
Pd-catalyzed aminocarbonylation of alkynes with amines using Co2(CO)8 as a carbonyl source†
Yaxi
Dong
a,
Suyan
Sun
a,
Fan
Yang
*a,
Yu
Zhu
a,
Weiguo
Zhu
a,
Huijie
Qiao
a,
Yusheng
Wu
*bc and
Yangjie
Wu
*a
aThe College of Chemistry and Molecular Engineering, Henan Key Laboratory of Chemical Biology and Organic Chemistry, Key Laboratory of Applied Chemistry of Henan Universities, Zhengzhou University, Zhengzhou 450052, China. E-mail: yangf@zzu.edu.cn; wyj@zzu.edu.cn
bTetranov Biopharm, LLC., 75 Daxue Road, Zhengzhou, 450052, China. E-mail: yusheng.wu@tetranovglobal.com
cCollaborative Innovation Center of New Drug Research and Safety Evaluation, Henan Province, China. E-mail: yusheng.wu@tetranovglobal.com
First published on 28th March 2016
Abstract
A simple and mild protocol for the synthesis of 2-ynamides by palladium-catalyzed aminocarbonylation of alkynes was developed. The reaction proceeded at room temperature in air, utilizing Co2(CO)8 as a carbonyl source. This aminocarbonylation reaction features mild reaction conditions and good tolerance of substrates especially including aryl amines and primary amines.
2-Ynamides have been extensively proven to be one of the most important building blocks in various biologically active molecules,1 heterocyclic derivatives2 and have wide application in medicine discovery and agricultural chemistry.3 For quite a long time, the most common synthetic route to 2-ynamides has been the Pd/Cu-catalyzed reaction of terminal alkynes with carbamoyl chlorides.4 Nevertheless, this protocol suffers from harsh reaction conditions and employs the not environmentally-benign carbamoyl chlorides as a carbonyl source, which does not conform to the demands of organic synthesis. To address these drawbacks and limitations, developing new and facile synthetic methodologies is undoubtedly urgent and challenging.
In recent decades, the carbonylation reaction has become a robust and reliable tool for the synthesis of esters, ketones and amides, and a series of transformations have been demonstrated.5–7 Among them, the aminocarbonylation version of alkynes with amines and CO gas was discovered by Hoberg and co-workers in 1983, although this stoichiometric Ni(II) complex promoted synthesis of 2-ynamides could not belong to the catalytic reaction category.8a Subsequently, the direct catalytic oxidative aminocarbonylation of terminal alkynes with secondary amines was realized under CO/air or CO/O2 atmosphere in the research groups of Gabriele, Bhanage and Xia (Scheme 1a–c).8b–d Recently, the Lee group also reported a palladium-catalyzed oxidative aminocarbonylation using arylpropiolic acid as the alkyne source under a CO (5 bar) atmosphere (Scheme 1d).8e However, these catalytic reactions still have their inevitable limitations. For example, using special pressure reaction setups, higher temperature and highly toxic, flammable and explosive CO gas makes these protocols inconvenient and unsafe. Moreover, the substrate scope cannot involve aryl amines and primary amines, greatly restricting the application of these methodologies. In addition, an external oxidant (e.g., O2 and Ag2O) is necessary to fulfill the catalytic cycle.
 | ||
| Scheme 1 Synthesis of 2-ynamides via aminocarbonylation of alkyne sources. | ||
In recent years, more and more attention has been paid to seeking safer carbonyl sources, and promisingly, metal carbonyls (e.g., [Mo(CO)6], [W(CO)6], [Cr(CO)6] and [Co2(CO)8]) as a carbonyl source have been introduced to carbonylation processes.9 Our research interest is to demonstrate a simple, mild and general route to 2-ynamides. In the end, we envisioned developing a catalytic aminocarbonylation of alkynes with amines using metal carbonyls as the carbonyl source at room temperature, and no external oxidants would be involved in this reaction.
Initially, the reaction of morpholine (1a) and (bromoethynyl)benzene (2a) was selected as the model reaction using Pd(OAc)2/Xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) as the catalyst system at room temperature in air, and a relatively low yield of 31% was obtained (Table 1, entry 1). Subsequently, the effect of other different ligands such as DPEphos (bis(2-diphenylphosphinophenyl)ether), PPh3, 1,10-phenanthroline and Xphos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) was checked, and fortunately, Xphos gave the product in higher yield of 41% (Table 1, entry 5). Then, some other bases (e.g., iPr2NEt, K2CO3, Na2CO3 and Cs2CO3) were screened, and Cs2CO3 led to an obviously higher yield of 65% (Table 1, entry 9 vs. entries 6–8). The results indicate that inorganic bases have a higher efficacy than organic bases (Table 1, entries 7–9 vs. entries 5 and 6). Subsequently, the solvent effect was also examined, and CH3CN as a solvent instead of 1,4-dioxane led to a high yield of 78% (Table 1, entry 13). Finally, some control experiments were performed. For example, some commercially available metal catalysts (e.g., PdCl2, Pd(PPh3)4, Pd2(dba)3, Cu(OAc)2 and NiCl2) were also evaluated in this aminocarbonylation reaction, and to our delight, the yield was enhanced to 92% when Pd2(dba)3 was employed as a catalyst (Table 1, entries 14–18). When the reaction time was decreased to 4 h, a lower yield of 72% was observed (Table 1, entry 19 vs. entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 2a (0.6 mmol), palladium catalyst (0.01 mmol Pd), ligand (0.02 mmol), Co2(CO)8 (0.075 mmol) and base (0.4 mmol) in solvent (1.0 ml) in air for 5 h in a sealed tube. b GC yield (isolated yield) based on the amount of morpholine. c For 4 h. | |||||
| 1 | Pd(OAc)2 | Xantphos | NEt3 | Dioxane | 31 |
| 2 | Pd(OAc)2 | DPEphos | NEt3 | Dioxane | 28 |
| 3 | Pd(OAc)2 | PPh3 | NEt3 | Dioxane | 24 |
| 4 | Pd(OAc)2 | 1,10-Phen | NEt3 | Dioxane | 19 |
| 5 | Pd(OAc)2 | Xphos | NEt3 | Dioxane | 41 |
| 6 | Pd(OAc)2 | Xphos | i Pr2NEt | Dioxane | 37 |
| 7 | Pd(OAc)2 | Xphos | K2CO3 | Dioxane | 51 |
| 8 | Pd(OAc)2 | Xphos | Na2CO3 | Dioxane | 47 |
| 9 | Pd(OAc)2 | Xphos | Cs2CO3 | Dioxane | 65 (61) |
| 10 | Pd(OAc)2 | Xphos | Cs2CO3 | THF | 51 |
| 11 | Pd(OAc)2 | Xphos | Cs2CO3 | Toluene | 20 |
| 12 | Pd(OAc)2 | Xphos | Cs2CO3 | 1,2-DCE | 46 |
| 13 | Pd(OAc)2 | Xphos | Cs2CO3 | CH3CN | 78 (73) |
| 14 | PdCl2 | Xphos | Cs2CO3 | CH3CN | 52 |
| 15 | Pd(PPh3)4 | Xphos | Cs2CO3 | CH3CN | 53 |
| 16 | Pd 2 (dba) 3 | Xphos | Cs 2 CO 3 | CH 3 CN | 92 (86) |
| 17 | Cu(OAc)2 | Xphos | Cs2CO3 | CH3CN | Trace |
| 18 | NiCl2 | Xphos | Cs2CO3 | CH3CN | 21 |
| 19c | Pd2(dba)3 | Xphos | Cs2CO3 | CH3CN | 72 (68) |
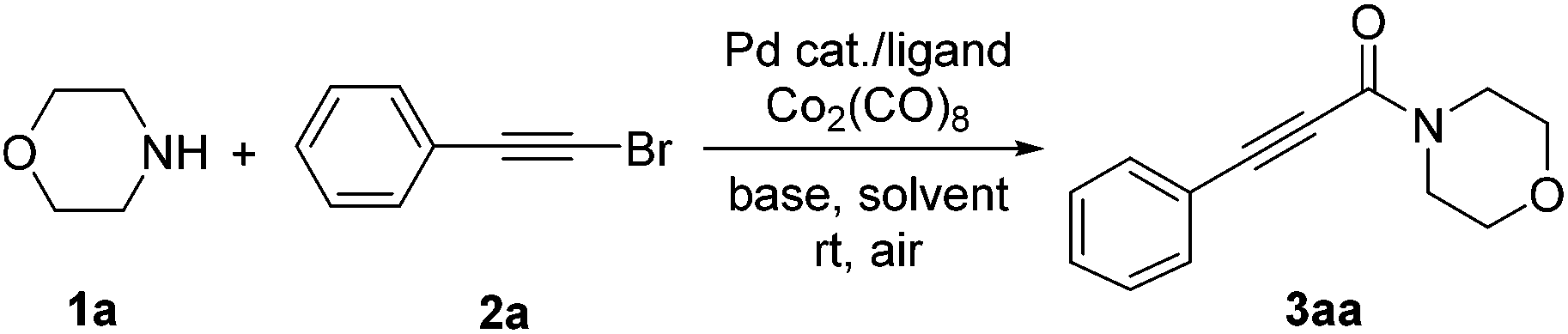
With the optimized conditions in hand, we explored the applicability of the present aminocarbonylation protocol for bromoalkynes and the results are summarized in Table 2. The results demonstrate that bromoalkynes bearing electron-withdrawing, electron-donating, or electron-neutral groups are all well tolerated in this reaction, and the desired products can be obtained in moderate to excellent yields (3aa–3ao). For example, bromoalkynes bearing a chlorine or fluorine atom generated the desired products in 84% and 94% yields, respectively (3ab and 3ac). Note that substrates containing other base-sensitive electron-withdrawing groups (e.g., CHO and CN) or other functional electron-withdrawing groups (e.g., NO2 and CF3) also provided the corresponding products in moderate yields (3ad–3ah). Electron-neutral bromoalkynes such as 1-(bromoethynyl)naphthalene could also afforded the desired product in 84% yield (3ai). Substrates containing electron-donating groups (e.g., p-Me, m-Me, o-Me, 3,5-dimethyl, m-MeO and o-MeO) afforded the corresponding products in a range of 59%–82% yields (3aj–3ao). In addition, the aminocarbonylation of aromatic heterocyclic bromoalkynes could also proceed smoothly, generating the corresponding product in 67% yield (3ap). However, the aliphatic bromoalkynes could not provide the desired products (3aq and 3ar).

Subsequently, the scope of amines was also explored under the optimized reaction conditions (Table 3). First, the secondary amines including aryl amines and open-chain or cyclic alkyl amines were all well tolerated in this reaction, providing the aminocarbonylative products in moderate to good yields (3ba–3ea). For example, the secondary aryl amine N-methylaniline (1b) could successfully afford the desired product in 51% yield (3ba), which has not been described in analogical reports yet. The reaction was also compatible with the open-chain secondary amine (1c) and cyclic secondary amines (1d and 1e), generating the desired products in 71%, 87% and 78% yields, respectively (3ca–3ea). Moreover, primary amines such as n-butylamine and cyclohexylamine also led to the desired products in 58% and 43% yields, respectively (3fa and 3ga), which is the first report for the aminocarbonylation involving primary amines. Unfortunately, the steric hindrance effect of amines is evident in this aminocarbonylation. For example, diisopropylamine (1h) only provided a 47% yield, while dicyclohexylamine (1i) could not even give the desired products (3ha and 3ia).

Some other alkynes were also further investigated and the results are shown in Table 4. Fortunately, when Ag2O was added into the catalyst system, both terminal alkynes and arylpropiolic acids successfully gave the desired products in moderate yields (3aa–3aq). The electronic effect did not have evident influence in this reaction. For example, the reaction of substrates containing various substituents (e.g., p-Cl, o-F, o-Me, o-MeO and p-tBu) proceeded smoothly, providing the corresponding products in mostly moderate yields (3ab, 3ac, 3al, 3ao and 3aq).
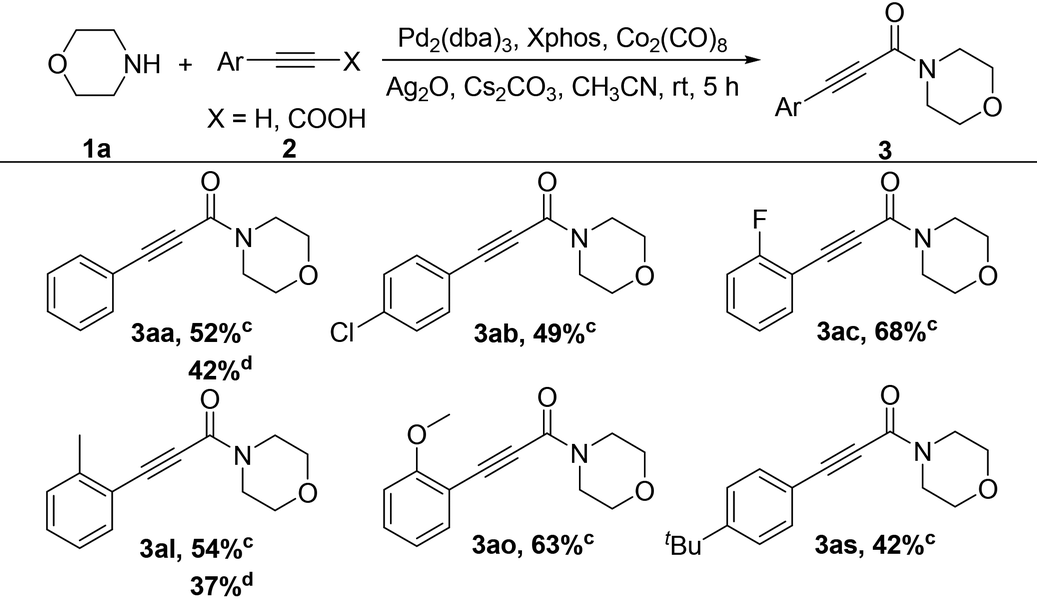
Some control experiments were performed to gain an insight into the mechanism (Scheme 2). For example, the reaction could not occur in the absence of Ag2O or a base. Taking oxygen instead of Ag2O could only generate the product in especially low yield; a dicarbonylated product could not be formed in the reaction of bromoalkyne with the mono-carbonylated product of primary amines, probably due to the steric hindrance effect.
 | ||
| Scheme 2 Control experiments. | ||
On the basis of the above-mentioned results and previous reports,10 a possible mechanism for Pd-catalyzed aminocarbonylation of bromoalkynes with amines is outlined in Scheme 3. The first step would be the formation of the catalytically active Pd(0)L2 (I) with the assistance of the alkyl phosphine ligand. Then, the oxidative addition of bromoalkynes (2) to Pd(0)L2 (I) would take place to form the intermediate II. Subsequently, the insertion of carbon monoxide generated in situ from Co2(CO)8 into the intermediate II afforded an acylpalladium intermediate III, which is susceptible to attack by nucleophiles. After the intermediate IV was generated from the interaction between the acylpalladium intermediate III and amines (1) in the presence of Cs2CO3, the reductive elimination of the intermediate (IV) would occur to deliver the corresponding products (3) and regenerate the catalytically active Pd(0) species to fulfill the catalytic cycle. On the other hand, in the case of terminal alkynes or arylpropiolic acids, the palladium (II) intermediate is the active palladium species (Scheme 4). The first step would be the oxidization of Pd(0)L (I) to the Pd(II)L2X2 (II) with the assistance of Ag2O. The ligand exchange of Pd(II)L2X2 (I) with an alkyne affords an alkynyl palladium complex (III). After a similar process to that of bromoalkynes, the reductive elimination provides the desired product, and Pd(0)L (I) would be regenerated to fulfil the catalytic cycle.
 | ||
| Scheme 3 Possible mechanism for aminocarbonylation of bromoalkynes. | ||
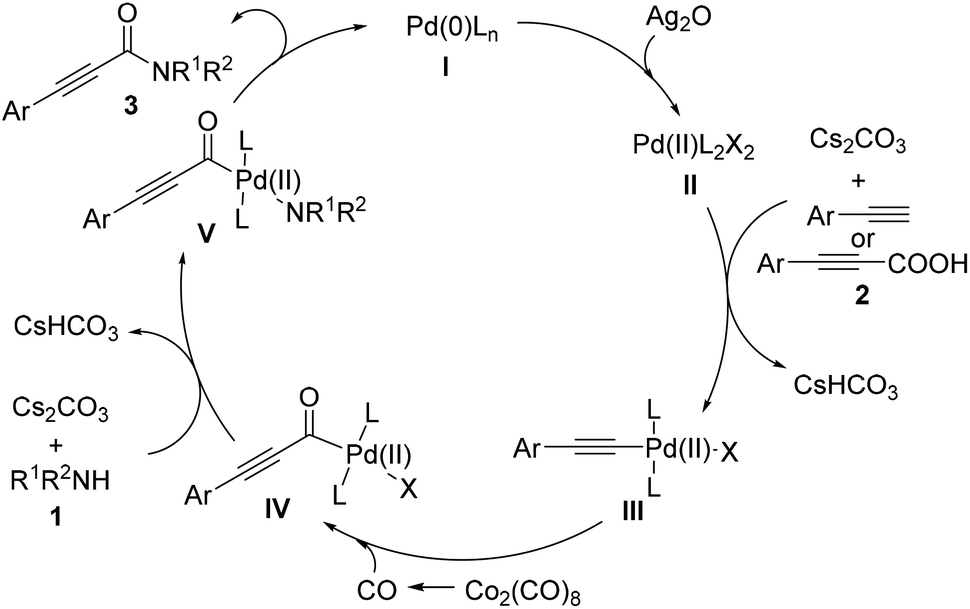 | ||
| Scheme 4 Possible mechanism for aminocarbonylation of terminal alkynes or alkynyl carboxylic acids. | ||
Conclusions
In conclusion, we have developed a palladium-catalyzed aminocarbonylation of alkynes with amines using Co2(CO)8 as a carbonyl source at room temperature in air, establishing a mild and convenient protocol for the synthesis of 2-ynamides. These remarkable features include taking Co2(CO)8 instead of high-pressure CO gas as an in situ generated carbonyl source, and good functional group tolerance for alkynes (e.g., bromoalkynes and terminal alkynes) and amines (e.g., primary and secondary amines).Acknowledgements
We are grateful to the Natural Science Foundation of China (no. 21102134, 21172200) for financial support.Notes and references
- (a) Z. T. Fomum, A. E. Nkengfack, G. W. P. Mpango, S. R. Landor and P. D. Landor, J. Chem. Res., Miniprint, 1985, 3, 901–924 Search PubMed; (b) W. D. Rudorf and R. Schwarz, Heterocycles, 1986, 24, 3459–3465 CrossRef CAS; (c) A. Pinto, L. Neuville, P. Retailleau and J. P. Zhu, Org. Lett., 2006, 8, 4927–4930 CrossRef CAS PubMed; (d) S. Eichner, T. Eichner, H. G. Floss, J. Fohrer, E. Hofer, F. Sasse, C. Zeilinger and A. Kirschning, J. Am. Chem. Soc., 2012, 134, 1673–1679 CrossRef CAS PubMed.
- (a) C. A. Zificsak, J. A. Mulder, R. P. Hsung, C. Rameshkumar and L. L. Wei, Tetrahedron, 2001, 57, 7575–7606 CrossRef CAS; (b) A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (c) W. T. Wu, Z. H. Zhang and L. S. Liebeskind, J. Am. Chem. Soc., 2011, 133, 14256–14259 CrossRef CAS PubMed; (d) H. Noda, G. Eros and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef CAS PubMed.
- (a) J. H. Ryan and P. J. Stang, J. Org. Chem., 1996, 61, 6162–6165 CrossRef CAS PubMed; (b) L. A. Hay, T. M. Koenig, F. O. Gina, J. D. Copp and D. Mitchell, J. Org. Chem., 1998, 63, 5050–5058 CrossRef CAS; (c) J. C. Lanter, Z. H. Sui, M. J. Macielag, J. J. Fiordeliso, W. Q. Jiang, Y. H. Qiu, S. Bhattacharjee, P. Kraft, T. M. John, D. H. Johnson, E. Craig and J. Clancy, J. Med. Chem., 2004, 47, 656–662 CrossRef CAS PubMed; (d) M. Perez, M. Lamothe, C. Maraval, E. Mirabel, C. Loubat, B. Planty, C. Horn, J. Michaux, S. Marrot, R. Letienne, C. Pignier, A. Bocquet, F. N. Wollbold, D. Cussac, L. Vries and B. L. Grand, J. Med. Chem., 2009, 52, 5826–5836 CrossRef CAS PubMed; (e) K. A. DeKorver, H. Y. Li, A. G. Lohse, R. Hayashi, Z. J. Lu, Y. Zhang and R. P. Hsung, Chem. Rev., 2010, 110, 5064–5106 CrossRef CAS PubMed.
- (a) Y. Tohda, K. Sonogashira and N. Hagihara, Synthesis, 1977, 777–778 CrossRef CAS; (b) T. Suda, K. Noguchi, M. Hirano and K. Tanaka, Chem. – Eur. J., 2008, 14, 6593–6596 CrossRef CAS PubMed; (c) H. C. Huang, G. J. Zhang and Y. Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 7872–7876 CrossRef CAS PubMed.
- For selected papers on the synthesis of esters, see: (a) H. N. Wang, B. Dong, Y. Wang, J. F. Li and Y. A. Shi, Org. Lett., 2014, 16, 186–189 CrossRef CAS PubMed; (b) W. F. Li and X. F. Wu, J. Org. Chem., 2014, 79, 10410–10416 CrossRef CAS PubMed; (c) K. Natte, A. Dumrath, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 10090–10094 CrossRef CAS PubMed; (d) W. Li, Z. L. Duan, R. Jiang and A. W. Lei, Org. Lett., 2015, 17, 1397–1400 CrossRef CAS PubMed; (e) L. P. Wu, Q. Liu, R. Jackstell and M. Beller, Org. Chem. Front., 2015, 2, 771–774 RSC.
- For selected papers on the synthesis of ketones, see: (a) T. M. Gogsig, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, Angew. Chem., Int. Ed., 2012, 51, 798–801 CrossRef CAS PubMed; (b) H. W. Lee, A. S. C. Chan and F. Y. Kwong, Chem. Commun., 2007, 2633–2635 RSC; (c) L. J. Gu, C. Jin and J. Y. Liu, Green Chem., 2015, 17, 3733–3736 RSC; (d) C. Tan, P. Wang, H. Liu, X. L. Zhao, Y. Lu and Y. Liu, Chem. Commun., 2015, 51, 10871–10874 RSC; (e) S. Sumino, T. Ui, Y. Hamada, T. Fukuyama and I. Ryu, Org. Lett., 2015, 17, 4952–4955 CrossRef CAS PubMed.
- For selected papers on the synthesis of amides, see: (a) M. Iizuka and Y. Kondo, Chem. Commun., 2006, 1739–1741 RSC; (b) Y. Du, T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 12074–12076 RSC; (c) C. Brancour, T. Fukuyama, Y. Mukai, T. Skrydstrup and I. Ryu, Org. Lett., 2013, 15, 2794–2797 CrossRef CAS PubMed; (d) P. L. Wang, Y. Li, Y. Wu, C. Li, Q. Lan and X. S. Wang, Org. Lett., 2015, 17, 3698–3701 CrossRef CAS PubMed; (e) C. Wang, L. Zhang, C. P. Chen, J. Han, Y. M. Yao and Y. S. Zhao, Chem. Sci., 2015, 6, 4610–4614 RSC.
- (a) H. Hoberg and H. J. Riegel, J. Organomet. Chem., 1983, 241, 245–250 CrossRef CAS; (b) B. Gabriele, G. Salerno, L. Veltri and M. Costa, J. Organomet. Chem., 2001, 622, 84–88 CrossRef CAS; (c) S. T. Gadge, M. V. Khedkar, S. R. Lanke and B. M. Bhanage, Adv. Synth. Catal., 2012, 354, 2049–2056 CrossRef CAS; (d) C. Y. Zhang, J. H. Liu and C. G. Xia, Catal. Sci. Technol., 2015, 5, 4750–4754 RSC; (e) J. Hwang, J. Choi, K. Park, W. Kim, K. H. Song and S. Lee, Eur. J. Org. Chem., 2015, 2235–2243 CrossRef CAS.
- For selected papers on carbonylation reaction using metal carbonyls as the carbonyl source, see: (a) P. A. Enquist, P. Nilsson and M. Larhed, Org. Lett., 2003, 5, 4875–4878 CrossRef CAS PubMed; (b) S. M. Dumbris, D. J. Dıaz and L. M. White, J. Org. Chem., 2009, 74, 8862–8865 CrossRef CAS PubMed; (c) J. B. Chen, K. Natte, A. Spannenberg, H. Neumann, M. Beller and X. F. Wu, Chem. – Eur. J., 2014, 20, 14189–14193 CrossRef CAS PubMed.
- (a) R. Grigg and S. P. Mutton, Tetrahedron, 2010, 66, 5515–5548 CrossRef CAS; (b) Q. Liu, H. Zhang and A. W. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788–10799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00075d |
| This journal is © the Partner Organisations 2016 |