
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the limits of amide–triazole isosteric substitution in bisamide-based physical gels†
Markus Tautza,
Juan Torras bc,
Santiago Grijalvode,
Ramón Eritjade,
César Saldíasf,
Carlos Alemánbc and
David Díaz Díaz*ag
bc,
Santiago Grijalvode,
Ramón Eritjade,
César Saldíasf,
Carlos Alemánbc and
David Díaz Díaz*ag
aInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de
bDepartament d'Enginyeria Química, EEBE, Universitat Politècnica de Catalunya, C/Eduard Maristany, 10-14, Ed. 12, 08019, Barcelona, Spain
cBarcelona Research Center for Multiscale Science and Engineering, Universitat Politècnica de Catalunya, C/Eduard Maristany, 10-14, 08019, Barcelona, Spain
dBiomedical Research Networking Centre in Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), Jordi Girona 18-26, 08034 Barcelona, Spain
eInstitute of Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, 08034 Barcelona, Spain
fDepartamento de Química Física, Facultad de Química, Pontificia Universidad Católica de Chile, Casilla 302, Correo 22, Santiago, Chile
gInstituto de Productos Naturales y Agrobiología del CSIC, Avda. Astrofísico Francisco Sánchez 3, 38206 La Laguna, Tenerife, Spain. E-mail: d.diaz.diaz@ipna.csic.es
First published on 3rd July 2019
Abstract
Gelation of organic solvents using N,N′-((1S,2S)-cyclohexane-1,2-diyl)didodecanamide (C12–Cyc) is driven by its self-assembly via antiparallel hydrogen bonds and van der Waals intermolecular interactions. In this work we carried out a dual isosteric substitution of the two amide groups with 1,2,3-triazole rings affording the corresponding isosteric gelator (click-C12–Cyc). A detailed comparative study in terms of the gelation ability and gel properties demonstrated that the 1,2,3-triazoles can take over all of the functions derived from the amide groups offering a versatile strategy for tuning the properties of the corresponding gels. This is not an obvious outcome because the directional amide groups in C12–Cyc constitute the source of the hydrogen bonds to build the 3D self-assembled network. Furthermore, theoretical calculations revealed that click-C12–Cyc can adopt a wide variety of interacting patterns, whose relative stability depends on the polarity of the environment, this is in good agreement with the experimental data obtained regarding its gelation ability. Other important features of click-C12–Cyc for potential practical applications are its non-cytotoxic character and its phase-selective gelation of water–oil mixtures.
Introduction
Hierarchical gel-based materials have received increasing attention over the past few decades1–3 owing to their fascinating molecular architectures and potential for high-tech applications in various fields including, among others,4,5 the preparation of sensors,6 liquid crystals,7 conductive scaffolds,8,9 templates for cell growth10 and inorganic structures,11 catalysis,12,13 cosmetics14 and food industries.15 In contrast to chemical gels16 that are formed by covalent bonds, (e.g. cross-linked polymers), physical (or supramolecular) gels17–19 are formed by self-assembly of low-molecular-weight (LMW) compounds in solution into 1D fibers that are nanometers in diameter and millimeters in length, followed by their physical entanglement generating a 3D scaffold. The solid-like appearance of the gels results from the immobilization of solvent molecules (the major component) into the interstices of the 3D network via surface tension and capillary forces, increasing the viscosity of the medium by factors up to 1010. Owing to the non-covalent nature of the interactions, mainly hydrogen-bonding, π-stacking and van der Waals interactions, most physical gels show reversible gel-to-sol transitions that can be triggered by different external stimuli (e.g. temperature, pH, ionic strength, irradiation).20–22In 1996 Hanabusa and co-workers23 reported the remarkable gelation ability of alkylamides derived from trans-1,2-diaminocyclohexane and the resulting chiral aggregates in organic solvents. Specifically, N,N′-((1S,2S)-cyclohexane-1,2-diyl)didodecanamide (C12–Cyc, Fig. 1) and its enantiomer were found to gel different organic solvents. Molecular modeling and mechanistic studies24 have shown that the two equatorial amide–NH and amide–CO groups direct themselves antiparallel to each other and perpendicular to the cyclohexyl ring, forming an extended asymmetric molecular tape stabilized by two intermolecular hydrogen bonds between each molecule. Subsequently, the tape-like molecular aggregates are interlocked through van der Waals interactions and transformed into helical fiber-like aggregates, which finally immobilize the organic fluids. The length of the aliphatic chains has been found to play a critical role in the gelation ability of this type of gelator, which should contain at least six carbons.25 Moreover, the chiral gelator should also be enantiomerically pure in order to maximize the gelation efficiency. The easy preparation and reliability of this LMW gelator has made it one of the most utilized and versatile systems in the field of physical gels.26,27
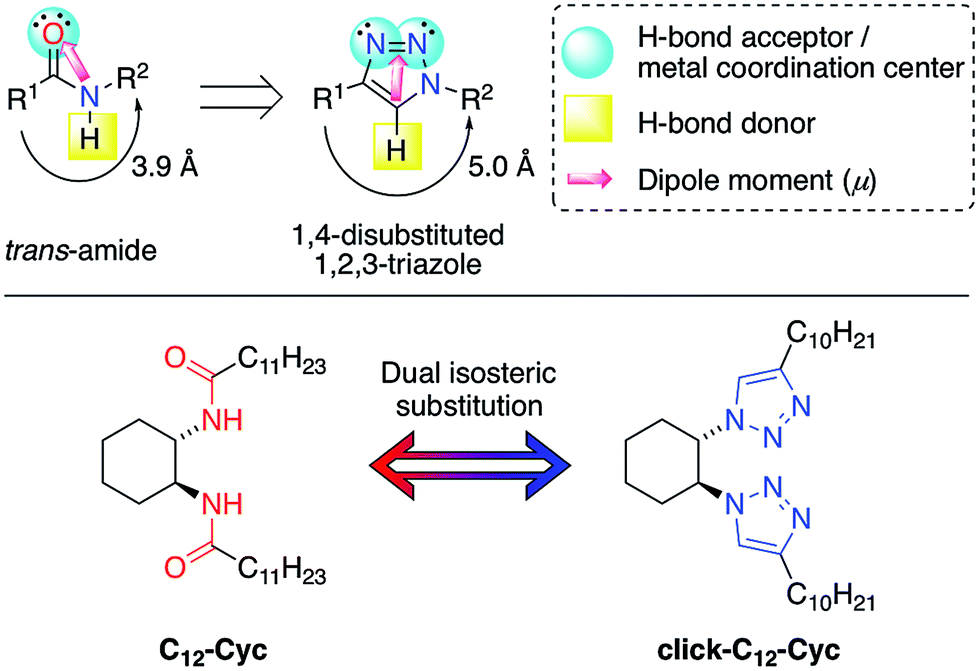 | ||
| Fig. 1 Top: isosteric relationship between trans-amides and 1,4-disubstituted 1,2,3-triazoles. Bottom: double isosteric replacement of the LMW gelator C12–Cyc investigated in this work. | ||
Among the large number of LMW gelators with different structures, those containing triazole rings have attracted considerable attention during the last few years.28–35 Recently, our group has demonstrated that the isosteric substitution of amides by 1,2,3-triazole groups in LMW gelators enables the modification of different gel properties.36,37 In general, isosteres possess an almost equal molecular shape and volume, approximately the same distribution of electrons and exhibit similar physical properties.38,39 Although this is still a recent concept in materials synthesis,36,37,40 it has a long and successful history in medicinal chemistry for drug development.41 In a previous and seminal work, we replaced an amide group with a 1,2,3-triazole in a glutamic acid-derived LMW gelator.36 Therein, both the amide group and the carboxylic acid groups played a key role in the aggregation pattern of the gelator via the hydrogen bonds. After this study we wondered whether 1,2,3-triazoles could replace all of the hydrogen bond centers in an LMW gelator without sacrificing its gelation capacity. To answer this question, we decided to carry out a dual isosteric replacement of both the amide groups in C12–Cyc with 1,2,3-triazole rings affording the corresponding isostere click-C12–Cyc (Fig. 1). In principle, the preservation of the gelation ability upon dual isosteric substitution is not obvious because the directional amide groups in this example constitute the source of hydrogen bonds to build the 3D network. Herein, we describe the preparation of both isosteres and a detailed comparative study in terms of the gelation ability and gel properties. The results of this research have revealed the remarkable potential of this approach for fine-tuning the functionality of physical gels.
Experimental
Materials
Unless otherwise indicated, all chemicals, reagents and solvents were of analytical grade, purchased from commercial sources and used as received without further purification. Specifically, (S,S)- and (R,R)-cyclohexanediamine were purchased from Sigma-Aldrich, 1-dodecyne from TCI Europe, and 1-tridecyne from Alfa Aesar. Additionally, double-distilled water was purified using a Millipore water-purifying system (Merck) prior to usage.Characterization methods
Low-resolution mass spectroscopy (MS) was carried out on a Varian MAT 311A. Elemental analyses were performed on a Heraeus Mikro-Rapid analyzer. Ultraviolet-visible (UV-vis) spectroscopy was performed using a Varian Cary 50 UV spectrophotometer and quartz–glass cuvettes of 0.5 cm thickness. Fourier transform infrared (FT-IR) spectra were recorded at room temperature using an Excalibur FTS 3000 FT-IR spectrometer (Biorad) equipped with a single reflection ATR (attenuated total reflection) accessory (Golden Gate, Diamond). Thin layer chromatography (TLC) analyses were performed using fluorescent-indicating plates (aluminum sheets precoated with silica gel 60 F254, thickness 0.2 mm, Merck), and visualization was achieved using UV light (λmax = 254 nm) and staining with phosphomolybdic acid and/or iodine. Melting points were measured on a Stuard SMP 10 apparatus. Optical activities were measured using an Anton Paar MCP500 polarimeter.
The thermal gel-to-sol transition temperature (Tgel) values were determined using a custom made set-up in which the sealed vial containing 100 μL of the gel was placed into a mold of an alumina block and heated at 1 °C/5 min using an electric heating plate equipped with a temperature control couple (Fig. S1†). The obtained values were verified using differential scanning calorimetry (DSC) measurements of one model gel (40 g L−1 C12–Cyc in cyclohexane). Herein, the temperature at which the gel started to break was defined as Tgel. Each measurement was repeated at least twice and the average value reported. Tgel values were found to be almost unaltered within a difference of 1–2 °C after several heating–cooling cycles.
Oscillatory rheology was performed with an AR 2000 Advanced rheometer (TA Instruments) equipped with a Julabo C cooling system. A 1000 μm gap setting and a torque of 5 × 10−4 N m−1 at 25 °C were used for the measurements in a plain-plate (40 mm, stainless steel). The following experiments were carried out for each sample, using ca. 2 mL total gel volume: (i) dynamic strain sweep (DSS): variation of G′ (storage modulus) and G′′ (loss modulus) with strain (from 0.01 to 100%); (ii) dynamic frequency sweep (DFS): variation of G′ and G′′ with frequency (from 0.1 to 10 Hz at 0.1% strain); and (iii) dynamic time sweep (DTS): variation of G′ and G′′ with time keeping the strain and frequency values constant and within the linear viscoelastic regime as determined by the DSS and DFS measurements (strain = 0.1% strain; frequency = 1 Hz).
Differential scanning calorimetry measurements were performed on a PerkinElmer DSC 8000 differential scanning calorimeter. The DSC thermogram was obtained under a dynamic argon atmosphere (gas flow rate = 25 mL min−1) at a heating rate of 5 °C min−1 from 25 to 50 °C and 1 °C min−1 from 50 to 80 °C. The measurement was carried out three times. An empty sample holder was used as the reference.
The FT-IR spectra were recorded as indicated above for the characterization of compounds. The CD spectra were recorded using a Jasco J-710 Spectropolarimeter. X-ray diffraction spectroscopy (XRD) was performed on a STOE STADI P powder diffractometer (StartFragment Transmission mode, flat samples, Dectris MYTHEN 1Kk microstrip solid-state detector) with CuKα radiation operating at 40 kV and 40 mA. Conditions were 0.015°, time 600 s per step, and a 2 theta range of 0–90°.
Field emission scanning electron microscopy (FESEM, resolution 0.8 nm) images of the xerogels were obtained using a Carl Zeiss Merlin, field emission scanning electron microscope equipped with a digital camera and operated at an accelerating voltage of 10 kV. For visualization, samples were prepared by the freeze-drying (FD) method: an Eppendorf tube containing the corresponding gel-material (100–200 μL) was frozen in liquid nitrogen or dry ice/acetone and the solvent was immediately evaporated under reduced pressure (0.6 mmHg) for 2 days at room temperature (RT). The obtained fibrous solid was placed on top of a tin plate and shielded by Pt (40 mA during 30 s; film thickness ≈ 5 nm).
Synthesis of compounds
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 100 mL) was added 1H-imidazole-4-sulfonyl azide (3) (379 mg, 2.2 mmol), CuSO4·5H2O (11.4 mg, 0.018 mmol) and K2CO3 (2.18 g, 15.8 mmol). The mixture was stirred for 3 days at RT and thereafter extracted with EtOAc (4 × 50 mL). The combined organic phases were dried over anhydrous Na2SO4 and most of the solvent was removed under reduced pressure. The concentrated solution was purified using column chromatography (eluent: hexane/Et2O/CHCl3 100:3:3 (ref. 45)) and dissolved in toluene for safe storage. Several batches afforded (1S,2S)-1,2-diazidocyclohexane (4) in yields ranging from 31% to 97% (yields were determined by 1H NMR by integral comparison using the toluene peaks as a reference). 1H NMR (400 MHz, CDCl3) δ (ppm) 3.20 (m, 2H), 2.07 (m, 2H), 1.79 (m, 2H), 1.29 (m, 4H).:1 v/v, 3.8 mL) a solution of 4 in toluene (123.5 mg, 0.743 mmol) was added. Subsequently, separate solutions of CuSO4·5H2O (249.7 mg, 1 mmol) and sodium ascorbate (198.1 mg, 1 mmol) in water (1 mL) were added alternately until the product precipitated with blurring of the clear solution. After stirring for 3 days, the reaction mixture was dissolved in a mixture of EtOAc and THF (1:1 v/v, 100 mL) and washed with aqueous Na2EDTA (0.1 mol L−1, 3 × 50 mL), brine (50 mL), water (50 mL) and dried over anhydrous Na2SO4. Finally, the solvent was removed under reduced pressure and recrystallized from acetone affording click-C12–Cyc as a light yellow solid in a 32% yield (117 mg, 0.236 mmol). Mp 129 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 6.79 (s, 2H), 4.73 (m, 2H), 2.52 (m, 4H), 2.31 (m, 4H), 2.04 (m, 2H), 1.59 (m, 2H), 1.49 (m, 4H), 1.24 (s, 28H), 0.88 (t, J = 6.64 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ (ppm) 32.46, 31.94, 29.65, 29.58, 29.38, 29.35, 29.25, 25.42, 24.65, 22.71, 14.13; FT-IR (ATR) νmax (cm−1) 3146, 3068, 2952, 2915, 2848, 1543, 1446, 1223, 1054, 809, 719; MS (ESI): m/z = 250.2 [M + 2H]2+, 499.4 [M + H]+, 997.9 [2M + H]+; [α]20D −44.7 ± 0.3° (c = 1; H2O). Elemental analysis calculated for C30H54N6: C, 72.24; H, 10.19; N, 16.85; found: C, 72.13; H, 10.61; N, 16.79. The other enantiomer (i.e. (1R,2R)-1,2-bis(4-decyl-1H-1,2,3-triazol-1-yl)cyclohexane) was prepared following the same route using (1R,2R)-1,2-diaminocyclohexane as the starting material.
:1 v/v, 100 mL) was added 1H-imidazole-4-sulfonyl azide (3) (379 mg, 2.2 mmol), CuSO4·5H2O (11.4 mg, 0.018 mmol) and K2CO3 (2.18 g, 15.8 mmol). The mixture was stirred for 3 days at RT and thereafter extracted with EtOAc (4 × 50 mL). The combined organic phases were dried over anhydrous Na2SO4 and most of the solvent was removed under reduced pressure. The concentrated solution was purified using column chromatography (eluent: hexane/Et2O/CHCl3 100:3:3 (ref. 45)) and dissolved in toluene for safe storage. Several batches afforded (1S,2S)-1,2-diazidocyclohexane (4) in yields ranging from 31% to 97% (yields were determined by 1H NMR by integral comparison using the toluene peaks as a reference). 1H NMR (400 MHz, CDCl3) δ (ppm) 3.20 (m, 2H), 2.07 (m, 2H), 1.79 (m, 2H), 1.29 (m, 4H).:1 v/v, 3.8 mL) a solution of 4 in toluene (123.5 mg, 0.743 mmol) was added. Subsequently, separate solutions of CuSO4·5H2O (249.7 mg, 1 mmol) and sodium ascorbate (198.1 mg, 1 mmol) in water (1 mL) were added alternately until the product precipitated with blurring of the clear solution. After stirring for 3 days, the reaction mixture was dissolved in a mixture of EtOAc and THF (1:1 v/v, 100 mL) and washed with aqueous Na2EDTA (0.1 mol L−1, 3 × 50 mL), brine (50 mL), water (50 mL) and dried over anhydrous Na2SO4. Finally, the solvent was removed under reduced pressure and recrystallized from acetone affording click-C12–Cyc as a light yellow solid in a 32% yield (117 mg, 0.236 mmol). Mp 129 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 6.79 (s, 2H), 4.73 (m, 2H), 2.52 (m, 4H), 2.31 (m, 4H), 2.04 (m, 2H), 1.59 (m, 2H), 1.49 (m, 4H), 1.24 (s, 28H), 0.88 (t, J = 6.64 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ (ppm) 32.46, 31.94, 29.65, 29.58, 29.38, 29.35, 29.25, 25.42, 24.65, 22.71, 14.13; FT-IR (ATR) νmax (cm−1) 3146, 3068, 2952, 2915, 2848, 1543, 1446, 1223, 1054, 809, 719; MS (ESI): m/z = 250.2 [M + 2H]2+, 499.4 [M + H]+, 997.9 [2M + H]+; [α]20D −44.7 ± 0.3° (c = 1; H2O). Elemental analysis calculated for C30H54N6: C, 72.24; H, 10.19; N, 16.85; found: C, 72.13; H, 10.61; N, 16.79. The other enantiomer (i.e. (1R,2R)-1,2-bis(4-decyl-1H-1,2,3-triazol-1-yl)cyclohexane) was prepared following the same route using (1R,2R)-1,2-diaminocyclohexane as the starting material.The synthetic procedure and characterization data for click-C13–Cyc, as well as the NMR and FT-IR spectra of the synthesized compounds, can be found in the ESI (Fig. S2–S6†).
General procedure for the preparation of organogels
Typically, a weighed amount of the corresponding solid gelator was placed into a 2 mL screw-capped vial (4 cm length × 1 cm diameter). The desired solvent was added on top and the mixture was gently heated with a heat-gun until the solid material was completely dissolved. Unless otherwise noted, the resulting isotropic solution was then spontaneously cooled to RT. Ultrasound treatment of the mixture was also applied in some cases as indicated in the main text. No control over temperature rate during the heating–cooling process was applied. The material was preliminarily classified as a “gel” if it did not exhibit gravitational flow upon turning the vial upside-down at RT. The state was further confirmed using rheological measurements.General procedure for phase selective gelation experiments
The desired organic solvent and water (1 mL) were placed into a glass vial forming a biphasic mixture. After adding the gelator as a solid or in solution, successful gelation was checked by turning the vial upside-down at RT.Cell viability studies
The metabolic activity of the cells was evaluated after 24 h of incubation at 37 °C using the MTT assay (MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). HeLa human tumor cells were exponentially passaged before they reached confluence. Cells (∼8000 cells per well) were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) for 24 h before the experiment in a 96-well cell culture plate. Prepared working concentrations of both gelators were placed in contact with HeLa cells in order to get a final concentration of 5, 10, 25, 50 and 100 μM in a final volume of 200 μL. Gelators were incubated in the presence of cells for 24 h at 37 °C. DMEM was removed and cells were washed with phosphate buffer saline (PBS). Fresh DMEM was added (200 μL) and cells were incubated for an additional 2 h. A solution of MTT (25 μL) in PBS was added and the resultant solution was incubated for 4 h. The solution was removed and formazan crystals were dissolved in 100 μL of DMSO. The resultant solution was shaken at RT for 15 min and the absorbance was measured on a Glomax Multimode Microplate reader at 560 nm. Results were expressed as a relative percentage to the untreated control cells.Statistical differences between the groups at significance levels greater than 95% were calculated using the Student's t test. In all cases, p values of less than 0.05 were regarded as significant. Numerical data are presented as mean ± SD.
Computational studies
All calculations were performed using the M06-2X functional,47 which was combined with the 6-31G(d,p) basis set48 in the Gaussian 09 computer package.49 The M06-2X functional describes medium-range (i.e. ≤5 Å) noncovalent interactions, such as conventional (e.g. N–H⋯O and H–O⋯H) and non-conventional (e.g. C–H⋯O) hydrogen bonds, and better than usual DFT functionals.50 Thus, the M06 group of functionals is able to describe the geometry and interaction energy of complexes stabilized by non-covalent interactions, including π–π stacking, with accuracy close to that of couple cluster calculations with both single and double substitutions.51 All geometry optimizations were performed in vacuum, and in implicit solvents of growing polarity such as cyclohexane (ε = 2.0165), methanol (ε = 32.613) and nitromethane (ε = 36.562), which was described using a simple self consistent reaction field (SCRF) method. More specifically, the polarizable continuum model52,53 (PCM) was used to represent bulk solvent effects. Frequency analyses were performed to verify the nature of the minimum state of the stationary points located during geometry optimizations, as well as to obtain zero-point energies and thermal corrections to the energy, which were used for the calculation of free energies. Binding energies (BE) were corrected with the basis set superposition error (BSSE) by means of the standard counterpoise (CP) method.54Results and discussion
Synthesis of isosteres
Low-molecular-weight C12–Cyc was rapidly synthesized with a modest yield (39%) in one step via the nucleophilic addition of (S,S)-1,2-cyclohexanediamine (1) to lauroyl chloride (2) in the presence of Et3N as a neutralization base under refluxing THF (Fig. 2). The isostere click-C12–Cyc was obtained with a modest yield (32% after recrystallization) via the Huisgen cycloaddition “click” reaction55,56 between (1S,2S)-1,2-diazidocyclohexane (4) and 1-dodecyne (5) catalyzed by CuI in a mixture of tBuOH/H2O. The diazide 4 was synthesized by reacting 1 with 1H-imidazole-4-sulfonyl azide (3) as an azide transfer agent in the presence of K2CO3 and CuII in a mixture of tBuOH/H2O (Fig. 2). The synthesis of the imidazole sulfonyl azide 3 was accomplished in a good yield (85%) by the addition of two equivalents of imidazole to chlorosulfonyl azide, which was prepared in situ by the reaction of equimolar quantities of sodium azide with thionyl chloride in acetonitrile (see ESI†).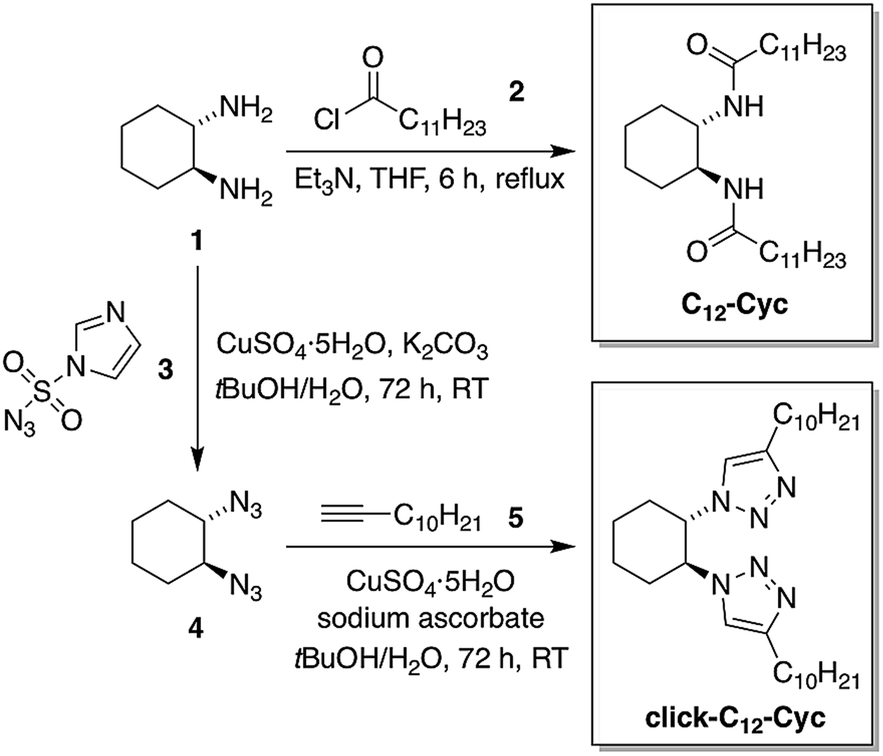 | ||
| Fig. 2 Synthetic scheme for the preparation of the LMW gelator C12–Cyc and its isostere click-C12–Cyc. The same procedures were used for the synthesis of the corresponding enantiomers as well as the analog click-C13–Cyc (see ESI†). | ||
Solubility properties and gelation ability
The gelation abilities of C12–Cyc and click-C12–Cyc were investigated in a large library of solvents using the classical heating–cooling cycle as described in the Experimental section (Tables S1 and S2, see ESI†). Materials that did not show gravitational flow upon turning the vial upside-down were initially classified as stable gels. The viscoelastic nature of the selected gels was later confirmed using oscillatory rheological experiments (vide infra). In agreement with previous studies,23 the chiral structure of the (C12–Cyc)-based aggregates in a model gel could be confirmed using CD spectroscopy (Fig. S7, see ESI†). Both isosteres were able to gel different non-polar (e.g. cyclohexane, hexane, oils) and polar-protic (e.g. ethylene glycol, methanol, nitromethane) solvents. However, C12–Cyc showed a broader solvent scope in terms of gelation (Table S2, see ESI†). For instance, significant nonpolar solvents, especially aromatic solvents such as benzene and toluene, and alcohols with more than two carbon atoms yielded clear solutions of click-C12–Cyc, which turned into suspensions above 120 g L−1. Thus, the intermolecular interactions responsible for the gel formation (e.g. gelator–gelator, gelator–solvent, aggregate–solvent interactions) were apparently more influenced by the nature of the solvent in the case of the click-isostere. Fig. 3 shows pictures of selected gels prepared at their corresponding CGC. In general, most gels were opaque except for those prepared in paraffin and silicon oil which were translucent. The opacity of the gels suggested the formation of aggregates larger than the wavelength of visible light (ca. 350–750 nm), which was later confirmed using electron microscopy imaging (vide infra). | ||
| Fig. 3 Digital photographs of upside-down vials containing organogels made of C12–Cyc and click-C12–Cyc in nine solvents at their corresponding critical gelation concentrations. | ||
It is worth mentioning that the click-analogue with only one extra methylene group (click-C13–Cyc) was able to form gels in a broader array of solvents compared to click-C12–Cyc, and in some cases (e.g. ethanol, propan-2-ol) even more efficiency than C12–Cyc (Table S2, see ESI†). This observation highlights the importance of the aliphatic chain length in the design of new LMW gelators. Moreover, triazole rings are chemically more robust than amides,57 which may offer alternative uses for click-gelators under harsh conditions.
Gel-to-sol transition temperature (Tgel), gelation time and CGC
Tgel, gelation kinetics and CGC were also compared for gels made from each isostere in nine solvents (Fig. 4). In general, gels made of C12–Cyc showed similar or higher Tgel values than those made of click-C12–Cyc, except in liquid paraffin in which the opposite was observed. All gels showed full thermo-reversibility with gelation kinetics ranging from seconds to 1 h. In general, gels made of click-C12–Cyc showed longer gelation times, except when ethylene glycol was used. The straight lines obtained in the ln–ln plots of Tgel and gelation time vs. gelator concentration indicated a power law-decay with similar values for both isosteres at high concentrations within the same incremental concentration interval, whereas gels made of C12–Cyc displayed a higher Tgel and lower gelation times at low concentrations (Fig. S8, see ESI†). In other words, one variable changes as a constant power of the other one for both isosteric systems. In terms of the CGC, similar values were found within the experimental error for both isosteres in cyclohexane, gasoline, liquid paraffin and silicone oil. In other solvents, click-C12-Cyc required higher concentrations for gelation, especially for polar-protic solvents (e.g. ethylene glycol, methanol, nitromethane).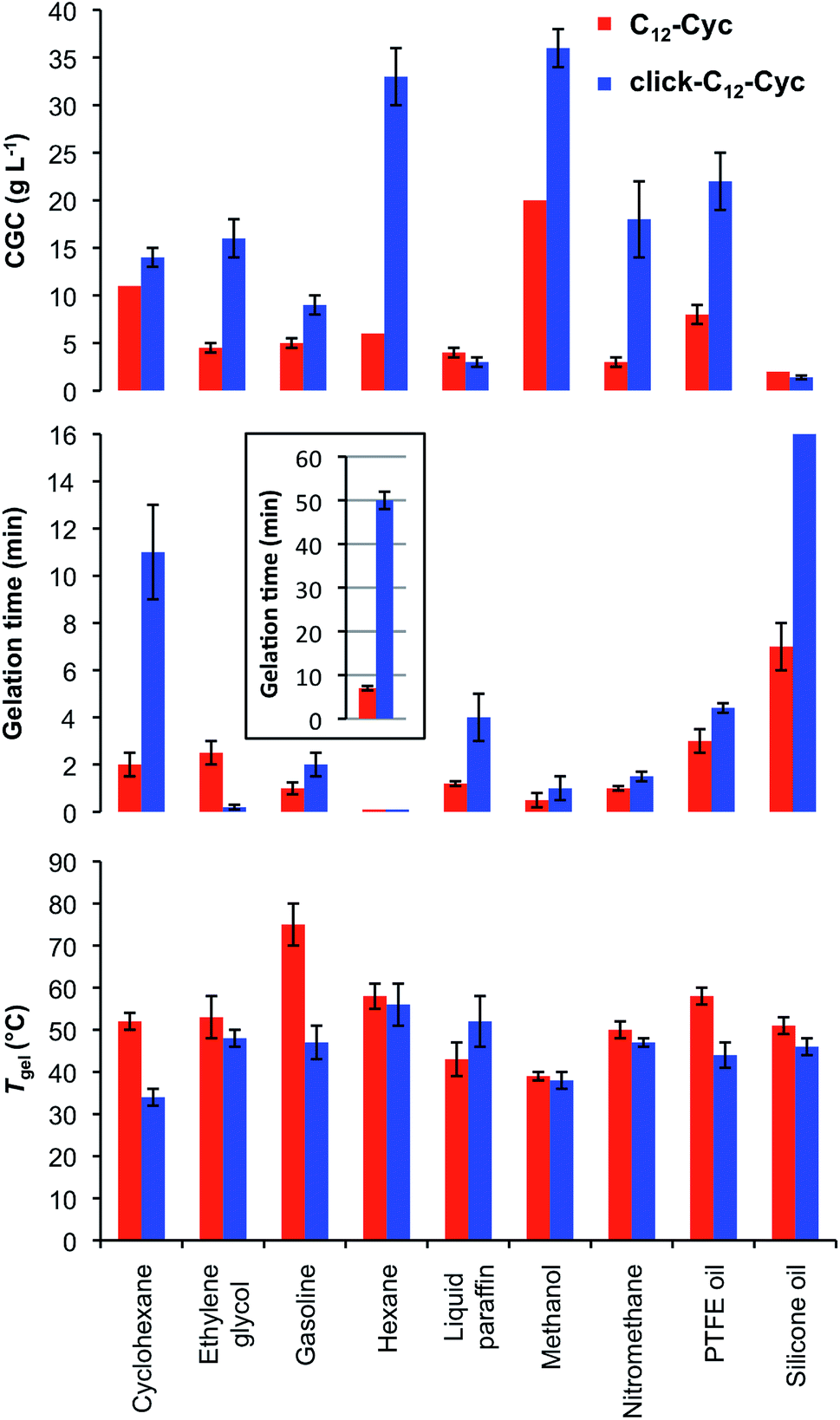 | ||
| Fig. 4 Comparative plots of the Tgel, gelation kinetics and CGC values for the organogels made of C12–Cyc and click-C12–Cyc in nine organic solvents. Both the Tgel and gelation time values were estimated at the corresponding CGC values. Inset (middle plot): gelation time in hexane. Each experiment was repeated at least three separate times and the error bars show the SDM. | ||
Stability and stimuli-responsive map
Unless otherwise indicated, the gels prepared from both gelators were stable for several months at room temperature without detriment of their consistency and visual appearance. As expected for physical (or supramolecular) gels, those made of C12–Cyc or click-C12–Cyc (taking cyclohexane as a model organic solvent) also showed clear responses to multiple external stimuli. Remarkably, the gels remained stable under UV irradiation, sonication, and upon addition of deionized water, seawater, phosphate buffer solution (0.1 mol L−1, pH 7.0), NaCl, HCl or NaOH solutions (1 mol L−1 each). However, both gels were easily dissolved after adding either methanol or acetic acid (1 M) (Fig. 5). The liquid phase obtained after addition of methanol could be transformed into a biphasic mixture with a stable gel on top by adding water to dissolve a part of the methanol and applying a heating–cooling cycle to the system. In contrast, the gel dissolved using acetic acid could not be restored by adding water or by neutralization with NaOH (1 M). These additives compete for hydrogen bonding interactions with the gelator molecules leading to the dissolution of the supramolecular aggregates. The addition of solid NaCl, AgNO3 or CuSO4·5H2O did not induce any phase transition. However, rapid gel-to-sol transitions were observed in both gels upon addition of solid PdCl2, which immediately began to diffuse into the gel phase. A similar response was observed with the gel made of C12–Cyc upon addition of FeCl3. However, the gel made of click-C12–Cyc remained stable in the presence of FeCl3 for 3 days. Afterward, this gel was also dissolved. Subsequent addition of metal complexation agents such as aqueous Na2EDTA (1 M) in the case of PdCl2 or aqueous oxalic acid (1 M) in the case of FeCl3 did not restore the gel phases. Manual mechanical agitation led to dissolution of the gel made of click-C12–Cyc, whereas the gel made of C12–Cyc remained stable. This qualitative observation was later correlated with the results obtained from rheological measurements (vide infra).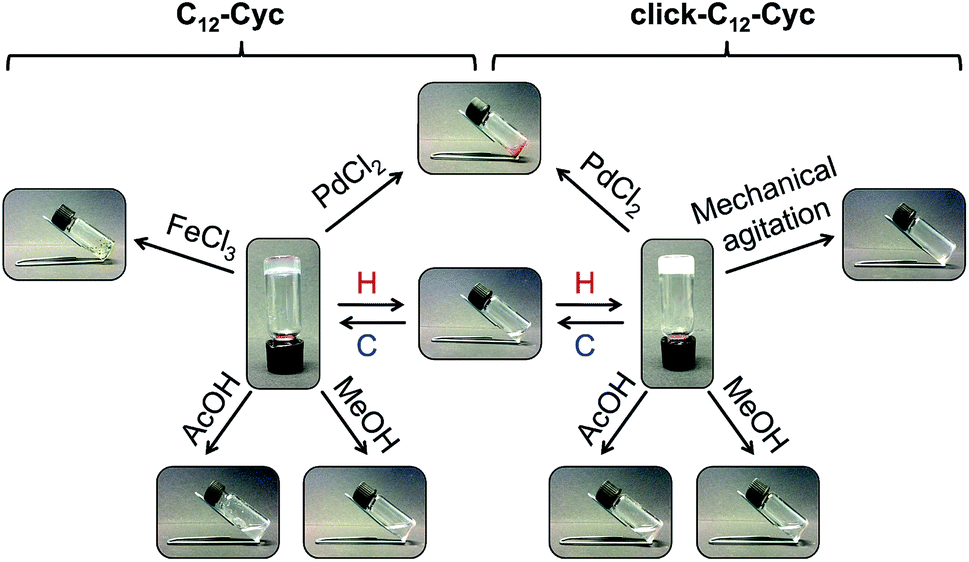 | ||
| Fig. 5 Representative stimuli responsive map of organogels made of C12–Cyc and click-C12–Cyc in cyclohexane (c = 15 g L−1). Abbreviation: H = heating; C = cooling. [AcOH] = 1 M. | ||
Morphological properties
In order to evaluate the possible effects of the isosteric replacement on the morphology of the corresponding gel networks, we recorded FESEM and TEM images of some xerogels (Fig. 6). Specifically, for these measurements we selected the gels made of C12–Cyc or click-C12–Cyc in cyclohexane (c = 20 g L−1) and in nitromethane (c = 30 g L−1). Entangled fibrillar networks were observed for the xerogels prepared in cyclohexane regardless of the gelator. In general, the fibers were 35–75 nm in diameter and several micrometers in length. Nevertheless, spherical aggregates integrated into the fibrillar scaffold reminiscent of a neural network were observed only in the case of click-C12–Cyc. The most obvious differences were observed with the xerogels prepared from the gels made in nitromethane. Specifically, coiled-coil helices were observed within the fibrillar network of C12–Cyc, whereas the xerogel made of Click-C12–Cyc displayed large planar structures assembled from a highly organized parallel network of gelator fibers. These fibers were much smaller in diameter than those formed by the isostere C12–Cyc. In general, the observed structures were also in agreement with those obtained by TEM (Fig. S9–S14, see ESI†). Although we have recorded representative images of the bulk materials using different techniques in order to recognize any possible artifacts, it should be emphasized that significant changes in the microstructures can take place during the preparation of freeze-dried samples and, hence, the interpretation of these images should always be performed carefully. | ||
| Fig. 6 Representative FESEM images of xerogels obtained upon freeze-drying the corresponding organogels made of (A) C12–Cyc in cyclohexane (c = 20 g L−1); (B) click-C12–Cyc in cyclohexane (c = 20 g L−1); (C) C12–Cyc in nitromethane (c = 30 g L−1); and (D) click-C12–Cyc in nitromethane (c = 30 g L−1). See ESI† for additional pictures. | ||
Rheological measurements
Standard dynamic oscillatory rheology was conducted in order to confirm the viscoelastic gel nature of selected gels and compare the overall mechanical stabilities towards external shear forces. DFS, DSS and DTS experiments for the organogels made of C12–Cyc or click-C12–Cyc in cyclohexane (c = 20 g L−1) revealed that the storage modulus G′ was one order of magnitude higher in the case of C12–Cyc (Table 1 and Fig. S15, see ESI†). This was in agreement with the visually stronger character observed for the gel made of C12–Cyc upon mechanical agitation (vide supra). Interestingly, the lowest tanδ was observed for the gel made of click-C12–Cyc, indicating that this gel acts in a more elastic way and has a greater potential to store the load rather than dissipating it. These results suggest that the amide-to-triazole isosteric replacement imposes, at least to some extent, restrictions against the molecular motion of the supramolecular polymer chains. Moreover, DSS experiments confirmed that the gel derived from C12–Cyc was more brittle in nature as suggested by the smaller maximum strain at break (γ) (i.e. 1.4% for the gel made of C12–Cyc vs. 2.3% for the gel made of click-C12–Cyc). Finally, the DTS experiments also confirmed the stability of both gels during aging at RT, 1 Hz frequency and 0.1% strain (Fig. S15, see ESI†).
| Gelator | G′ (kPa) | G′′ (kPa) | tanδ |
γ (%) |
|---|---|---|---|---|
| C12–Cyc | 15 | 1.4 | 0.25 | 1.4 |
| click-C12–Cyc | 3.5 | 0.7 | 0.12 | 2.3 |
In vitro cytotoxicity studies
Toxicity studies of both gelators (C12–Cyc and click-C12–Cyc) were assessed via in vitro studies using the HeLa human tumor cell line. The cellular viability percentage was studied for concentrations that ranged from 5 to 100 μM and was compared to the negative control (blank) in which HeLa cells were incubated in the absence of the aforementioned gelators.Both hydrophobic derivatives displayed cellular viabilities greater than 80% in all concentrations tested. Specifically, click-C12–Cyc did not affect the proliferation process of HeLa cells after 24 h of incubation when compared with the results obtained in the presence of C12–Cyc (Fig. S16 and Table S3, see ESI†). In this regard, a non-toxic behavior and significant values were found for concentrations 10, 25 and 50 μM (100% of cellular viability in all cases) if we compare these results with the toxicity values obtained for C12–Cyc using the same conditions at 10 and 25 μM (88% of cellular viability with *p < 0.05 and 85% of cellular viability with **p < 0.01, respectively). Interestingly, when the gelator concentration was increased to 50 and 100 μM, the same non-toxic tendency was also exhibited (100 and 89%, respectively) for click-C12–Cyc, whereas the cellular viabilities were slightly lower in the case of C12–Cyc (85 and 82%, respectively). These values did not show any significant difference. The aforementioned results suggest the gels made of click-C12–Cyc could potentially be used as non-toxic composites in release experiments of active substances or in environmental applications.
Phase selective gelation
The non-toxic behavior of the isosteric gelators prompted us to investigate their phase selective gelation capacity, which constitutes an important property of some gelators with potential for use in oil spill treatment.58–60 In this study we found that both gelators were able to selectively gel the organic phases of biphasic mixtures made of seawater and different organic solvents such as gasoline, hexane, cyclohexane and PTFE oil. This observation indicated that the amide–triazole isosteric replacement preserved this important feature of the gelator while allowing fine-tuning of other functional properties of the gels. In particular, the oil phase of a mixture of seawater and PTFE oil was successfully gelled by adding click-C12–Cyc (c = 30 g L−1) on top of the system and applying a classical heating–cooling cycle (Fig. 7). Importantly, clean seawater could easily be recovered after gelation was complete by simple filtration of the gelled oil phase.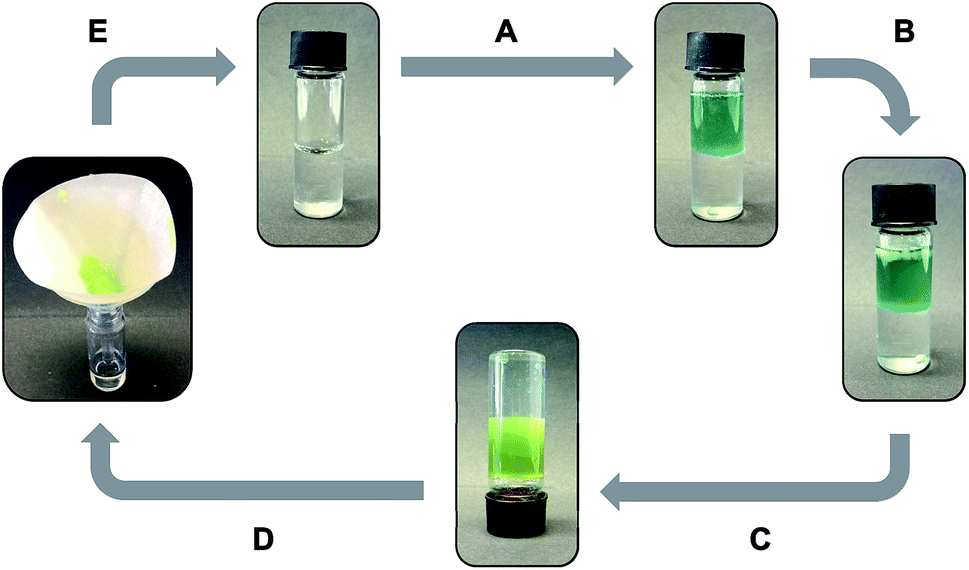 | ||
| Fig. 7 Phase selective gelation cycle of PTFE oil on seawater: (A) addition of 2 mL of PTFE oil to 2 mL of seawater; (B) addition of 60 mg click-C12–Cyc (c = 30 g L−1); (C) application of a heating–cooling cycle leads to selective gelation of the PTFE oil phase; (D) filtration of the gelled oil phase; and (E) recovery of clean seawater. | ||
In other cases in which a heating–cooling cycle is not recommended, the gelator can be sprayed on top of the biphasic mixture as a highly concentrated solution. For instance, the organic phase of a mixture of seawater and gasoline was successfully gelled by spraying a pre-formed solution of click-C12–Cyc in gasoline (c = 200 g L−1) (Fig. 8). As in the previous example, the gelled phase could be rapidly separated by filtration. In addition, both gasoline and gelator could be recycled multiple times upon distillation of the separated gel phase.
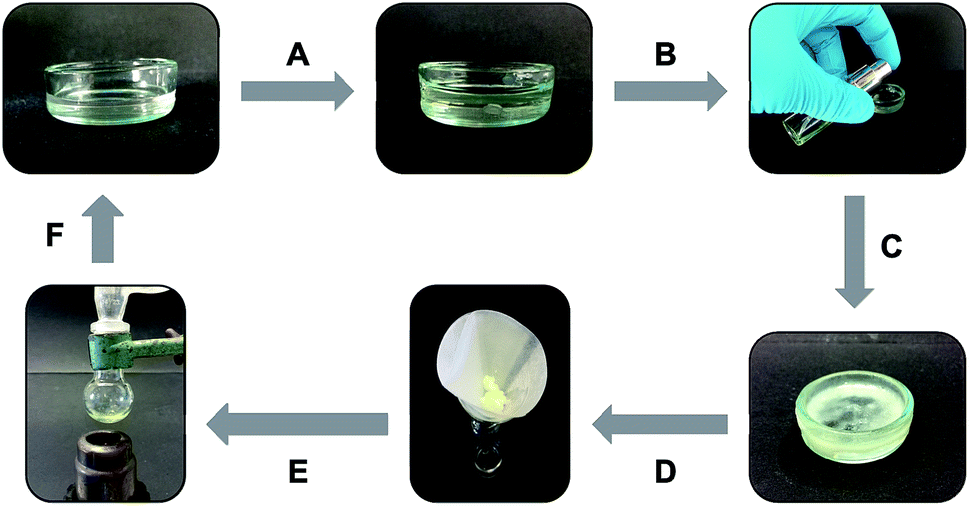 | ||
| Fig. 8 Phase selective gelation cycle of gasoline on seawater: (A) addition of 1.8 mL of gasoline to 10 mL of seawater; (B) addition of 0.2 mL of a warm solution containing 120 mg of click-C12–Cyc; (C) gelation of the system without external treatment; (D) filtration of the gelled gasoline phase; (E) transfer of the gel to distillation apparatus; and (F) recycling of both the gelator and solvent is possible. | ||
Aggregation mode and computational studies
In order to get preliminary insights into the differences in the aggregation mode of both isosteres, we conducted comparative FT-IR studies in different solvents (i.e. cyclohexane, ethylene glycol and nitromethane). The analysis of the solid gelators and the corresponding xerogels revealed shifted bands suggesting the involvement of non-covalent molecular interactions of different strengths during the formation of the gel networks. In particular, the carbonyl C–O stretching vibration associated to C12–Cyc gave an identical value of 1636 cm−1 for the solid gelator and the xerogels prepared in cyclohexane and nitromethane. However, the N–H stretching vibration was shifted 4 cm−1 to a higher wavenumber suggesting weaker intermolecular hydrogen bonding (Fig. S17 and S18, see ESI†). In the case of click-C12–Cyc, the C–H stretching vibration of the triazole hydrogen atom was shifted 4 cm−1 to a higher wavenumber, but only for the cyclohexane xerogel. This suggested a higher electron density inside the π-system and, presumably, a different aggregation pattern compared to the raw gelator and the xerogel in nitromethane, in which it remained unaltered. These results were in good agreement with the XRD data, which revealed a slight decrease in the crystallinity of the xerogel aggregates formed in cyclohexane compared to that of the solid gelator (Fig. S19–S21, see ESI†). However, in the case of nitromethane, C12–Cyc was found to decrease the crystallinity, whereas click-C12–Cyc showed a stronger crystalline character compared to the solid gelator in concordance with the more solid-like vibrational frequencies observed using FT-IR.At this point, we decided to perform theoretical calculations to visualize the key stabilizing interactions in each case. Thus, the intermolecular interactions of click-C12–Cyc were modelled in different environments by building complexes formed by two interacting model molecules. More specifically, the two C10H21 groups of each click-C12–Cyc molecule were replaced by C2H5 groups. In order to consider different non-covalent interactions (i.e. stabilizing π–π stacking, dispersion, hydrogen bonding and dipole–dipole interactions), a total of 30 complexes were built varying the relative position between the two interacting model molecules. All of these structures were used as starting points for complete geometry optimizations in a vacuum, cyclohexane, methanol and nitromethane, with 23–25 different minima being obtained depending on the environment (i.e. the rest of the starting structures converged after geometry optimization to a minimum already obtained from other starting structures).
The minimum energy complexes were ranged and followed an increasing order of free energies in a vacuum and labeled using a letter (i.e. from A to Z). Fig. 9 displays the relative free energies obtained for each structure in the four studied environments. As it can be seen, the polarity of the medium drastically affects not only the stability of the different minima, but also the spread of free energies (i.e. the free energy difference between the most and least favored complexes is 18.4, 13.6, 11.2, and 10.1 kcal mol−1 in a vacuum, cyclohexane, nitromethane and methanol, respectively). The lowest free energy complexes in a vacuum, cyclohexane, methanol and nitromethane corresponds to structures A, B, T and C, respectively, which are depicted in Fig. 10.
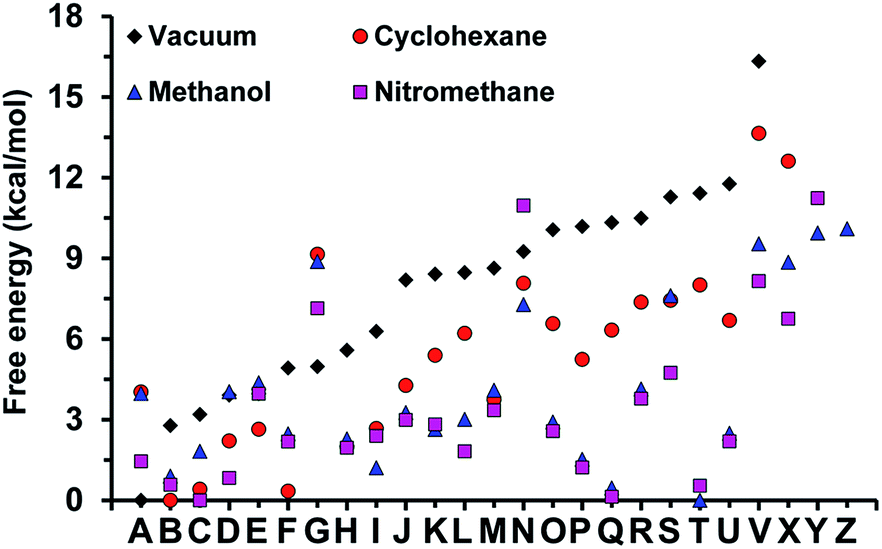 | ||
| Fig. 9 Relative free energy of model complexes calculation in a vacuum, cyclohexane, methanol and nitromethane with respect to the lowest free energy minimum of the most stable complex in each environment. Complexes have been labeled from A to Z, each letter referring to the complex optimized using the same starting point for each environment. | ||
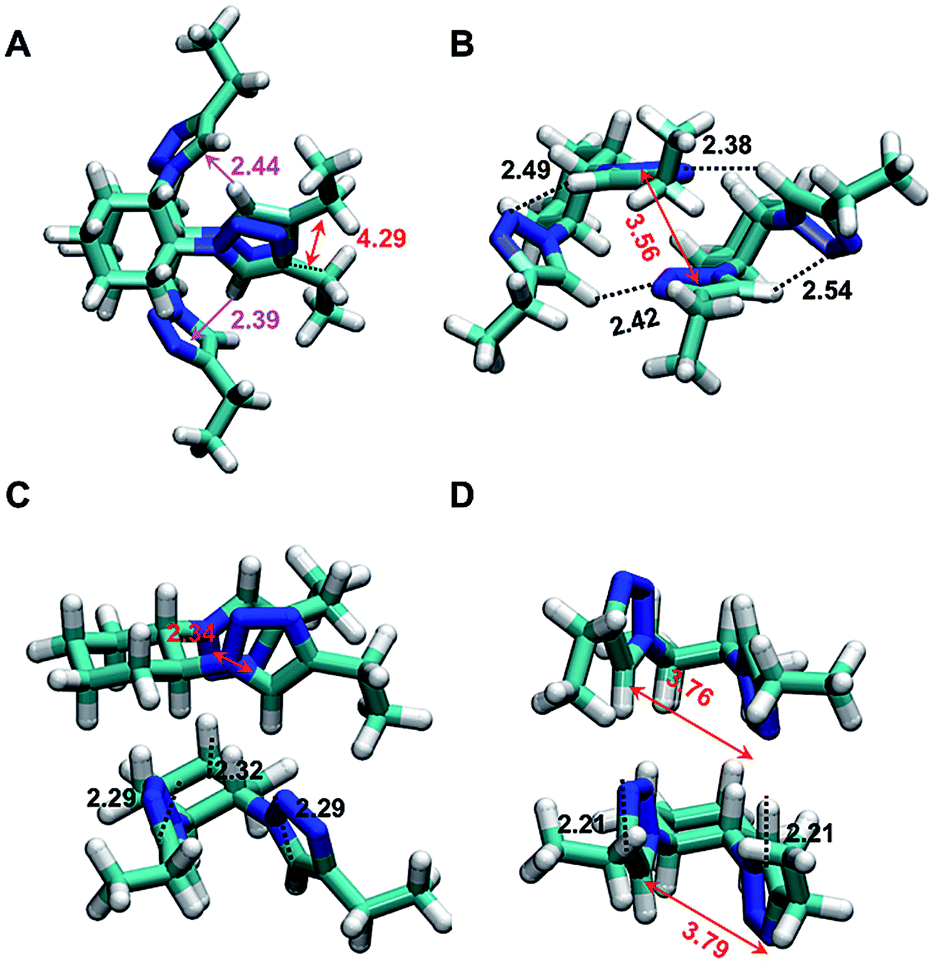 | ||
| Fig. 10 Representation of the lowest free energy minima obtained in (A) a vacuum (⊗G = −1749.090117 au, BE = −19.33 kcal mol−1); (B) cyclohexane (⊗G = −1749.09237 au, BE = −16.35 kcal mol−1); (C) methanol (⊗G = −1749.106305 au, BE = −15.57 kcal mol−1); and (D) nitromethane (⊗G = −1749.106021 au, BE = −13.06 kcal mol−1). Hydrogen bonds are represented by dotted black lines, N–H⋯π interactions as pink arrows and π–π stacking is shown as red double arrows. Labels refer to the distances (in Å) found for each stabilizing interaction: H⋯O distance in the hydrogen bonds; H···center of masses in N–H⋯π interactions; and the center of masses to the center of masses in π–π stacking. | ||
Complex A (Fig. 10A) is mainly stabilized by a π–π stacking interaction between the two triazole rings, two N–H⋯π interactions and the van der Waals interactions induced by the almost canonical stacking of the two cyclohexyl rings. Complex A is unfavored by 4.0 kcal mol−1 in cyclohexane solution, while complex B is destabilized by 2.8 kcal mol−1 in a vacuum. Complex B, which is the most favored in cyclohexane (Fig. 10B) exhibits two strong intermolecular N–H⋯N hydrogen bonds and π–π stacking between the two triazole rings, while the two N–H⋯π interactions disappear. In methanol, the most favored structure corresponds to complex T (Fig. 10C), which preserves the π–π stacking and one N–H⋯N hydrogen bond. In addition, two non-conventional C–H⋯N hydrogen bonds, which has been proven to be important in assemblies involving small organic molecules,61,62 are identified. It is worth noting that complex T is destabilized by 11.4 and 8.0 kcal mol−1 in a vacuum and in cyclohexane solution, respectively, while complexes A and B are unfavored by 4.0 and 0.9 kcal mol−1, respectively, in methanol. In nitromethane the most favored complex is complex C, which is destabilized by 3.2, 0.4 and 1.8 kcal mol−1 in a vacuum, cyclohexane and methanol, respectively. Complex C is stabilized by two intramolecular π–π stacking interactions and two intermolecular N–H⋯N hydrogen bonds (Fig. 10D). Overall, the results presented in Fig. 10A and B indicate that click-C12–Cyc molecules can adopt a wide variety of interacting patterns, whose relative stability depends on the polarity of the environment. This explains the noticeable tendency of click-C12–Cyc to form gels in a remarkably wide array of solvents.
Inspection of intermolecular BE calculated for the four considered environments (Fig. S22, see ESI†) provides similar conclusions. The BE expand from −8.9 to −19.3 kcal mol−1 in a vacuum, from −5.6 to −18.7 kcal mol−1 in cyclohexane, from −4.7 to −17.4 kcal mol−1 in methanol, and from −4.8 to −17.2 kcal mol−1 in nitromethane. These wide intervals are consistent with a large amount of stabilizing interactions that, as discussed above, include hydrogen bonds, N–H⋯π, π–π stacking and van der Waals interactions. As can be seen from comparing Fig. 9 and S22 (see ESI†), complexes with more favorable BE do not necessarily correspond to the structures with lower free energies. This is attributed to the key role played by intramolecular interactions in the stability of model complexes, as these contributions are not included in the BE.
Conclusions
The aforementioned results prove that the application of isosteric substitution for fine-tuning of the functional properties of physical gels is a promising approach for materials synthesis. Herein, we have demonstrated that 1,2,3-trizole rings can replace all amide groups existing in an LMW organogelator, while preserving the most important features associated with a precise hydrogen-bonding network. In particular, the gelation of organic solvents using the bis-amide C12–Cyc is driven by self-assembly via antiparallel hydrogen bonds and van der Waals intermolecular interactions in isotropic solutions. Thus, the substitution of the two amide groups, which in this example constitutes the source of the hydrogen bonds, by isosteric 1,2,3-triazoles (click-C12–Cyc) is not an obvious outcome to maintain and/or tune the properties of the physical network. This study demonstrates that 1,2,3-triazoles can take over all of the functions derived from the amides groups offering a versatile strategy to tune the properties of the corresponding bulk gels in terms of gelation kinetics and stimuli-responsive behavior. Modifications in the critical gelation concentrations, as well as in the gel-to-sol transition temperatures, were also derived from the dual isosteric substitution. Theoretical calculations revealed that click-C12–Cyc molecules can adopt a wide variety of interacting patterns, whose relative stability depends on the polarity of the environment, which is in good agreement with the experimental data obtained regarding the gelation ability. Other important features of click-C12–Cyc for potential practical applications are its non-cytotoxic character and its phase-selective gelation of water–oil mixtures. Moreover, the increased chemical robustness of triazoles compared to amides under different conditions also confers practical importance to this strategy for the synthesis of new functional materials beyond the field of gel materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Regensburg, the Deutsche Forschungsgemeinschaft (DFG), Generalitat de Catalunya (SGR/264), Instituto de Salud Carlos III (ISCIII) (CB06_01_0019); Ministerio de Economía, Industria y Competitividad (MINECO) (CTQ2014-52588-R and CTQ2017-84415-R) and European FEDER funds (MAT2015-69367-R), the Agència de Gestió d'Ajuts Universitaris i de Recerca (2017SGR359 and XRTQC), and Fondecyt iniciación 11160707. Support for the research of C. A. was received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. D. D. D. thanks the DFG for the Heisenberg Professorship Award.Notes and references
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS PubMed.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed.
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC.
- R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664–675 RSC.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- S. Li, V. T. John, G. C. Irvin, S. H. Bachakonda, G. L. McPherson and C. J. O'Connor, J. Appl. Phys., 1999, 85, 5965–5967 CrossRef CAS.
- T. Kato, Science, 2002, 295, 2414–2418 CrossRef CAS PubMed.
- J. Puigmarti-Luis, V. Laukhin, A. P. del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238–241 CrossRef CAS PubMed.
- W. Kubo, S. Kambe, S. Nakade, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2003, 107, 4374–4381 CrossRef CAS.
- N. A. Peppas, P. Bures, W. Leobandung and H. Ichikawa, Eur. J. Pharm. Biopharm., 2000, 50, 27–46 CrossRef CAS PubMed.
- A. Friggeri, K. J. C. van Bommel and S. Shinkai, in Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, Dordrecht, 2006, pp. 857–893 Search PubMed.
- D. D. Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
- E. M. Schön, E. Marqués-López, R. P. Herrera, C. Alemán and D. D. Díaz, Chem.–Eur. J., 2014, 20, 10720–10731 CrossRef PubMed.
- T. Patrick, Semin. Cutaneous Med. Surg., 2004, 23, 233–235 CrossRef PubMed.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- S.-k. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- F. Vögtle, Supramolecular Chemistry, Wiley, Chichester, 1991 Search PubMed.
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH Weinheim, 1995 Search PubMed.
- R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, New York, 2006 Search PubMed.
- J. van Esch and S. D. Feyter, Chem.–Eur. J., 2006, 3, 1238–1243 CrossRef.
- K. Yoza, N. Amanokura, Y. Ono, T. Akao, H. Shinmori, M. Takeuchi, S. Shinkai and D. N. Reinhoudt, Chem.–Eur. J., 1999, 5, 2722–2729 CrossRef CAS.
- J. Chen and A. J. McNeil, J. Am. Chem. Soc., 2008, 130, 16496–16497 CrossRef CAS PubMed.
- K. Hanabusa, M. Yamada, M. Kimura and H. Shirai, Angew. Chem., Int. Ed., 1996, 35, 1949–1951 CrossRef CAS.
- N. Zweep, A. Hopkinson, A. Meetsma, W. R. Browne, B. L. Feringa and J. H. van Esch, Langmuir, 2009, 25, 8802–8809 CrossRef CAS PubMed.
- H. Sato, T. Nakae, K. Morimoto and K. Tamura, Org. Biomol. Chem., 2012, 10, 1581–1586 RSC.
- B. T. Kato, T. Kutsuna, K. Hanabusa and M. Ukon, Liq. Cryst., 1998, 606–608 Search PubMed.
- S. Yajima, K. Takami, R. Ooue and K. Kimura, Analyst, 2011, 136, 5131–5133 RSC.
- B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226–4251 RSC.
- L. Latxague, A. Gaubert, D. Maleville, J. Baillet, M. A. Ramin and P. Barthélémy, Gels, 2016, 2, 25 CrossRef PubMed.
- I. S. Okafor and G. Wang, Carbohydr. Res., 2017, 451, 81–94 CrossRef CAS PubMed.
- Y. Huang, S. Liu, Z. Xie, Z. Sun, W. Chai and W. Jiang, Front. Chem. Sci. Eng., 2017, 2, 252–261 Search PubMed.
- Y. Huang, H. Li, Z. Li, Y. Zhang, W. Cao, L. Wang and S. Liu, Langmuir, 2017, 33, 311–321 CrossRef CAS PubMed.
- G. Wang, A. Chen, H. P. R. Mangunuru and J. R. Yerabolu, RSC Adv., 2017, 7, 40887–40895 RSC.
- K. Ghosh, A. Panja and S. Panja, New J. Chem., 2019, 40, 3476–3483 RSC.
- I. Torres-Moya, B. Saikia, P. Prieto, J. R. Carrillo and J. W. Steed, CrystEngComm, 2019, 21, 2135–2143 RSC.
- J. Bachl, J. Mayr, F. J. Sayago, C. Cativiela and D. D. Díaz, Chem. Commun., 2015, 51, 5294–5297 RSC.
- M. Häring, S. K. Nandi, J. Rodríguez-López, D. Haldar, V. S. Martín, A. D. Lozano-Gorrín, C. Saldías and D. D. Díaz, ACS Omega, 2019, 4, 2111–2117 CrossRef.
- A. Burger, Prog. Drug Res., 1991, 37, 287–371 CAS.
- I. Langmuir, J. Am. Chem. Soc., 1919, 41, 1543–1559 CrossRef CAS.
- A. Panja and K. Ghosh, New J. Chem., 2019, 43, 934–945 RSC.
- Bioisosteres in Medicinal Chemistry, ed. N. Brown, Wiley-VCH, Weinheim, 2012, vol. 54 Search PubMed.
- H. Sato, T. Nakae, K. Morimoto and K. Tamura, Org. Biomol. Chem., 2012, 10, 1581–1586 RSC.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- H. Sato, T. Nakae, K. Morimoto and K. Tamura, Org. Biomol. Chem., 2012, 10, 1581–1586 RSC.
- S. Goksu, H. Secen and Y. Sutbeyaz, Synthesis, 2002, 1–6 Search PubMed.
- H. Sato, T. Nakae, K. Morimoto and K. Tamura, Org. Biomol. Chem., 2012, 10, 1581–1586 RSC.
- Y. Zhao and D. Truhlar, Theor. Chim. Acta, 2008, 120, 215–241 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas,J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1., Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS PubMed.
- K. Remya and C. H. Suresh, J. Comput. Chem., 2013, 34, 1341–1353 CrossRef CAS PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 114, 2708–2711 CrossRef.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- A. Brik, J. Alexandratos, Y. C. Lin, J. H. Elder, A. J. Olson, A. Wlodawer, D. S. Goodsell and C. H. Wong, ChemBioChem, 2005, 6, 1167–1169 CrossRef CAS PubMed.
- A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250–5252 RSC.
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS PubMed.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- E. Navarro, C. Alemán and J. Puiggalí, J. Am. Chem. Soc., 1995, 117, 7307–7310 CrossRef CAS.
- C. Alemán, E. Navarro and J. Puiggalí, J. Org. Chem., 1995, 60, 6135–6140 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra (NMR, FT-IR, XRD, CD), tabular data, additional plots and electron microscopy images. See DOI: 10.1039/c9ra03316e |
| This journal is © The Royal Society of Chemistry 2019 |