
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of low-temperature plasma treatment of electrospun polycaprolactone fibrous scaffolds on calcium carbonate mineralisation†
Anna A. Ivanova a,
Dina S. Syromotinaa,
Svetlana N. Shkarinaa,
Roman Shkarinbc,
Angelica Ceciliad,
Venera Weinhardtbde,
Tilo Baumbachbd,
Mariia S. Savelevaf,
Dmitry A. Gorinf,
Timothy E. L. Douglasgh,
Bogdan V. Parakhonskiyi,
Andre G. Skirtachi,
Pieter Coolsi,
Nathalie De Geyteri,
Rino Morenti,
C. Oehrj,
Maria A. Surmenevaa and
Roman A. Surmenev*a
a,
Dina S. Syromotinaa,
Svetlana N. Shkarinaa,
Roman Shkarinbc,
Angelica Ceciliad,
Venera Weinhardtbde,
Tilo Baumbachbd,
Mariia S. Savelevaf,
Dmitry A. Gorinf,
Timothy E. L. Douglasgh,
Bogdan V. Parakhonskiyi,
Andre G. Skirtachi,
Pieter Coolsi,
Nathalie De Geyteri,
Rino Morenti,
C. Oehrj,
Maria A. Surmenevaa and
Roman A. Surmenev*a
aPhysical Materials Science and Composite Materials Centre, Research School of Chemistry & Applied Biomedical Sciences, National Research Tomsk Polytechnic University, 634050 Tomsk, Russia. E-mail: rsurmenev@gmail.com; Tel: +7 903 953 09 69
bLaboratory for Applications of Synchrotron Radiation, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen, Germany
cInstitute for Applied Computer Science, Karlsruhe Institute of Technology, Karlsruhe, Germany
dInstitute for Photon Science and Synchrotron Radiation, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen, Germany
eCentre for Organismal Studies, University of Heidelberg, Heidelberg, Germany
fSkolkovo Institute of Science and Technology, 3 Nobelya Str., Moscow 121205, Russian Federation
gUK Engineering Department, Lancaster University, UK
hMaterials Science Institute (MSI), Lancaster University, UK
iDepartment of Molecular Biotechnology, Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, Ghent 9000, Belgium
jFraunhofer IGB, Stuttgart, Germany
First published on 22nd November 2018
Abstract
This article reports on a study of the mineralisation behaviour of CaCO3 deposited on electrospun poly(ε-caprolactone) (PCL) scaffolds preliminarily treated with low-temperature plasma. This work was aimed at developing an approach that improves the wettability and permeability of PCL scaffolds in order to obtain a superior composite coated with highly porous CaCO3, which is a prerequisite for biomedical scaffolds used for drug delivery. Since PCL is a synthetic polymer that lacks functional groups, plasma processing of PCL scaffolds in O2, NH3, and Ar atmospheres enables introduction of highly reactive chemical groups, which influence the interaction between organic and inorganic phases and govern the nucleation, crystal growth, particle morphology, and phase composition of the CaCO3 coating. Our studies showed that the plasma treatment induced the formation of O- and N-containing polar functional groups on the scaffold surface, which caused an increase in the PCL surface hydrophilicity. Mineralisation of the PCL scaffolds was performed by inducing precipitation of CaCO3 particles on the surface of polymer fibres from a mixture of CaCl2- and Na2CO3-saturated solutions. The presence of highly porous vaterite and nonporous calcite crystal phases in the obtained coating was established. Our findings confirmed that preferential growth of the vaterite phase occurred in the O2-plasma-treated PCL scaffold and that the coating formed on this scaffold was smoother and more homogenous than those formed on the untreated PCL scaffold and the Ar- and NH3-plasma-treated PCL scaffolds. A more detailed three-dimensional assessment of the penetration depth of CaCO3 into the PCL scaffold was performed by high-resolution micro-computed tomography. The assessment revealed that O2-plasma treatment of the PCL scaffold caused CaCO3 to nucleate and precipitate much deeper inside the porous structure. From our findings, we conclude that O2-plasma treatment is preferable for PCL scaffold surface modification from the viewpoint of use of the PCL/CaCO3 composite as a drug delivery platform for tissue engineering.
Introduction
One of the most effective strategies for bone regeneration is the use of composite scaffolds that mimic the structure and biological features of extracellular matrices (ECM) of bone. Porous electrospun scaffolds offer significant advantages over various other candidate materials, mainly because of their high surface-to-volume ratios, high porosity, and spatial interconnectivity, which are beneficial for cell migration, drug loading, and nutrient distribution. A wide range of polymers can be employed for scaffold fabrication, such as poly(lactic acid) (PLA),1–4 poly(lactic-co-glycolic acid) (PLGA),1,5,6 and poly(ε-caprolactone) (PCL).1,7–11 PCL is beneficial for tissue regeneration and drug delivery applications on account of its biocompatibility, processability, degradability, and mechanical properties. However, the surface wettability and osteoconductivity of PCL materials require improvement given the low cell attachment and proliferation on untreated PCL scaffolds.7,8 Several surface modification techniques such as chemical treatment, film deposition, blending, ion beam radiation, and plasma treatment have been developed to overcome these shortcomings.2,3,7,9,12 The most promising techniques are plasma treatment and biomineralisation of polymer scaffolds.12–14 Plasma treatment is an effective surface modification method for improving the surface and biological properties of PCL materials without altering the properties of the bulk. Plasma treatment is a non-solvent process that involves the use of a smaller amount of chemical reagents and facilitates changes in the chemical composition, wettability, surface energy, refractive index, hardness, chemical inertness, and biocompatibility of a PCL material.14 The weak chemical bonds of PCL are replaced with highly reactive amine, carboxy, hydroxy, and aldehyde groups by the use of glow discharges of non-deposition gases such as NH3, O2, and Ar.15–17 A previous study reported that these chemical groups could act as binding sites for cell adhesion and protein adsorption.13 Because of the presence of functional groups, plasma-modified polymer surfaces have applicability to biomolecular immobilisation.18,19 Introduction of inorganic particles into the polymer scaffold and deposition of a thin film on the scaffold surface to achieve superior features for bone tissue substitution are highly promising approaches for biomineralisation of the scaffold surface or bulk. Various inorganic species have been used for this purpose, such as hydroxyapatite (HAp), bioactive glass, CaCO3, and TiO2.20,21 Among these species, CaCO3 is advantageous because of its biocompatibility, simple synthesis method, and straightforward decomposition conditions. CaCO3 has three anhydrous crystallographic polymorphs: vaterite, aragonite, and calcite. Many approaches have been developed to control the morphology and polymorphs of CaCO3.22–27 Specifically, vaterite is the least stable, and thus the most challenging, phase of CaCO3, which possesses an orthorhombic or hexagonal crystal structure. Savelyeva et al. studied the deposition of porous CaCO3 coatings on electrospun PCL fibres;7 they obtained various structures and phase compositions of the CaCO3 coatings by varying the growth conditions (incubation time and ultrasonic treatment). The vaterite-calcite transformation in CaCO3 was induced by an increase in the number of consecutive treatments, which could be explained by the Ostwald ripening process. Their study suggested that as the size of the vaterite crystallites increased beyond a critical value, the process of recrystallisation of vaterite to calcite occurred. However, Andreassen showed that spherical and polycrystalline particles of vaterite precipitated from solution and grew by a spherulitic growth mechanism, and not by aggregation of nanosized precursor crystals.22 This work suggested that the transformation of vaterite to calcite was not a solid-state transformation but a surface-controlled process.22 This implies that the dissolution of vaterite and the nucleation of calcite are two parallel processes. Thus, three stages of CaCO3 formation are differentiated: occurrence and disappearance of an unstable amorphous CaCO3 phase, crystallisation and dissolution of the metastable vaterite phase, and precipitation of the thermodynamically stable calcite phase.22,25–27 Vaterite CaCO3 usually occurs in the form of highly porous spherical particles, which are beneficial from the viewpoint of bonding of the scaffold with bone tissue and for the use of the scaffold as a drug delivery system. Saveleva et al. recently tested a PCL/CaCO3 scaffold loaded with tannic acid as a model substance in vivo and demonstrated beneficial effects of the implanted scaffold on the stabilisation of blood vessels in rats by the release of loaded drugs, which, in turn, was induced by the recrystallisation of vaterite to calcite.28 It was also shown that the introduction of vaterite particles as a filler or coating onto polymer scaffolds induced the biomimetic precipitation of HAp from simulated body fluid.21,29A number of studies demonstrated that the nucleation and growth processes of CaCO3 crystals are affected by the microenvironment.30,31 As described above, plasma treatment can change the surface properties of PCL fibres. It is well established that plasma treatment causes activation of the surface layer of the polymer scaffold and the formation of some dangling bonds or free radicals,15–17 which may attract oxygen or moisture when the plasma-treated polymer scaffolds are exposed to air.32 To the best of our knowledge, no data are yet available on the formation of a CaCO3 coating on plasma-treated PCL and the correlation of CaCO3 film properties with the gas atmosphere of the plasma discharge. In the present study, therefore, we demonstrate that PCL/CaCO3 scaffolds with modified surface properties can be developed by combining plasma treatment of PCL with wet precipitation methods. The effect of plasma treatment on the CaCO3 mineralisation process is investigated by several analytical techniques, including X-ray diffraction (XRD), scanning electron microscopy (SEM), and X-ray photoelectron spectroscopy (XPS), and high-resolution micro-computed tomography (μCT).
Experimental section
Electrospinning of PCL scaffolds
Micro-fibrous scaffolds were produced using the electrospinning instrument at the National Research Tomsk Polytechnic University, Russia.33 PCL (Mn = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Sigma-Aldrich, USA) was dissolved in chloroform (CHCl3) at a concentration of 9% (w/v). The solutions were loaded into a 10 mL plastic syringe with a stainless-steel needle (diameter: 0.58 mm). The flow rate of the solution was 3.6 mL h−1. The needle was connected to a high-voltage power supply, which generated a voltage of 8 kV. Randomly oriented fibres were deposited on a grounded drum collector (rotation speed: 600 rpm) located at a distance of 8 cm from the needle tip.
000 g mol−1, Sigma-Aldrich, USA) was dissolved in chloroform (CHCl3) at a concentration of 9% (w/v). The solutions were loaded into a 10 mL plastic syringe with a stainless-steel needle (diameter: 0.58 mm). The flow rate of the solution was 3.6 mL h−1. The needle was connected to a high-voltage power supply, which generated a voltage of 8 kV. Randomly oriented fibres were deposited on a grounded drum collector (rotation speed: 600 rpm) located at a distance of 8 cm from the needle tip.
Plasma treatment of PCL scaffolds
Plasma treatment of the PCL scaffolds was performed in a dielectric barrier discharge plasma system in a direct mode (Fraunhofer Institute for Interfacial Engineering and Biotechnology, Germany) by use of an RF power generator (13.56 MHz). Different working gases were used for processing the PCL scaffolds with a constant gas flow of 30 cm3 min−1: 99.99% O2, 99.99% Ar, and 99.99% NH3. All plasma treatments were performed for 120 s under a pressure of 30 Pa with applied discharge powers of 30 W and 50 W for Ar plasma and O2 and NH3 plasmas, respectively. Electrospun fibre mats 2 × 2 cm2 in size were treated with plasma in each of the three gas atmospheres and then cut into 8 samples 0.5 × 1 cm2 in size each for further investigation and mineralisation.Enrichment of PCL scaffolds with CaCO3
The procedure for the formation of CaCO3 on the PCL scaffolds was similar to that described elsewhere.7 Briefly, CaCO3 particles were crystallised on the surface of polymer fibres from a mixture of 1 M-CaCl2-saturated and 1 M-Na2CO3-saturated solutions. The CaCl2 and Na2CO3 solutions were prepared using calcium chloride dihydrate (CaCl2·2H2O; Sigma-Aldrich Chemie, Germany) and sodium carbonate (Na2CO3; Sigma-Aldrich Chemie, Germany), respectively. First, the PCL scaffold samples (0.5 × 1 cm2) were treated with 1 mL of CaCl2 solution in an ultrasonic bath until complete wetting was achieved. Complete wetting of scaffolds that were pretreated with the O2 plasma was achieved immediately, whereas that of the other scaffolds was achieved in about 10 min. Subsequently, 1 mL of the Na2CO3 solution was added to the system upon ultrasonication, after which the system was subjected to ultrasound for 30 s. Finally, the samples immersed in the solution were kept out for 60 s to complete the crystallisation process and washed twice with deionised (DI) water. The mineralisation of the PCL scaffolds was performed twice and completed with drying of the samples at 40–45 °C for 30–40 min.Characterisation of scaffolds
The reconstructed datasets were filtered using a median filter with a radius of 1.8 μm in a slice-by-slice manner. Then, each slice was segmented using entropy-based and Otsu thresholding algorithms implemented with the NumPy and scikit-image packages of the Python programming language.36 The segmentation process generates binary datasets in which only the material has non-zero values. Then, the segmented slices were analysed using the Quanfima package,37 mainly by means of the connected component algorithm provided by the scikit-image package.38 The size and orientation of the fibres, the porosity of the PCL/CaCO3 scaffold, and the ratio of the CaCO3 volume to the total volume of the PCL scaffold were calculated for each sample. The 3D visualisation was performed using the Amira 5.4.1 software (FEI Visualization Sciences Group). The Matplotlib package was used for plotting 2D charts.39
Results and discussion
Previous studies demonstrated that plasma treatment of PCL samples induced formation of O-containing and N-containing polar functional groups on the surface, which caused an increase in the surface energy and thus enhanced the hydrophilic properties of the samples.12–19 This is because polymer mineralisation reactions between organic and inorganic phases are significant and can control the crystal growth, particle morphology, and spatial arrangement of CaCO3 agglomerates. Liu et al. and Ma et al. studied the effect of grafting of different functional groups on substrates on the crystallisation process of CaCO3.23,24 As changes in the functional groups and wettability of the surface are expected to affect the mineralisation behaviour of CaCO3, we used XPS and contact angle measurements to characterise the surface of PCL samples treated with plasma in different atmospheres prior to enrichment with CaCO3.Surface properties of electrospun PCL scaffolds before and after plasma treatment
The wettability of PCL scaffold surfaces before and after plasma treatment in O2, NH3, and Ar atmospheres was evaluated by measuring the contact angle of DI water. The change in the surface hydrophobicity of the plasma-treated samples is shown in Fig. 1.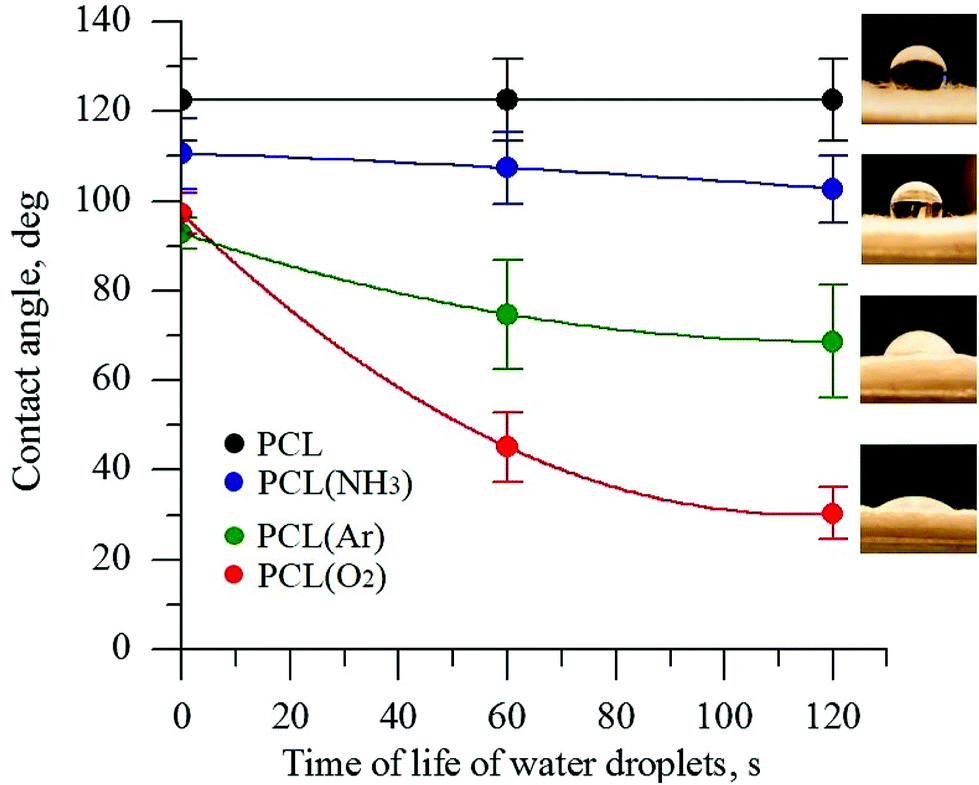 | ||
| Fig. 1 Contact angle of PCL scaffolds before and after plasma treatment under different atmospheres. | ||
The untreated PCL scaffold was hydrophobic, with a water contact angle of 123° ± 10°. After plasma treatment, depending on the gas atmosphere used in the plasma treatment, the PCL surface became more hydrophilic. The DI water contact angles for the PCL scaffolds treated in the NH3- and Ar-plasma atmospheres decreased from 123° ± 10° to 102° ± 5° and to 69° ± 10°, respectively. The lowest contact angle was observed for the PCL scaffold treated with O2 plasma (30° ± 6°). The decrease in the contact angle of a water drop is considered to be referred mainly to the enhancement of the total surface energy of the fiber substrate as well as the generation of new binding states by the plasma treatment.40,41 The obtained data indicate that the plasma treatment with oxygen considerably increases the formation of polar species on the PCL surface. These findings are consistent with previous studies.32,42 These results obviously indicate that the direct plasma treatment provides an effective improvement of surface hydrophilicity that is essential for deep penetration of reagents for deposition of CaCO3.
All the untreated and plasma-treated PCL scaffolds were further analysed by XPS, as shown in Fig. 2a. From the XPS survey spectra, we calculated the atomic ratios (at%) of C, O, and N on the surface of the PCL scaffolds (Table 1). The surface of the untreated PCL scaffold sample contained 81.5 at% C and 18.5 at% O. This C content was slightly higher than that expected for PCL (C6H10O2), because of the presence of adventitious hydrocarbon contaminants, which is inevitable in a laboratory environment.
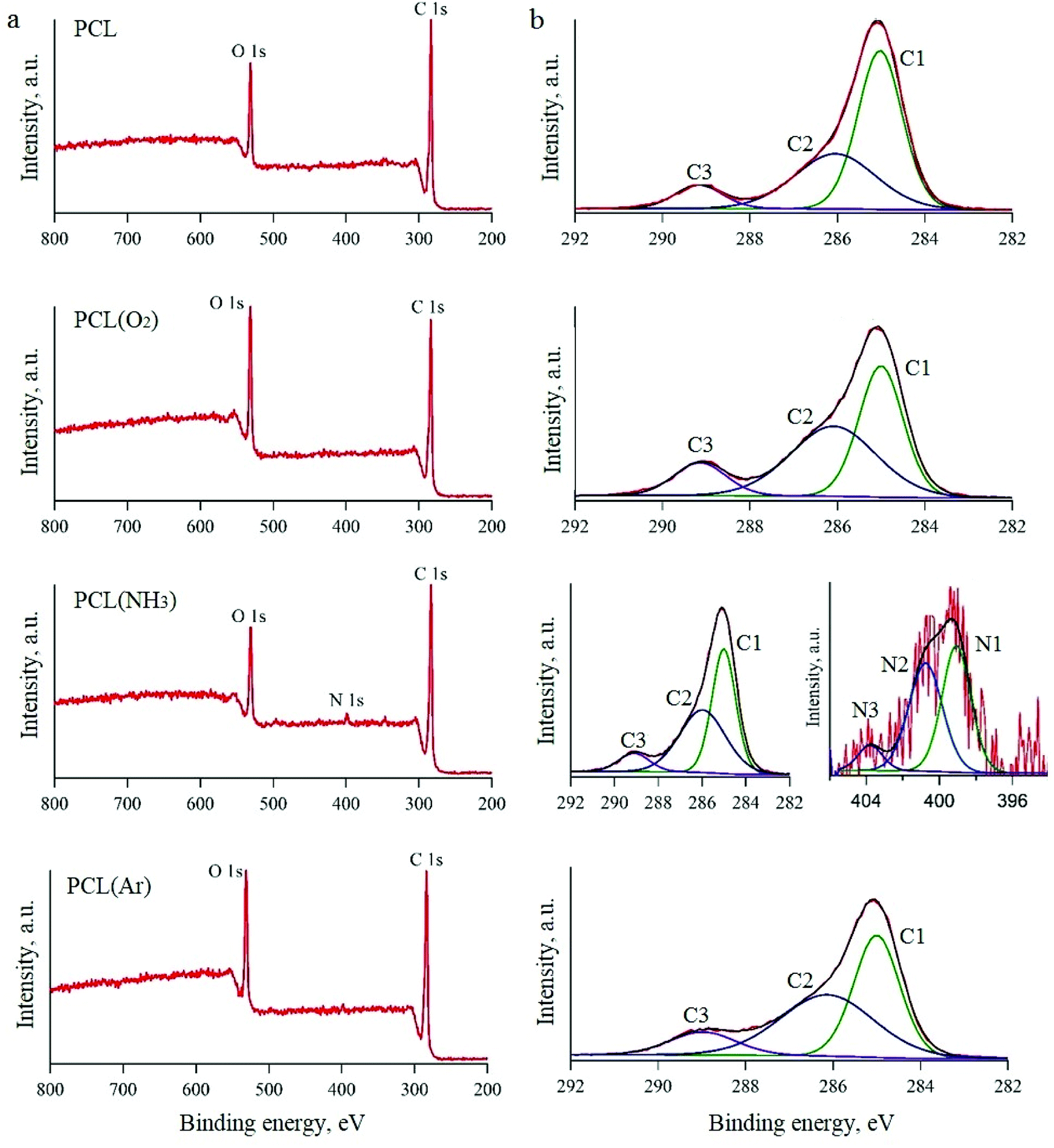 | ||
| Fig. 2 XPS spectra of PCL scaffolds before and after plasma treatment: survey spectra (a) and high-resolution spectra (b). | ||
| Processing condition | Atomic percentage | C/O ratio | ||
|---|---|---|---|---|
| C1s | O1s | N1s | ||
| PCL | 81.5 | 18.5 | 4.4 | |
| PCL(O2) | 75.4 | 24.6 | 3.1 | |
| PCL(NH3) | 81.1 | 17.5 | 1.25 | 4.6 |
| PCL(Ar) | 77.1 | 23.0 | 3.4 | |
After Ar- and O2-plasma treatments, the O content of the PCL surfaces increased to 23.0 at% and 24.6 at%, respectively. However, after the NH3-plasma treatment, the O content of the PCL surface decreased slightly to 17.5 at% (from 18.5 at%, i.e. the O content of the untreated PCL scaffold). This result can be explained by the dissociation of NH3 in the plasma into hydrogen atoms and its reaction with the O on the polymer surface.43 After NH3-plasma treatment, the N1s peak was observed in the XPS survey spectra, which indicates the incorporation of 1.25 at% N into the polymer surface. Higher-resolution spectra of the C1s and N1s regions are shown in Fig. 2b. The C1s line can be resolved into three peaks, which reflect the local environments of the carbon atoms. For instance, the peak at 285 eV corresponds to the hydrocarbon environment (the C1 group), that at 285.9 eV corresponds to the carbon monoxide environment (the C2 group), and that at 289.1 eV corresponds to other carbon oxide environments (the C3 group). The N1s curve of the PCL sample treated with NH3 plasma was fitted using three peaks, two of which could be assigned to the amine (N1, 398.9 eV) and imide (N2, 400.5 eV) groups.32,44 The third peak, located at a higher binding energy (403.8 eV), was ascribed to oxidised nitrogen species (N3).
While the XPS spectra of all the studied PCL scaffolds were similar in terms of the overall content, they differed in terms of the proportions of carbon environments. After O2-plasma treatment, there was an evident increase in the amount of C3 groups, which was attributed to the oxidation of the PCL surface. A previous study showed that these oxygen-containing groups improve surface hydrophilicity.45 The XPS spectra of the samples treated with Ar plasma also showed higher amounts of the C2 and C3 groups in comparison to the untreated sample. There are two possible explanations for this observation. Either the plasma chamber with Ar gas contains a very small amount of oxygen or highly active groups are formed in the Ar plasma, which subsequently react with oxygen when exposed to air.46
Influence of plasma treatment on deposited CaCO3-based coating
Calcium carbonate enriched PCL scaffolds without and after plasma treatment are shown in Fig. 3. There are visible differences in the particle dispersion and CaCO3 coating homogeneity on the PCL surface between the plasma-treated and untreated scaffolds. For the untreated PCL scaffold and the sample exposed under Ar atmosphere many CaCO3 agglomerates protrude out (the areas enclosed by a circle in Fig. 3). For the coating deposited on the O2-plasma treated PCL a rather smooth and homogeneous surface is observed. The CaCO3 coating consists of agglomerates of spherical porous CaCO3 microparticles referred to vaterite (white arrows in Fig. 3). A clear difference in the particles morphology of the CaCO3 coating deposited on the PCL scaffold treated in the NH3-plasma atmosphere compared with other samples is displayed. It seems that the coating on this sample consists of cubic particles (black arrows in Fig. 3), which indicate the presence of the calcite phase. | ||
| Fig. 3 SEM images of PCL/CaCO3 scaffolds before and after plasma treatment at different magnifications. | ||
The XRD spectra of the untreated and plasma-treated PCL scaffolds after mineralisation are shown in Fig. 4. The deposited coating is composed of the vaterite and calcite phases. The diffractograms show three diffraction peaks assigned to the (112), (114), and (300) planes of the vaterite phase (ICDD card no. 86-0174) and one peak assigned to the (104) plane of the calcite phase resolved at 29° (ICDD card no. 86-0174). The presence of calcite is established in all the studied samples. The observed XRD peaks are quite broad, which leads to the conclusion that the CaCO3 coatings deposited on the surface of the PCL scaffolds are nanocrystalline. The integrated intensities of vaterite and calcite were measured from the corresponding reflections of the X-ray diffractograms by means of IGOR Pro software. The ratio of the integrated intensity of the calcite phase (Ic) to the total integrated intensity (IΣ) represents the relative proportions of these two phases in the deposited coatings. The lower the Ic/IΣ ratio, the higher is the number of vaterite crystals in the coating. The results showed that the vaterite phase grew more preferentially in the O2-plasma-treated PCL scaffold sample than in the untreated and Ar- and NH3-plasma-treated PCL scaffold samples.
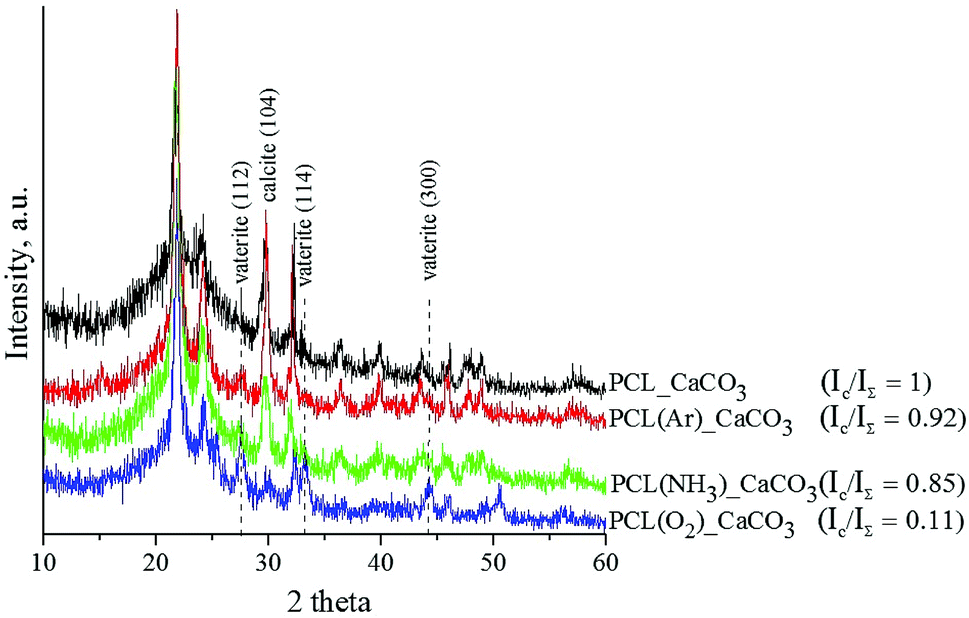 | ||
| Fig. 4 XRD spectra of PCL/CaCO3 scaffolds before and after plasma treatment. The ratio of the integrated intensity of the calcite phase (Ic) to the total integrated intensity (IΣ) represents the relative proportion of the calcite phase in the deposited coatings. | ||
The differences in the CaCO3 coating morphology and phase composition observed in our study are attributed to the different effects of the three plasma treatments of the PCL scaffolds. We assume that the O2-plasma treatment leads to the formation of a large number of active growth sites for the nucleation of vaterite particles, which results in a more homogenous and smoother coating morphology than that achieved with the Ar- and NH3-plasma treatments. In the NH3 atmosphere, the PCL surface becomes saturated with amino groups, which induce crystallisation of CaCO3 into calcite. Ma et al. reported that the interaction between –NH3+ groups in a chitin film and CO32− affects the nucleation of CaCO3 and leads to the irregular formation of calcite.24
The homogenous CaCO3 coating deposited on the O2-plasma-treated PCL scaffold sample showed the best results in terms of wettability and vaterite phase content. Thus, this sample has the highest potential for bone engineering. Therefore, only the PCL/CaCO3 scaffolds treated with O2 plasma were used for further analysis.
Synchrotron μCT imaging of PCL/CaCO3 scaffolds after O2-plasma treatment
We used synchrotron μCT for a complementary 3D analysis of the internal characteristics of the scaffolds and the influence of O2 plasma on the calcification process of the fibre structure. Fig. 5a and b shows 3D renderings of the reconstructed volumes from the μCT data. The PCL fibres and CaCO3 particles are rendered in blue and red, respectively. To obtain a more representative view, regions of interest (ROI) with a 3D area of 550 × 440 × 880 μm3 and magnified regions with a 3D area of 100 × 100 × 100 μm3 were extracted. Reconstructed volumes enable visualisation of the CaCO3 present within the scaffold at different depths (Fig. 5c and d, 3D rendering of CaCO3 only). Spatial arrangement of the CaCO3 agglomerates in the 3D reconstructions corresponds to the plasma propagation direction. The O2-plasma-treated scaffold shows larger agglomerates and deeper penetration of CaCO3 than does the untreated PCL scaffold (see Fig. 5d).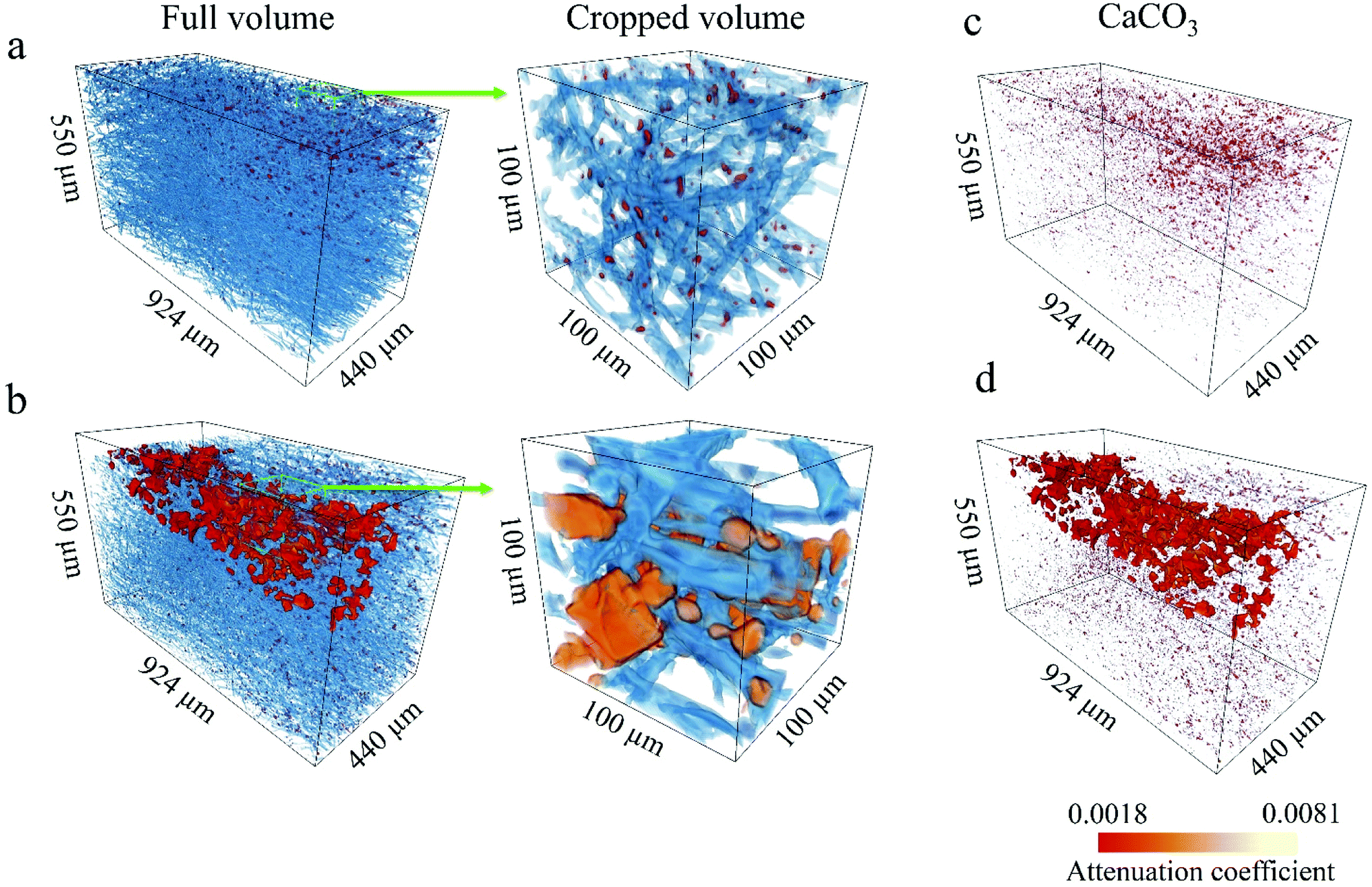 | ||
| Fig. 5 X-ray-μCT-based visualisation of full and cropped volumes of untreated PCL/CaCO3 scaffolds (a and c) and PCL/CaCO3 scaffolds after O2-plasma treatment (b and d) and CaCO3 distribution within the volume of 3D scaffolds (c and d). The polymer and inorganic phases are shown in blue and red-yellow colours, respectively. The colour bar represents the X-ray linear attenuation coefficient in μm−1. | ||
From the high-resolution 3D volume, we calculated the PCL fibre diameter, ratio of the CaCO3 volume to the total sample volume, and porosity for both the PCL/CaCO3 samples (ESI Fig. S1†). For both the samples, the fibre diameters are found to range from 1 μm to 15 μm, with very similar distributions. Pie charts show that the predominant fibre diameter in both the samples is in the range of 1–5 μm. The O2-plasma-treated scaffold sample shows a higher ratio of the CaCO3 volume to the total volume (0.82% ± 0.11%) than the untreated scaffold sample (0.63% ± 0.11%).
Because of the higher volume of CaCO3 agglomerates in the O2-plasma-treated PCL/CaCO3 scaffold, the porosity of this scaffold is lower (∼82%) than that of the untreated scaffold (∼88%). Such deep penetration of CaCO3 into the O2-plasma-treated PCL scaffold can be explained by its higher surface wettability. Moreover, values of the X-ray linear attenuation coefficient reveal that the mineral density of the scaffold surface pretreated with O2 plasma is higher than that of the untreated scaffold. The highly reactive chemical groups on the surface of the O2-plasma-treated PCL scaffold potentially act as nucleation sites for CaCO3 and control the formation of crystals.
Plasma treatment is a basic approach to modify the electrospun scaffold surface prior to biomineralization. There are a number of reports on using plasma pre-treated PCL fibers as an active substrate for biomimetic growth of the calcium phosphates.47–49 For instance, Yang et al. achieved a homogeneous coating of CaP coating onto polymer scaffolds after Ar-plasma treatment and a long period (2–6 h and 7 days) of immersion in simulated body fluid (SBF).49 Lebourg et al. reported the deposition of CaP layers grown on PCL treated with air plasma and subsequently immersed in SBF for 14 days.48 An improvement in the wettability of the electrospun PCL scaffolds and osteoblast-like cell adhesion was observed. Despite promising results, which were obtained, sufficient period of time was required for the surface-modified samples to promote the nucleation of the CaP with the desired phase composition. Moreover, during the biomimetic coating deposition, the pore size of the polymer scaffolds was reduced significantly. The latter fact may be a limiting factor since the porous structure is essential for cell migration, it is important to keep this structure to the most extent. The processing of the PCL scaffolds applied in this work does not have the mentioned disadvantages. The deposition of the dense CaCO3 porous layer took 3 min in total and the structure of the fiber web remained unchanged. Nevertheless, the main advantage of the vaterite deposits on the PCL scaffold surface is that they can serve as a drug delivery vehicle. Despite excellent osteoconductivity and ease of applicability, use of CaPs as a drug delivery coating is limited. It is known that the development of a coating with high total porosity does not ensure drug molecules penetration and elution, unless there is an adequate pore size distribution.50 As shown in ref. 47–49, the biomimetic CaP coating is formed by a mesh of interlocking of flack-like crystals, which allows to create an open microporosity. This structure is suitable for the short time drug administration, because the incorporated drug release is diffusion-controlled. However, a sustained drug delivery has attracted more interest. There is still a lot to be done in terms of adjusting the coating features to different therapeutical needs and obtaining predictable drug delivery systems. But, since the vaterite particles are the least stable polymorph form of calcium carbonate, they tend to transform into calcite in aqueous environment following by the dissolution and crystallization.51 The dissolution-precipitation process of the CaCO3 particles has been shown to allow controlling the drug release time, which makes CaCO3 the best candidate for polymer scaffold mineralization.28
Conclusions
In summary, we investigated the dependence of the mineralisation behaviour of CaCO3 deposited on a plasma-pretreated PCL scaffold surface on the working gas atmosphere. We found that the type and number of active groups on the surface of the plasma-treated PCL scaffolds affect the subsequent nucleation and growth of CaCO3 crystals. The obtained results demonstrate that among the gas atmospheres used in this study (O2, NH3, and Ar), O2 plasma is the most favourable for promoting a positive influence of biomaterials on CaCO3 formation. Preliminary exposure of the surface of the PCL scaffold to O2 plasma promotes the formation of a homogeneous and smooth coating that consists mainly of CaCO3 in a highly porous vaterite crystalline phase. Because of its unique structure and high specific surface area, the vaterite coating has potential for use in various biomedical applications, especially for drug delivery. Functional groups formed on the polymer surface by the O2-plasma treatment enhance the surface hydrophilicity and permeability, thus inducing deeper penetration of CaCO3 into the sample surface, as revealed by μCT. Thus, O2-plasma treatment of PCL scaffolds results in a higher mineralisation efficiency, which is required to ensure their high drug-loading capacity; this high capacity is, in turn, necessary for their use as implantable drug delivery systems in orthopaedic devices. In our future work, we intend to investigate whether the properties of the PCL/CaCO3 composite can be controlled by altering the process parameters of the O2-plasma treatment and the crystallisation conditions of CaCO3. The extent to which the biological performance of the PCL/CaCO3 scaffolds—particularly in terms of their effects on the loading capacity and release kinetics of different drugs—can be improved by O2-plasma treatment, remains to be investigated.Statement of contributions
R. S. prepared hybrid scaffolds; M. S. investigated properties of hybrid composites; A. I. and D. S. investigated composition of the scaffolds; T. B., A. C., R. S., S. S. and V. W. revealed ultrastructure of the samples using synchrotron radiation; M. S. and D. G. mineralized scaffolds in different regimes; T. D. and B. P. investigated different mineralization mechanisms; A. S. and R. M. characterized the obtained scaffolds chemistry, C. O. performed analysis of the composites fabrication mechanisms; P. C. and N. G. performed literature analysis and characterization of the obtained hybrid samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was conducted at Tomsk Polytechnic University within the framework of the Tomsk Polytechnic University Competitiveness Enhancement Program. B. P. acknowledges the FWO (Vlaanderen). V. W. was supported by the German Research Foundation Research Fellowship WE 6221/1-1. We thank the Institute for Photon Science and Synchrotron Radiation, Karlsruhe Institute of Technology, for the allocated beam time at the synchrotron source and the valuable support provided during our experiments.References
- D. I. Braghirolli, D. Steffens and P. Pranke, Drug discovery today, 2014, 19, 743–753 CrossRef CAS PubMed.
- T. Haddad, S. Noel, B. Liberelle, R. El Ayoubi, A. Ajji and G. De Crescenzo, Biomatter, 2016, 6, e1231276 CrossRef PubMed.
- E. Hoveizi, M. Nabiuni, K. Parivar, S. Rajabi-Zeleti and S. Tavakol, Cell biology international, 2014, 38, 41–49 CrossRef CAS PubMed.
- C. Zhou, Q. Shi, W. Guo, L. Terrell, A. T. Qureshi, D. J. Hayes and Q. Wu, ACS Appl. Mater. Inter., 2013, 5, 3847–3854 CrossRef CAS PubMed.
- P. Gentile, V. Chiono, I. Carmagnola and P. V. Hatton, Inter. J. Molec. Sci., 2014, 15, 3640–3659 CrossRef CAS PubMed.
- W. Zhao, J. Li, K. Jin, W. Liu, X. Qiu and C. Li, Mater. Sci. Eng. C, 2016, 59, 1181–1194 CrossRef CAS PubMed.
- M. S. Savelyeva, A. A. Abalymov, G. P. Lyubun, I. V. Vidyasheva, A. M. Yashchenok, T. E. Douglas, D. A. Gorin and B. V. Parakhonskiy, J. Biome. Mater. Res. A, 2017, 105, 94–103 CrossRef CAS PubMed.
- S. N. Gorodzha, A. R. Muslimov, D. S. Syromotina, A. S. Timin, N. Y. Tcvetkov, K. V. Lepik, A. V. Petrova, M. A. Surmeneva, D. A. Gorin, G. B. Sukhorukov and R. A. Surmenev, Coll. Surf. B: Biointerfaces, 2017, 160, 48–59 CrossRef CAS PubMed.
- S. Khorshidi, A. Solouk, H. Mirzadeh, S. Mazinani, J. M. Lagaron, S. Sharifi and S. Ramakrishna, J. Tissue Eng. Regener. Med., 2016, 10, 715–738 CrossRef CAS PubMed.
- I. Rajzer, E. Menaszek, R. Kwiatkowski, J. A. Planell and O. Castano, Mater. Sci. Eng. C, 2014, 44, 183–190 CrossRef CAS PubMed.
- D. Li, T. Wu, N. He, J. Wang, W. Chen, L. He, C. Huang, H. A. EI-Hamshary, S. S. Al-Deya, Q. Ke and X. Mo, Coll. Surf. B:Biointerfaces, 2014, 121, 432–443 CrossRef CAS PubMed.
- Q. Cheng, B. L.-P. Lee, K. Komvopoulos, Z. Yan and S. Li, Tissue Eng. A, 2013, 19, 1188–1198 CrossRef CAS PubMed.
- M. Domingos, F. Intranuovo, A. Gloria, R. Gristina, L. Ambrosio, P. Bártolo and P. Favia, Acta biomaterialia, 2013, 9, 5997–6005 CrossRef CAS PubMed.
- M. Asadian, H. Declercq, M. Cornelissen, R. Morent and N. De Geyter, in 31st International conference on surface modification technologies, 2017 Search PubMed.
- C. López-Santos, A. Terriza, J. Portolés, F. Yubero and A. R. González-Elipe, J. Phys. Chem. C, 2015, 119, 20446–20452 CrossRef.
- R. Ghobeira, N. De Geyter and R. Morent, Biodegradable polymers: recent developments and new perspectives, IAPC Publishing, 2017, pp. 191–236 Search PubMed.
- P. Cools, R. Morent and N. De Geyter, Plasma modified textiles for biomedical applications, Intech, 2016 Search PubMed.
- E. D. Yildirim, H. Ayan, V. N. Vasilets, A. Fridman, S. Guceri and W. Sun, Plasma Processes Polymers, 2008, 5, 58–66 CrossRef CAS.
- L. C. Lopez, M. R. Belviso, R. Gristina, M. Nardulli, R. d'Agostino and P. Favia, Plasma Processes Polymers, 2007, 4, S402–S405 CrossRef.
- Z. Cao, D. Wang, L. Lyu, Y. Gong and Y. Li, RSC Advances, 2016, 6, 10641–10649 RSC.
- H. Maeda, V. Maquet, Q. Chen, T. Kasuga, H. Jawad and A. Boccaccini, Mater. Sci. Eng. C, 2007, 27, 741–745 CrossRef CAS.
- J.-P. Andreassen, J. Crystal Growth, 2005, 274, 256–264 CrossRef CAS.
- X. Liu, Y. Gao and Q. Feng, Fuhe Cailiao Xuebao, 2012, 29, 21–27 CAS.
- Y. Ma and Q. Feng, Cryst. Eng. Comm., 2015, 17, 32–39 RSC.
- M. Ø. Olderøy, M. Xie, B. L. Strand, E. M. Flaten, P. Sikorski and J.-P. Andreassen, Crystal Growth Design, 2009, 9, 5176–5183 CrossRef.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, Nanoscale, 2011, 3, 265–271 RSC.
- G. Zhang, J. Ge, M. Sun, B. Pan, T. Mao and Z. Song, Science in China Series B: Chemistry, 2007, 50, 114–120 CrossRef CAS.
- M. Saveleva, A. Ivanov, M. Kurtukova, V. Atkin, A. Ivanova, G. Lyubun, A. V. Martyukov, E. I. Cherevko, A. K. Sargsyan, A. S. Fedonnikov, A. I. Norkin, A. G. Skirtach, D. A. Gorin and B. V. Parakhonskiy, Mater. Sci. Eng. C, 2018, 85, 57–67 CrossRef CAS PubMed.
- T. Kasuga, H. Maeda, K. Kato, M. Nogami, K.-i. Hata and M. Ueda, Biomaterials, 2003, 24, 3247–3253 CrossRef CAS PubMed.
- A. Neira-Carrillo, R. Gentsch, H. G. Börner, D. F. Acevedo, C. A. Barbero and H. Cölfen, Langmuir, 2016, 32, 8951–8959 CrossRef CAS PubMed.
- L. Liu, D. He, G.-S. Wang and S.-H. Yu, Langmuir, 2011, 27, 7199–7206 CrossRef CAS PubMed.
- D. Yan, J. Jones, X. Yuan, X. Xu, J. Sheng, J. M. Lee, G. Q. Ma and Q. S. Yu, J. Biomed. Materi. Res. A, 2013, 101, 963–972 CrossRef CAS PubMed.
- S. Shkarina, R. Shkarin, V. Weinhardt, E. Melnik, G. Vacun, P. Kluger, K. Loza, M. Epple, S. I. Ivlev, T. Baumbach, M. A. Surmeneva and R. A. Surmenev, Sci. Rep., 2018, 8, 8907 CrossRef PubMed.
- S. Gorodzha, T. E. Douglas, S. K. Samal, R. Detsch, K. Cholewa-Kowalska, K. Braeckmans, A. R. Boccaccini, A. G. Skirtach, V. Weinhardt, T. Baumbach, M. A. Surmeneva and R. A. Surmenev, J. Biomed. Mater. Res. A, 2016, 104, 1194–1201 CrossRef CAS PubMed.
- M. Vogelgesang, S. Chilingaryan, T. dos_Santos Rolo and A. Kopmann, High Performance Computing and Communication & 2012 IEEE 9th International Conference on Embedded Software and Systems (HPCC-ICESS), 2012 IEEE 14th International Conference on: IEEE, 2012, pp. 824–9 Search PubMed.
- S. V. D. Walt, S. C. Colbert and G. Varoquaux, Computing in Sci. Eng., 2011, 13, 22–30 Search PubMed.
- R. Shkarin, rshkarin/quanfima: quanfima-v0.1, 2018, DOI:10.5281/zenodo.1212971.
- S. Van der Walt, J. L. Schönberger, J. Nunez-Iglesias, F. Boulogne, J. D. Warner, N. Yager, E. Gouillart and T. Yu, Scikit-Image: Image Processing in Python, PeerJ, 2014, 2, e453 CrossRef PubMed.
- J. D. Hunter, Matplotlib: A 2D graphics environment, Computing In Sci. Eng., 2007, 9, 90–95 Search PubMed.
- J. Hyun, Polymer, 2001, 42, 6473–6477 CrossRef CAS.
- H.-U. Lee, Y.-S. Jeong, S.-Y. Jeong, S.-Y. Park, J.-S. Bae and H.-G. Kim, Appl. Surf. Sci., 2008, 254, 5700–5705 CrossRef CAS.
- A. Martins, E. D. Pinho, S. Faria, I. Pashkuleva, A. P. Marques and R. L. Reis, Small, 2009, 5, 1195–1206 CAS.
- N. Recek, M. Resnik, H. Motaln, T. Lah-Turnšek, R. Augustine, N. Kalarikkal, N. Kalarikkal, S. Thomas and M. Mozetič, Int. J. Polymer Sci., 2016, 2016, 7354396 Search PubMed.
- M. C. Chang and J. Tanaka, Biomaterials, 2002, 23, 3879–3885 CrossRef CAS PubMed.
- A. M. Pappa, V. Karagkiozaki, S. Krol, S. Kassavetis, D. Konstantinou, C. Pitsalidis, L. Tzounis, N. Pliatsikas and S. Logothetidis, Beilstein J. Nanotech., 2015, 6, 254 CrossRef PubMed.
- R. Morent, N. De Geyter, T. Desmet, P. Dubruel and C. Leys, Plasma Processes and Polymers, 2011, 8, 171–190 CrossRef CAS.
- P. A. Tran, H. T. Nguyen, P. J. Hubbard, H. P. Dang and D. W. Hutmacher, Materials Letters, 2018, 230, 12–15 CrossRef CAS.
- M. Lebourg, J. S. Antón and J. G. Ribelles, Compos. Sci. Tech., 2010, 70, 1796–1804 CrossRef CAS.
- F. Yang, J. Wolke and J. Jansen, Chem. Eng. J., 2008, 137, 154–161 CrossRef CAS.
- M. Espanol, R. Perez, E. Montufar, C. Marichal, A. Sacco and M. Ginebra, Acta Biomaterialia, 2009, 5, 2752–2762 CrossRef CAS PubMed.
- A. Trofimov, A. Ivanova, M. Zyuzin and A. Timin, Pharmaceutics, 2018, 10, 167–236 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07386d |
| This journal is © The Royal Society of Chemistry 2018 |