
Diastereoselective and regiodivergent oxa-[3 + 2] cycloaddition of Achmatowicz products and cyclic 1,3-dicarbonyl compounds†
Jingxun
Yu‡
a,
Haichen
Ma‡
a,
Hongliang
Yao
a,
Hang
Cheng
a and
Rongbiao
Tong
*ab
aDepartment of Chemistry, Hong Kong University of Science and Technology, Clearwater Bay, Kowloon, Hong Kong, China. E-mail: rtong@ust.hk
bHKUST Shenzhen Research Institute, Shenzhen, 518057, China
First published on 25th March 2016
Abstract
Development of two new protocols for oxa-[3 + 2] cycloaddition reactions of Achmatowicz products with 1,3-dicarbonyl compounds for rapid and highly efficient assembly of polycyclic furopyranones is described. Plausible mechanisms were proposed to involve either Pd-catalyzed Tsuji–Trost allylation and concomitant oxa-Michael cyclization or quinine-promoted cascade Michael addition and SN2-type cycloacetalization.
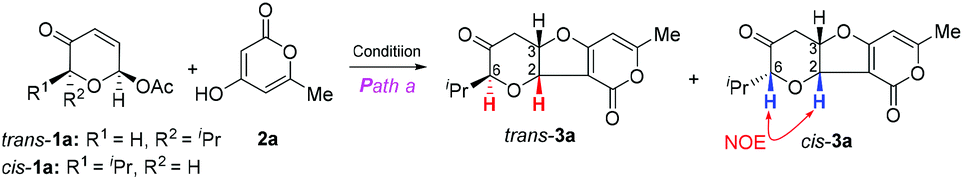
Achmatowicz rearrangement (AchR),1 an oxidative ring expansion process of furfuryl alcohols to pyranone acetals (also known as pyranuloses), has received growing interest in organic synthesis.2 The expanding synthetic utility of AchR lies in the versatile reactivity of pyranuloses or their direct derivatives under different chemical conditions. For example, acylated pyranulose is a glycosyl donor for glycosylation via palladium catalysis,3 a 1,3-dipole for [5 + 2] cycloaddition with alkenes under basic conditions,4 and an excellent substrate for phosphine-catalyzed [3 + 2]-cycloaddition with 2,3-butadienoates.5 Our continuing interest in exploitation of AchR for natural product synthesis6 and discovery of new reaction modes7 prompted us to re-examine the fundamental reactivity of AchR products. On the basis of the electrophilic properties at both C2 (acetal) and C3 (enone), we anticipated that the AchR product could serve as a bis-electrophile for cycloaddition reaction if the appropriate bis-nucleophile could be identified. In this regard, we fully recognized that 1,3-dicarbonyl compounds have been widely used in domino or multicomponent reactions due to their intrinsic bis-nucleophilic nature.8 Therefore, we proposed that an oxa-[3 + 2] cycloaddition reaction of the AchR product (or its derivative, 1) with a 1,3-dicarbonyl compound (2) might occur to provide the bicyclic furopyranone (3 or 4), a valuable building block embodied in many natural products or bioactive compounds such as chafurosides A and B9 and pittosporatobiraside A (Scheme 1).10 The inherent challenge posed by this hypothetic cyclization is the regioselectivity (path a versus path b), which has not been fully addressed although individual similar processes were reported previously (path a by Füstner11 with only single example and path b by Ramasastry12) under different conditions. In particular, the diastereoselectivity (if R1 ≠ H) remains unexplored. Herein, we describe two new protocols for these two interesting oxa-[3 + 2] cycloaddition reactions, their substrate scope, and mechanistic hypothesis for rationalization of the observed regioselectivity and diastereoselectivity. In addition, an unexpected cascade involving Michael/decarboxylation/acetalization was discovered (Table 3).
 | ||
| Scheme 1 Achmatowicz rearrangement and our hypothesis. | ||
The cyclization via path a11 might conceptually involve cascade Tsuji–Trost allylation13 and oxa-Michael14 cyclization while intermolecular Michael addition15 followed by acetalization might operate via path b.12 This mechanistic postulate guided us to examine the first step of the hypothetic cyclization via palladium catalysis (Tsuji–Trost allylation) or base/acid catalysis (Michael addition) since the second step was expected to occur concomitantly through a favourable 5-exo-trig or 5-exo-tet cyclization.16 Therefore, we first investigated the palladium-catalyzed reaction of readily available acetoxy pyranone trans-1a (or cis-1a) and 2-pyrone 2a (Table 1). To our delight, 1 mol% Pd(PPh3)4 was found to be effective for oxa-[3 + 2] cycloaddition, providing the desired cis-fused furopyranone trans-3a as the single diastereomer (dr > 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 83% yield (entry 1). Surprisingly, the cascade cyclization of the corresponding cis-1a and 2a delivered an inseparable mixture of trans-3a and cis-3a (trans/cis 7:10) under the identical reaction conditions (entry 2). Since it is well known that Tsuji–Trost allylation with soft nucleophiles (such as 1,3-dicarbonyl compounds) typically proceeds with a net retention of stereochemistry,17 we speculated that the poor diastereoselectivity for cis-1a might have arisen from the facile epimerization of the α chiral center of the carbonyl group in the presence of base via enol–keto tautomerization (Scheme 2). To suppress this potential epimerization and gain mechanistic insights into this unusual stereochemistry-dependent diastereoselectivity, we carried out the oxa-[3 + 2] cycloaddition reaction at variable temperature and time (entries 3–6). Apparently, at a lower reaction temperature (0 °C or −20 °C) and within a shorter reaction time (quenching the reaction within 2 minutes) the reaction of cis-1a or trans-1a with 2a proceeded with the expected retention of configuration and provided the corresponding products cis-3a and trans-3a, respectively, with excellent diastereoselectivity but low conversion. When cis-3a was subjected to heating conditions (entry 6), trans-3a was obtained exclusively (dr > 30:1) with 91% yield, which suggested that trans-3a was the thermodynamically more stable product. This finding prompted us to search for mild conditions that could exclusively produce trans-3a from both cis-1a and trans-1a. Preliminary screening of solvents (entries 7–9: DMF, THF and toluene), bases (entries 12–14: DBU, quinine and Et3N) and palladium catalysts [entries 9–12: Pd(PPh3)4, PdCl2, Pd2(dba)3 and Pd(OAc)2] led us to identify the optimal conditions for both substrates (entries 9 and 15): triethyl amine (1 eq.) as the base, Pd(PPh3)4 (1 mol%) as the catalyst and toluene as the solvent at room temperature for 30 min, which afforded trans-3a with an excellent yield (90% from cis-1a, 92% from trans-1a) and diastereoselectivity (trans-3a, dr > 30:1). The structures of cis-3a and trans-3a were confirmed by careful analysis of the spectral data (cis-fused furopyranone with a distinctive high value of J = 8 Hz and the nOe observed at H2 and H6).
:1) with 83% yield (entry 1). Surprisingly, the cascade cyclization of the corresponding cis-1a and 2a delivered an inseparable mixture of trans-3a and cis-3a (trans/cis 7:10) under the identical reaction conditions (entry 2). Since it is well known that Tsuji–Trost allylation with soft nucleophiles (such as 1,3-dicarbonyl compounds) typically proceeds with a net retention of stereochemistry,17 we speculated that the poor diastereoselectivity for cis-1a might have arisen from the facile epimerization of the α chiral center of the carbonyl group in the presence of base via enol–keto tautomerization (Scheme 2). To suppress this potential epimerization and gain mechanistic insights into this unusual stereochemistry-dependent diastereoselectivity, we carried out the oxa-[3 + 2] cycloaddition reaction at variable temperature and time (entries 3–6). Apparently, at a lower reaction temperature (0 °C or −20 °C) and within a shorter reaction time (quenching the reaction within 2 minutes) the reaction of cis-1a or trans-1a with 2a proceeded with the expected retention of configuration and provided the corresponding products cis-3a and trans-3a, respectively, with excellent diastereoselectivity but low conversion. When cis-3a was subjected to heating conditions (entry 6), trans-3a was obtained exclusively (dr > 30:1) with 91% yield, which suggested that trans-3a was the thermodynamically more stable product. This finding prompted us to search for mild conditions that could exclusively produce trans-3a from both cis-1a and trans-1a. Preliminary screening of solvents (entries 7–9: DMF, THF and toluene), bases (entries 12–14: DBU, quinine and Et3N) and palladium catalysts [entries 9–12: Pd(PPh3)4, PdCl2, Pd2(dba)3 and Pd(OAc)2] led us to identify the optimal conditions for both substrates (entries 9 and 15): triethyl amine (1 eq.) as the base, Pd(PPh3)4 (1 mol%) as the catalyst and toluene as the solvent at room temperature for 30 min, which afforded trans-3a with an excellent yield (90% from cis-1a, 92% from trans-1a) and diastereoselectivity (trans-3a, dr > 30:1). The structures of cis-3a and trans-3a were confirmed by careful analysis of the spectral data (cis-fused furopyranone with a distinctive high value of J = 8 Hz and the nOe observed at H2 and H6).
 | ||
| Scheme 2 Proposed mechanism of cascade Tsuji–Trost allylation and oxa-Michael cyclization. | ||
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Pd cat. (1 mol%) | Base (1 eq.)/solvent | Temp (°C)/time (min) | Yieldc (%) (trans:cis)b |
| a The reaction was run with 0.1 mmol of 1a. b Ratio was determined by NMR analysis of the crude reaction mixture. c Combined yield after flash column chromatography on silica gel. DMF: N,N-dimethylformamide; THF, tetrahydrofuran; DCM: dichloromethane; Tol: toluene; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene. | |||||
| 1 | trans-1a | Pd(PPh3)4 | Et3N/DCM | rt/30 | 83 (>30:1) |
| 2 | cis-1a | Pd(PPh3)4 | Et3N/DCM | rt/30 | 86 (7:10) |
| 3 | cis-1a | Pd(PPh3)4 | Et3N/DCM | 0/30 | 80 (1:10) |
| 4 | cis-1a | Pd(PPh3)4 | Et3N/DCM | −20 °C/2 | 10 (1:25) |
| 5 | trans-1a | Pd(PPh3)4 | Et3N/DCM | −20 °C/2 | 12 (>30:1) |
| 6 | cis-3a | Pd(PPh3)4 | Et3N/DCM | Reflux/300 | 91 (>30:1) |
| 7 | cis-1a | Pd(PPh3)4 | Et3N/DMF | rt/30 | 85 (8:1) |
| 8 | cis-1a | Pd(PPh3)4 | Et3N/THF | rt/30 | 60 (1:1) |
| 9 | cis- 1a | Pd(PPh 3 ) 4 | Et 3 N/Tol | rt/30 |
90 (>30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1)
|
| 10 | cis-1a | Pd(OAc)2 | Et3N/Tol | rt/30 | 59 (>30:1) |
| 11 | cis-1a | PdCl2 | Et3N/Tol | rt/30 | 8 (>30:1) |
| 12 | cis-1a | Pd2(dba)3 | Et3N/Tol | rt/30 | 35 (>30:1) |
| 13 | cis-1a | Pd(PPh3)4 | DBU/Tol | rt/30 | 85 (>30:1) |
| 14 | cis-1a | Pd(PPh3)4 | Quinine/Tol | rt/30 | 89 (>30:1) |
| 15 | trans- 1a | Pd(PPh 3 ) 4 | Et 3 N/Tol | rt/30 |
92 (>30:1)
|
On the basis of these results (Table 1), we proposed plausible mechanistic pathways for the oxa-[3 + 2] cycloaddition reaction (Scheme 2). The reaction of trans-1a and 2a (Nu−) was initiated by oxidative addition of palladium followed by a SN2-type nucleophilic substitution and subsequent intramolecular oxa-Michael cyclization (Scheme 2a). The oxa-[3 + 2] cycloaddition reaction of cis-1a with 2a involved similar oxidative addition of palladium and oxa-Michael cyclization, but it might operate differently in the course of substitution through path (I), probably due to the unfavourable steric interaction of the isopropyl group with the incoming nucleophile (2a). In path (I) the nucleophilic substitution occurs at the palladium followed by reductive elimination (III → IV → II), which resulted in a net inversion of stereochemistry.17 However, the reaction temperature and solvent (e.g., THF) might make path (II) a competitive pathway as shown in Table 1. It is less likely that epimerization of cis-3a to trans-3a occurred through enol–keto tautomerization under the mild reaction conditions within a short time because subjection of a mixture of cis-3a and trans-3a (cis/trans = 5/4) to the standard reaction conditions resulted in a small increase of the ratio of trans-3a (cis/trans 3/7).
Next, we turned our attention to explore the possibility of an oxa-[3 + 2] cycloaddition reaction via path b involving intermolecular Michael addition and concomitant acetalization under non-aqueous conditions (different from Ramasastry's conditions). We first investigated the reaction of trans-1a and 2a (Table 2). Fortunately, treatment of trans-1a and 2a with triethyl amine in DCM at reflux for 12 h provided the expected cis-fused furopyranone cis-4a in 60% yield with excellent diastereoselectivity (dr = 15:1). Encouraged by this result, we began to examine different bases (entries 1–7, 14 and 15) and solvents (entries 16–18) in order to identify the optimal reaction conditions (Table 2). It was found that in the presence of one equivalent of quinine18 (entry 8) the reaction proceeded cleanly and gave the best yield and stereoselectivity, which was in contrast to the inorganic base (entries 14 and 15) that could not promote the cycloaddition reaction. Interestingly, the extended reaction time and/or increased reaction temperature resulted in isomerization of cis-4a to trans-4a (entries 8–10). However, similar isomerization was not observed in the oxa-[3 + 2] cycloaddition reaction of cis-1a and 2a under the identical conditions via path b, which led to the exclusive formation of trans-4a (entries 11–13). These seemingly contradictory results might be attributed to the formation of the thermodynamically more stable trans-4a through the enol–keto tautomerization in the presence of a base at reflux (Scheme 3). The structures of cis-4a and trans-4a were unambiguously substantiated by single crystal X-ray diffraction analysis (Fig. 1). Notably, the reaction of trans-1a and 2a under previously reported conditions12 (entry 19) was very sluggish (25% conversion at rt for 12 h) with poor diastereoselectivity (dr 3:1), which was in sharp contrast to the reported observation (NaHCO3, H2O, rt, 1 h, 88%).12
 | ||
| Scheme 3 Proposed mechanism of cascade Michael addition and acetalization. | ||
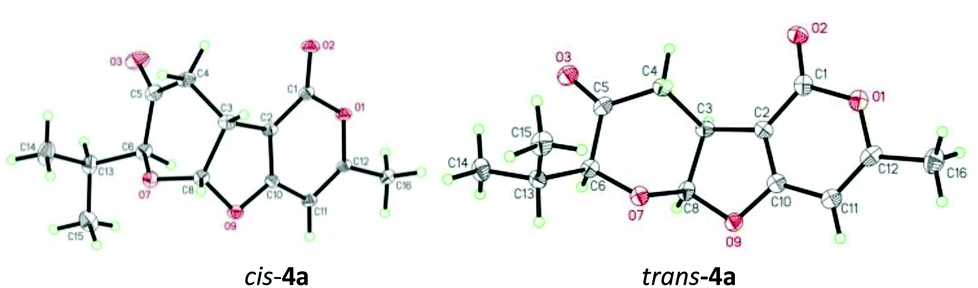 | ||
| Fig. 1 ORTEP diagrams of trans-4a and cis-4a. | ||
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Base (1 eq.) | Solvent | Temp (°C)/time (h) | Yieldc (%) (trans:cis)b |
| a The reaction was run with 0.1 mmol of 1a. b Ratio was determined by NMR analysis of the crude reaction mixture. c Combined yield after flash column chromatography on silica gel. d The reaction was performed in tube sealing at 70 °C for 36 h in DCM. TFA: trifluoroacetic acid. DABCO: triethylenediamine; DMAP: 4-dimethylaminopyridine. | |||||
| 1 | trans-1a | Et3N | DCM | Reflux/12 | 60 (1:15) |
| 2 | trans-1a | DBU | DCM | Reflux/12 | 53 (1:15) |
| 3 | trans-1a | (iPr)2NEt | DCM | Reflux/12 | 75 (1:1) |
| 4 | trans-1a | DABCO | DCM | Reflux/12 | 70 (1:10) |
| 5 | trans-1a | Pyridine | DCM | Reflux/12 | NR |
| 6 | trans-1a | Pyrrolidine | DCM | Reflux/12 | NR |
| 7 | trans-1a | DMAP | DCM | Reflux/12 | 75 (1:1) |
| 8 | trans- 1a | Quinine | DCM | Reflux/12 |
90 (1:15)
|
| 9 | trans-1a | Quinine | DCM | Reflux/24 | 88 (1:10) |
| 10 | trans-1a | Quinine | DCM | Reflux/36d | 85 (>30:1) |
| 11 | cis-1a | Quinine | DCM | rt/5 | 21 (>30:1) |
| 12 | cis-1a | Quinine | DCM | rt/12 | 49 (>30:1) |
| 13 | cis- 1a | Quinine | DCM | Reflux/12 |
91 (>30:1)
|
| 14 | trans-1a | NaHCO3 | DCM | Reflux/12 | NR |
| 15 | trans-1a | NaOH | DCM | Reflux/12 | NR |
| 16 | trans-1a | Quinine | Tol | 50 °C/12 | 80 (3:1) |
| 17 | trans-1a | Quinine | THF | 50 °C/12 | 75 (1:3) |
| 18 | trans-1a | Quinine | MeOH | rt/12 | trace |
| 19 | trans-1a | NaHCO3 (2 eq.) | H2O | rt/1 | 25 (3:1) |

Mechanistically, we speculated that the oxa-[3 + 2] cycloaddition reaction started with Michael addition,19 in which diastereoselectivity was controlled by the acetoxy group to avoid the otherwise steric interaction developed between the acetoxy group and the incoming nucleophile. The second step of the cascade sequence might involve transacetalization20 through an intramolecular SN2 substitution, which under basic conditions using CH2Cl2 as the aprotic solvent was promoted by dual activation21 through double hydrogen bonding interactions with quinine.22 Although transacetalization via the SN1 substitution (via oxonium ions)23 could not be ruled out and more experimental work is needed to further elucidate the detailed mechanism, we choose at this stage to further expand the substrate scope of these two novel oxa-[3 + 2] cycloaddition reactions.
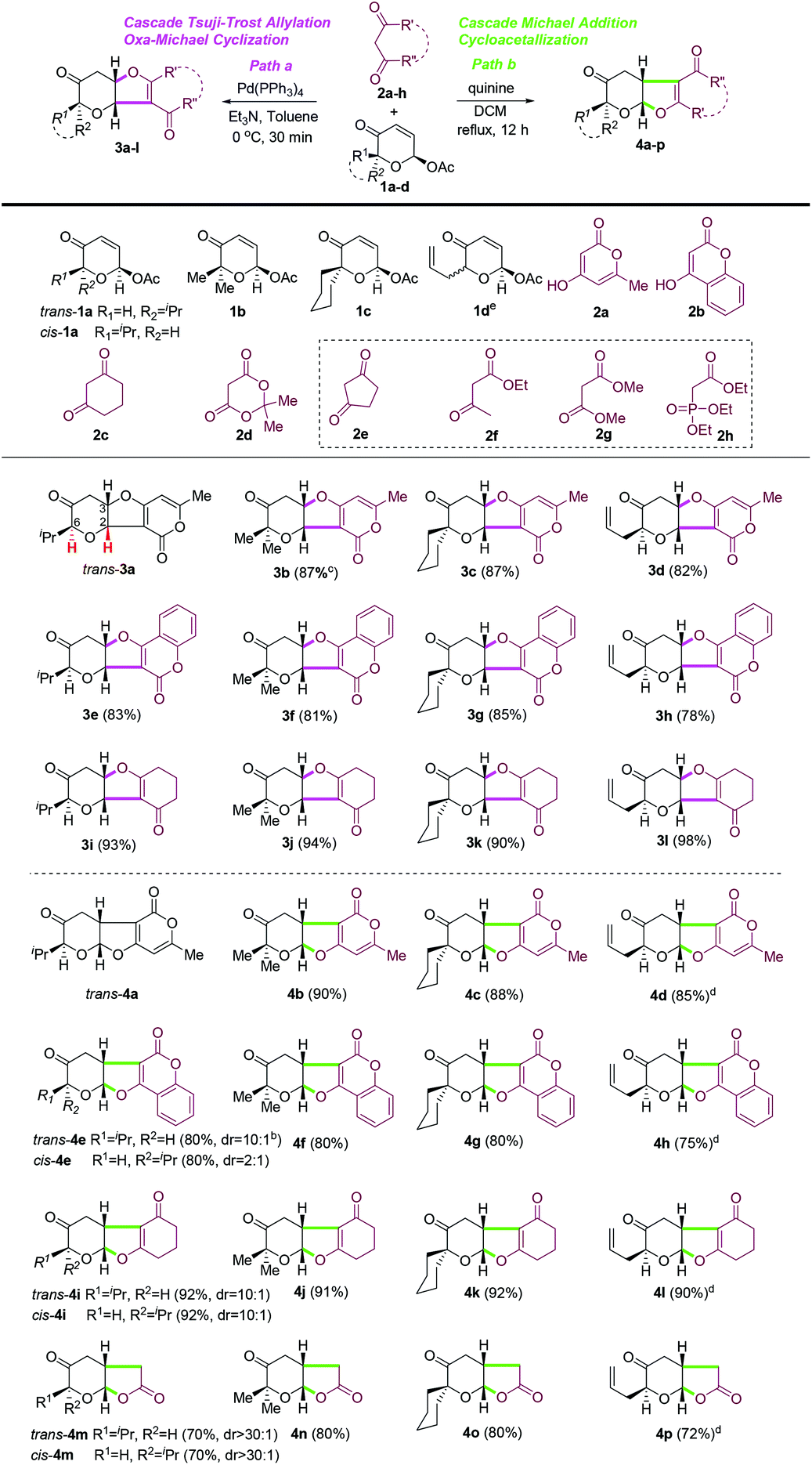
With the optimized conditions in hand, the scope and limitations of both oxa-[3 + 2] cycloaddition reactions (path a versus path b) were examined with a small set of acetoxy-2-pyranones 1a–d and a series of 1,3-dicarbonyl compounds (2a–h) (Table 3). In general, both oxa-[3 + 2] cycloaddition reactions of acetoxy-2-pyranones with different substitutions at C6 (1a–d) and most six-membered cyclic 1,3-dicarbonyl compounds (2a–d)24 could proceed smoothly under our optimized conditions to provide the desired furopyranones (3b–l and 4b–p) in good to excellent yields (72–98%) with excellent diastereoselectivity (dr ≥ 10:1). Unexpectedly, 1,3-cyclopentanedione (2e), acyclic 1,3-dicarbonyl compounds (e.g., 2f and 2g) and triethyl phosphonoacetate (2h) did not react with any acetoxy-2-pyranones (2a–d) under various conditions, which could not be well rationalized at this point. It is noteworthy that a mixture of diastereomeric acetoxypyranones 1d could be employed for both the oxa-[3 + 2] cycloaddition reactions to provide the corresponding cyclization products as the single diastereomer with excellent yields (3d, 3h, 3l, 4d, 4h, 4l, 4p). Interestingly, the reaction of acetoxy-2-pyranones with Meldrum's acid25 (2d) under our quinine-mediated reaction conditions delivered the unexpected decarboxylation products (4m–n) in excellent yields, while Meldrum's acid was not reactive towards acetoxy-2-pyranones with palladium catalysis. Further exploration of this finding is ongoing and will be reported in due course.
a
|
a The reaction was run with 0.1 mmol of 1a–d.
b Ratio was determined by NMR analysis of the crude reaction mixture.
c Combined yield after flash column chromatography on silica gel.
d All cascade reactions of 1d and 2a–d were carried out in tube sealing at 70 °C for 36 h in DCM.
e The allylic substrate is a mixture of diastereomers (trans:cis = 3:2).
|
|---|
|
Conclusions
In summary, we have developed two new protocols for oxa-[3 + 2] cycloaddition reactions, which allowed a rapid and highly efficient assembly of structurally interesting polycyclic furopyranones. Importantly, we have demonstrated for the first time that Achmatowicz products could be employed as bis-electrophiles for diastereoselective and regiodivergent oxa-[3 + 2] cycloaddition reactions with 1,3-dicarbonyl compounds, which greatly expands the synthetic utility of Achmatowicz rearrangement. Plausible mechanistic pathways for both oxa-[3 + 2] cycloaddition reactions were proposed on the basis of our new results and findings to rationalize the regiodivergence and diastereoselectivity: palladium catalysis involves Tsuji–Trost allylation followed by intramolecular oxa-Michael cyclization; quinine-mediated cascade cyclization occurs through a diastereoselective intermolecular Michael addition and a subsequent SN2-type cycloacetalization by dual activation. In addition, we discovered an unexpected new cascade sequence: Michael addition/decarboxylation/acetalization. These two novel oxa-[3 + 2] cycloaddition reactions may find applications in drug discovery and natural product synthesis.Acknowledgements
This research was financially supported by HKUST and Research Grant Council of Hong Kong (ECS 605912, GRF 605113, and GRF 16305314) and partially supported by NSFC (Project No. 21472160).Notes and references
- (a) O. Achmatowicz Jr., P. Bukowski, B. Szechner, Z. Zwierzcho-wska and A. Zamojski, Tetrahedron, 1971, 27, 1973–1996 CrossRef; (b) L. Zhu, L. Song and R. Tong, Org. Lett., 2012, 14, 5892–5895 CrossRef CAS PubMed; (c) J. Ren, Y. Liu, L. Song and R. Tong, Org. Lett., 2014, 16, 2986–2989 CrossRef CAS PubMed.
- J. Deska, D. Thiel and E. Gianolio, Synthesis, 2015, 3435–3450 CrossRef CAS.
- (a) R. S. Babu and G. A. O'Doherty, J. Am. Chem. Soc., 2003, 125, 12406–12407 CrossRef CAS PubMed; (b) R. S. Babu, M. Zhou and G. A. O'Doherty, J. Am. Chem. Soc., 2004, 126, 3428–3429 CrossRef CAS PubMed; (c) A. C. Comely, R. Felkema, A. J. Minnaard and R. L. Feringa, J. Am. Chem. Soc., 2003, 125, 8714–8715 CrossRef CAS PubMed.
- (a) J. B. Hendrickson and J. S. Farina, J. Org. Chem., 1980, 45, 3359–3361 CrossRef CAS. For enantioselective [5 + 2] cycloaddition of Achmatowicz adducts, see: (b) N. Z. Burns, M. R. Witten and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed; (c) M. R. Witten and E. N. Jacobsen, Angew. Chem., Int. Ed., 2014, 53, 5912–5916 CrossRef CAS PubMed. For selected reviews, see: (d) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed; (e) V. Singh, U. M. Krishna and G. K. Trivedi, Tetrahedron, 2008, 64, 3405–3428 CrossRef CAS; (f) H. Pellissier, Adv. Synth. Catal., 2011, 353, 189–218 CrossRef CAS.
- R. A. Jones and M. J. Krische, Org. Lett., 2009, 11, 1849–1851 CrossRef CAS PubMed.
- (a) L. Zhu and R. Tong, Org. Lett., 2015, 17, 1966–1969 CrossRef CAS PubMed; (b) J. Ren, J. Wang and R. Tong, Org. Lett., 2015, 17, 744–747 CrossRef CAS PubMed; (c) L. Zhu, Y. Liu, R. Ma and R. Tong, Angew. Chem., Int. Ed., 2015, 54, 627–632 CAS; (d) J. Ren and R. Tong, J. Org. Chem., 2014, 79, 6987–6995 CrossRef CAS PubMed; (e) J. Ren, Y. Liu, L. Song and R. Tong, Org. Lett., 2014, 16, 2986–2989 CrossRef CAS PubMed.
- (a) L. Zhu, L. Song and R. Tong, Org. Lett., 2012, 14, 5892–5895 CrossRef CAS PubMed; (b) Z. Li, F. C. F. Ip, N. Y. Ip and R. Tong, Chem. – Eur. J., 2015, 21, 11152–11157 CrossRef CAS PubMed.
- (a) D. Bonne, Y. Coquerel, T. Constantieux and J. Rodriguez, Tetrahedron: Asymmetry, 2010, 21, 1085–1109 CrossRef CAS.
- T. Furuta, M. Nakayama, H. Suzuki, H. Tajimi, M. Inai, H. Nukaya, T. Wakimoto and T. Kan, Org. Lett., 2009, 11, 2233–2236 CrossRef CAS PubMed , and references therein.
- K. Munesada, K. Ogihara and K. Suga, Phytochemicals, 1991, 30, 4158–4159 CrossRef CAS.
- (a) A. Fürstner, F. Feyen, H. Prinz and H. Waldmann, Tetrahedron, 2004, 60, 9543–9558 CrossRef; (b) M. J. Bartlett, C. A. Turner and J. E. Harvey, Org. Lett., 2013, 15, 2430–2433 CrossRef CAS PubMed.
- S. Kasare, S. K. Bankar and S. S. V. Ramasastry, Org. Lett., 2014, 16, 4284–4287 CrossRef CAS PubMed.
- (a) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387–4388 CrossRef; (b) B. M. Trost and T. J. Fullerton, J. Am. Chem. Soc., 1973, 95, 292–294 CrossRef CAS; (c) B. M. Trost and D. A. Thaisrivongs, J. Am. Chem. Soc., 2008, 130, 14092–14093 CrossRef CAS PubMed; (d) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC.
- (a) C. F. Nising and S. Bräse, Chem. Soc. Rev., 2008, 37, 1218–1228 RSC; (b) C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988–999 RSC.
- (a) Y. Zhang and W. Wang, Catal. Sci. Technol., 2012, 2, 42–53 RSC; (b) J. Comelles, M. Moreno-Mañas and A. Vallribera, ARKIVOC, 2005, 207–238 CAS.
- (a) J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC; (b) J. E. Baldwin, R. C. Thomas, L. I. Kruse and L. Silberman, J. Org. Chem., 1977, 42, 3846–3852 CrossRef CAS.
- (a) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631–12635 CrossRef CAS PubMed. For excellent reviews, see: (b) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS PubMed; (c) B. M. Trost, T. Zhang and J. D. Sieber, Chem. Sci., 2010, 1, 427–440 RSC; (d) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed.
- For recent quinine-promoted Michael additions, see: (a) C. De Fusco and A. Lattanzi, Eur. J. Org. Chem., 2011, 3728–2731 CrossRef CAS; (b) J. Wang, L. D. Heikkinen, H. Li, L. Zu, W. Jiang, H. Xie and W. Wang, Adv. Synth. Catal., 2007, 349, 1052–1056 CrossRef CAS; (c) E. Sekino, T. Kumamoto, T. Tanaka, T. Ikeda and T. Ishikawa, J. Org. Chem., 2004, 69, 2760–2767 CrossRef CAS PubMed.
- For an excellent example of Michael additions of enones with 1,3-dicarbonyl compounds under basic conditions, see: F. Wu, H. Li, R. Hong and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 947–950 CrossRef CAS PubMed.
- (a) J.-Y. Ortholand, N. Vicart and A. Greiner, J. Org. Chem., 1995, 60, 1880–1884 CrossRef CAS; (b) R. G. Cooks, H. Chen, M. N. Eberlin, X. Zheng and W. A. Tao, Chem. Rev., 2006, 106, 188–211 CrossRef CAS PubMed.
- I. Čorić, S. Vellalath and B. List, J. Am. Chem. Soc., 2010, 132, 8536–8537 CrossRef PubMed.
- B.-D. Cui, S.-W. Li, J. Zuo, Z.-J. Wu, X.-M. Zhang and W.-C. Yuan, Tetrahedron, 2014, 70, 1895–1902 CrossRef CAS.
- For reviews, see: (a) F. Perron and K. F. Albizati, Chem. Rev., 1989, 89, 1617–1661 CrossRef CAS; (b) J. Sperry, Z. E. Wilson, D. C. K. Rathwell and M. A. Brimble, Nat. Prod. Rep., 2010, 27, 1117–1137 RSC.
- For selected Michael additions of cyclic 1,3-dicarbonyl compounds, see: (a) N. Halland, T. Hansen and K. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 4955–4957 CrossRef CAS PubMed; (b) H. Kim, C. Yen, P. Preston and J. Chin, Org. Lett., 2006, 8, 5239–5242 CrossRef CAS PubMed; (c) J. Xie, L. Yue, W. Chen, W. Du, J. Zhu, J. Deng and Y. Chen, Org. Lett., 2007, 9, 413–415 CrossRef CAS PubMed; (d) Z. Dong, L. Wang, X. Chen, X. Liu, L. Lin and X. Feng, Eur. J. Org. Chem., 2009, 5192–5197 CrossRef CAS. For selected oxa-[3 + 3] cycloaddition of cyclic 1,3-dicarbonyl compounds, see: (e) K. P. Cole and R. P. Hsung, Org. Lett., 2003, 5, 4843–4846 CrossRef CAS PubMed; (f) G.-Y. Luo, H. Wu, Y. Tang, H. Li, H.-S. Yeom, K. Yang and R. P. Hsung, Synthesis, 2015, 2713–2720 CAS; (g) Y. Tang, J. Oppenheimer, Z. Song, L. You, X. Zhang and R. P. Hsung, Tetrahedron, 2006, 62, 10785–10813 CrossRef CAS.
- For Meldrum's acid in organic synthesis, see: (a) B.-C. Chen, Heterocycles, 1991, 32, 529–597 CrossRef CAS; (b) A. M. Dumas and E. Fillion, Acc. Chem. Res., 2010, 43, 440–454 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization methods and copies of the 1H- and 13C-NMR spectra of new compounds. CCDC 1444735 (trans-4a) and 1444736 (cis-4a). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00034g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |