, 17, 976 CrossRef CAS PubMed.
- B. Vinayak, A. Ashok and M. Chandrasekharam, Eur. J. Org. Chem., 2017, 2017, 7127 CrossRef CAS.
- L. S. Konstantinova, O. I. Bol'shakov, N. V. Obruchnikova, H. Laborie, A. Tanga, V. Sopéna, I. Lanneluc, L. Picot, S. Sablé, V. Thiéry and O. Rakitin, Bioorg. Med. Chem. Lett., 2009, 19, 136 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C spectra of the products. See DOI: 10.1039/d0ra00808g |
| ‡ P. H. P. and K. X. N. equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |