
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
γ-Lactams and furan bispyrrolidines via iodine mediated cyclisation of homoallylamines†
Mariwan A.
Hama Salih
a,
Louise
Male
b,
Neil
Spencer
c and
John S.
Fossey
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK. E-mail: j.s.fossey@bham.ac.uk
bX-Ray Crystallography Facility, School of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK
cNMR Facility, School of Chemistry, University of Birmingham, Edgbaston, Birmingham, West Midlands B15 2TT, UK
First published on 27th August 2015
Abstract
1,3,5-Substituted pyrrolidin-2-ones were synthesised via an iodine mediated cyclisation of 3-methyl substituted homoallylamines in good to excellent yield, as mixtures of diastereoisomers. These were separable and their identity confirmed by techniques including single crystal X-ray diffraction. When 3-phenyl substituted homoallylamines were cyclised intriguing fused tricyclic motifs were obtained as C2-symmetric racemates, whose structures were also confirmed by techniques including single crystal X-ray diffraction analysis.
 John S. Fossey | Dr John S. Fossey, University of Birmingham, UK. Obtained an MChem degree from Cardiff University of Wales in 2000, and a PhD, in early 2004, from Queen Mary University of London. After a JSPS postdoctoral position with Professor S. Kobayashi at the University of Tokyo and time as a temporary faculty member at the University of Bath he took up his present post at the University of Birmingham. He holds a number of visiting positions including a guest Professor at East China University of Science and Technology (ECUST) and enjoys working with others to develop new catalysts and sensors. |
Introduction
Pyrrolidin-2-ones are an important class of compounds, the lactam core is found in many natural products such as antimicrobial agent pramanicin 1.1 Cotinine 2 is a metabolite of nicotine and is found in tobacco,2 it can cross the blood brain barrier and has interesting medicinal properties (Fig. 1).3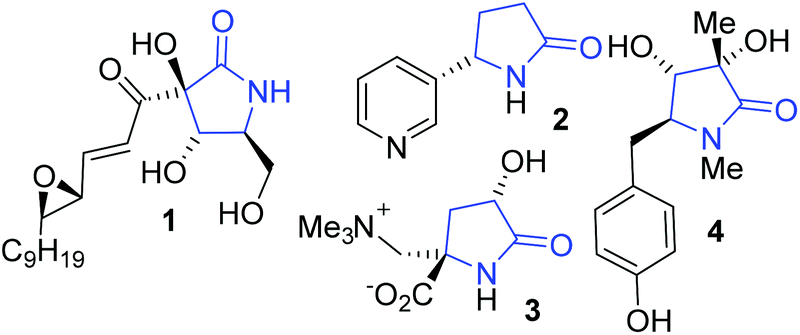 | ||
| Fig. 1 Representative γ-lactams. | ||
γ-Lactams with quaternary stereogenic centres at C5, such as dysibetaine 3, displays neuroexcitotoxine activity.4 Rigidiuculamide A (4) shows cytotoxic effects against selected cell lines,5 and 1,3,5-trisubstituted pyrrolin-2-ones act as CCR4 antagonists.6 Katritzky et al.7 and Sun et al.6 have also reported on the importance of this calss of compound.
Herein, an expeditious route to 1,3,5-trisubstituted γ-lactams bearing a tertiary alcohol at the 3-position is reported. Divergent behaviour of homoallyl starting materials is explored and a new class of furan bispyrrolidines is revealed.
We have previously shown that homoallylamine derivatives may be transformed into 2,4-cis-azetidines, which may in turn be thermally isomerised to 4-cis-pyrrolidines (Scheme 1, R3 = H).8 Following directly on from that work we next chose to investigate substitution at the R3 position, since we envisaged this may provide a convenient route to substituted azetidines and pyrrolidines.
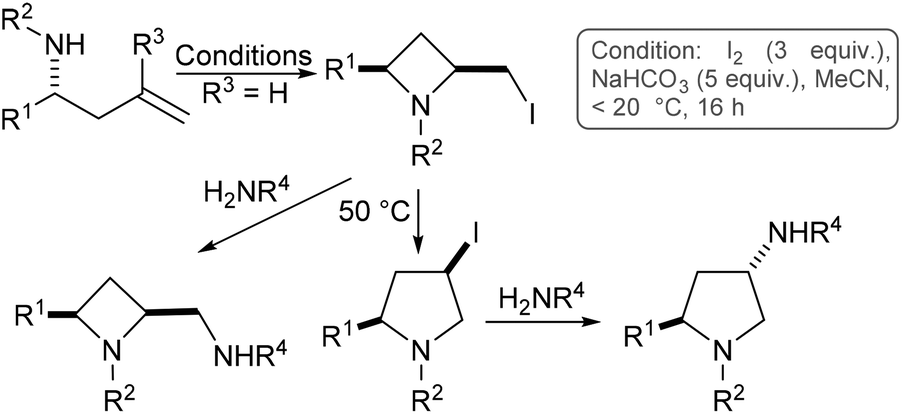 | ||
| Scheme 1 Azetidine and pyrrolidine synthesis via iodine mediated cyclisation of homoallylamines (R3 = H). | ||
Compound 5a (R3 = Me) was prepared as a racemate and exposed to conditions derived from our previous work that we expected to deliver an azetidine product. To our surprise a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers of γ-lactam 6a was formed (35% yield, Scheme 2). Compounds similar to this have been previously accessed by reaction of nitrones, reported in 1968 by Huisgen et al.9 and elegantly revisited by Yang et al. where a chromium catalysed cyclisation of ketoenamides delivered cyclic compounds containing C
:1 mixture of diastereoisomers of γ-lactam 6a was formed (35% yield, Scheme 2). Compounds similar to this have been previously accessed by reaction of nitrones, reported in 1968 by Huisgen et al.9 and elegantly revisited by Yang et al. where a chromium catalysed cyclisation of ketoenamides delivered cyclic compounds containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds that could be converted to γ-lactam related to 6.10 Ficarra et al. studied the separation of secondary alcohol analogues of 6 (i.e.6, R3 = H).11 At least three equivalents of iodine were required to form the product (6a), any less and only by-products were observed, this led us to speculate a number of possible mechanistic explanations. In the absence of computational support for a mechanistic hypothesis our current tentative model is that iodine activates the double bond of the homoallyl amine (see ESI† for suggested mechanism). Unlike the case where R3 = H, the present hypothesised intermediate may be too sterically encumbered to cyclise to a 4-membered ring and instead delivers an intermediate iodo-pyrrolidine, a sequence of oxidations12 and hydrations deliver 6a as a mixture of diastereoisomers. With a route to tertiary alcohol bearing γ-lactams in hand we next looked at optimising the reaction conditions to improve yield and explore the potential for obtaining 6a as a single diastereoisomer. Table 1 details a range of solvents and bases, inspection of crude proton NMR spectrums revealed the ratio of starting material 5a to lactam product 6a and a by-product imine I derived from oxidation of starting material.12 After an initial purification step cis- and trans-6a were isolated as a mixture and the combined yield and diastereoisomer ratios (proton NMR) are also given in the table.
C double bonds that could be converted to γ-lactam related to 6.10 Ficarra et al. studied the separation of secondary alcohol analogues of 6 (i.e.6, R3 = H).11 At least three equivalents of iodine were required to form the product (6a), any less and only by-products were observed, this led us to speculate a number of possible mechanistic explanations. In the absence of computational support for a mechanistic hypothesis our current tentative model is that iodine activates the double bond of the homoallyl amine (see ESI† for suggested mechanism). Unlike the case where R3 = H, the present hypothesised intermediate may be too sterically encumbered to cyclise to a 4-membered ring and instead delivers an intermediate iodo-pyrrolidine, a sequence of oxidations12 and hydrations deliver 6a as a mixture of diastereoisomers. With a route to tertiary alcohol bearing γ-lactams in hand we next looked at optimising the reaction conditions to improve yield and explore the potential for obtaining 6a as a single diastereoisomer. Table 1 details a range of solvents and bases, inspection of crude proton NMR spectrums revealed the ratio of starting material 5a to lactam product 6a and a by-product imine I derived from oxidation of starting material.12 After an initial purification step cis- and trans-6a were isolated as a mixture and the combined yield and diastereoisomer ratios (proton NMR) are also given in the table.
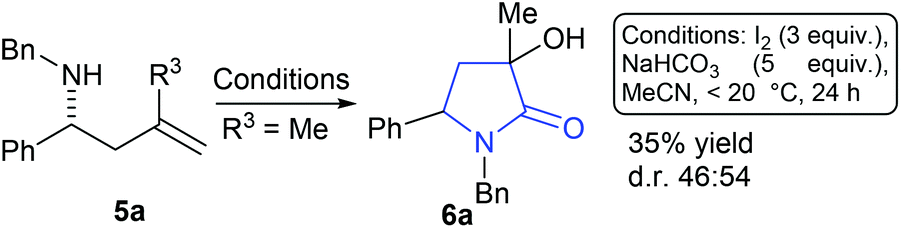 | ||
| Scheme 2 When R3 = Me, γ-lactam 6a is formed as a 1:1 mixture of diastereoisomers. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Base | Solvent | 5a | 6a | I | 6a cis + trans | d.r.c |
|
a Determined by inspection of the crude 1H NMR spectrums reported as a ratio of whole numbers totalling 100 to aid comparison. 1H NMR dd peaks at 4.16 and 4.53 ppm, corresponding to CH of diastereoisomers 6a and 6b, were compared to an apparent singlet at 4.84 ppm or an apparent singlet at 4.88 ppm corresponding to CCH2 of 5 along with a singlet at 8.10 ppm corresponding to imine CHN of compound I.
b Isolated combined yield of 6-cis + 6b-trans after initial purification.
c Determined by inspection of the 1H NMR spectra of 6-cis + 6b-trans.
d CH3CN:H2O (1:1).
|
|||||||
| 1 | NaHCO3 | CH3CN | 35 | 35 | — | 35 | 46:54 |
| 2 | Cs 2 CO 3 | CH 3 CN | 27 | — | 73 | — | — |
| 3 | NaOAc | CH3CN | 60 | 20 | — | 20 | 50:50 |
| 4 | NaOH | CH3CN | 75 | 25 | — | 18 | 51:49 |
| 5 | Na2CO3 | CH3CN | 33 | 27 | — | 20 | 60:40 |
| 6 | K2CO3 | CH3CN | 33 | 30 | 42 | 25 | 43:57 |
| 7 | Li2CO3 | CH3CN | 48 | 22 | 30 | 22 | 56:44 |
| 8 | LiOH | CH3CN | 34 | 47 | 19 | 17 | 57:43 |
| 9 | KOH | CH3CN | 53 | 22 | 26 | 21 | 54:46 |
| 10 | NaHCO3 | CH3CN:H2Od |
48 | 30 | 21 | 21 | 33:67 |
| 11 | NaHCO 3 | EtOAc | — | >99 | — | 89 |
46:54 |
| 12 | NaHCO3 | THF anhydrous | 10 | 60 | 30 | 60 | 38:62 |
| 13 | NaHCO3 | MeOH | 43 | 40 | 17 | 10 | 45:55 |
| 14 | NaHCO3 | DCM | 90 | 10 | — | 10 | 50:50 |
| 15 | NaHCO3 | CH3CN anhydrous | 60 | 20 | 20 | 20 | 48:52 |
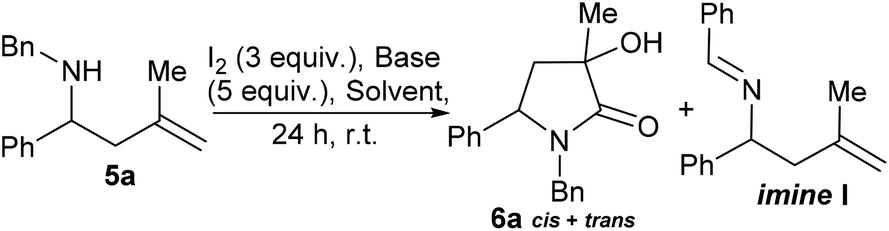
In a parallel screen of bases the best conversion to 6a was achieved using LiOH (47% conversion), but problems purifying 6a from unreacted stating material and by-product I compromised isolated yield (Table 1, entry 8). This trend was observed in other reactions, where by-product and unreacted starting material remain, isolated yields of 6a often suffered from multiple purification steps required and loss of material as a result. As expected Cs2CO3 served to oxidise starting material under these conditions (Table 1, entry 2), giving by-product I (73% conversion) and unreacted starting material only. Diastereoisomer ratios were not improved across bases employed, Na2CO3 (Table 1, entry 5) gave the best ratio of only 1.5:1 and isolated yield was low (20%). As judged by isolated yield of a mixture of cis- and trans-6a, NaHCO3 (Table 1, entry 1) remained the base of choice for further investigation of various solvents. Some divergence (albeit small) in which diastereoisomer was formed in preference was noticed, but no solvent (of those tried) gave one diastereoisomer as a significant major product. The use of anhydrous acetonitrile gave a lower conversion to the desired product, and this was accompanied by an increased prevalence of the unwanted by-product (Table 1, entry 15). Addition of water to acetonitrile also did not improve the reaction ourtcome (Table 1, entry 10). Methanol and dichloromethane (Table 1, entries 13 and 14 respectively) also did not improve reaction outcomes. Tetrahydrofuran (Table 1, entry 12) gave a jump in isolate yield to 60%. To our surprise the use of ethyl acetate as reaction solvent gave quantitative conversion to desired product (Table 1, entry 11), 89% isolated yield in an approximate 1:1 ratio of diastereoisomers. Next scope of functionality at R1 and R2 was investigated (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yielda/% | d.r.b |
| a Isolated yield. b Determined by inspection of the corresponding 1H NMR spectrums, reported as a ratio of whole numbers totalling 100 to aid comparison, after initial purification to a mixture of cis and trans products, see ESI for details. | |||||
| 1 | Ph | Bn | 6a | 89 | 46:54 |
| 2 | Ph | 3-Picolyl | 6b | 91 | 57:43 |
| 3 | Ph | n-Pr | 6c | 34 | 56:44 |
| 4 | Ph | 2-Me-benzyl | 6d | 43 | 60:40 |
| 5 | 3-Furyl | Bn | 6e | 72 | 54:46 |
| 6 | t-Bu | Bn | 6f | 67 | 55:45 |
| 7 | 3,4-Di-MeO-Ph | Bn | 6g | 65 | 56:44 |
| 8 | Ph | PMB | 6h | 78 | 61:39 |
| 9 | Ph | Admantyl | 6i | 57 | 30:70 |
| 10 | 2-Thio | Bn | 6j | 73 | 36:64 |
| 11 | 2-Br-Ph | Bn | 6k | 55 | 67:33 |
| 12 | Ph | 4-Cl-benzyl | 6l | 64 | 60:40 |
| 13 | 4-NO2-Ph | Bn | 6m | 55 | 45:55 |
| 14 | 1-Naph | Bn | 6n | 72 | 24:76 |
| 15 | Ph | Me | 6o | 43 | 56:44 |
| 16 | 3-Pyr | PMB | 6p | 99 | 52:48 |
| 17 | 3-Pyr | Me | 6q | 68 | 53:47 |
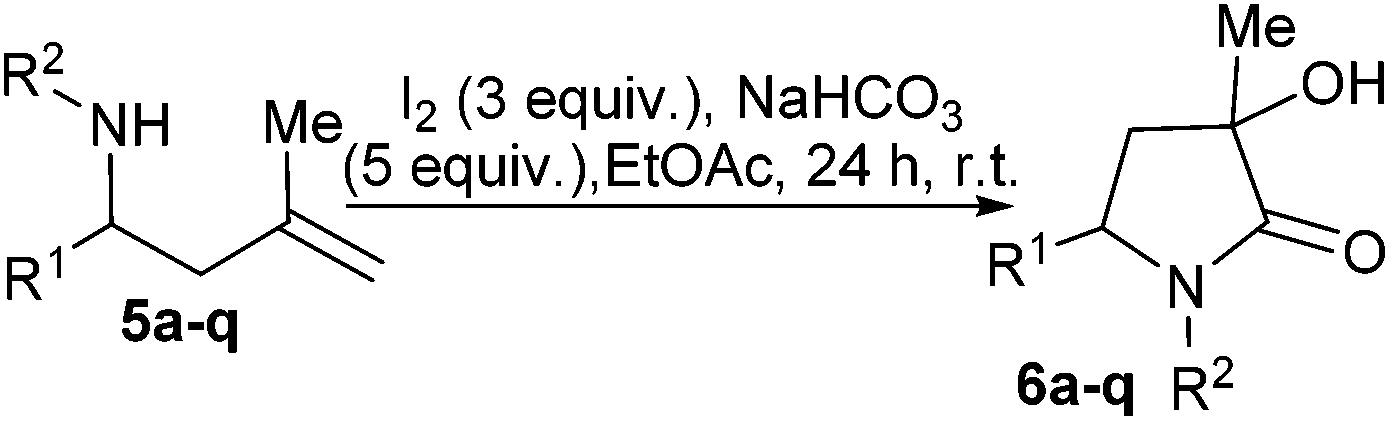
At the R1 position of compound 5 aromatic and heteroaromatic groups can be accommodated (Table 2, entries 1, 5 10, 14 and 16). Electron donating and withdrawing groups (Table 2, entries 7 and 13 respectively) were employed and yield was slightly compromised for the para-nitro substituted variant (55% yield). tert-Butyl at R1 also allowed for lactam products to be formed (67% yield Table 2, entry 6). A range of benzylic groups could be incorporated at R2, including 3-picolyl, para-methoxybenzyl and para-chlorobenzyl (Table 2, entries 2, 8 and 12 respectively) with para-chlorobenzyl giving the lowest yield amongst them (64%). ortho-Tolylbenzyl at R2 led to a much reduced yield (43%, Table 2, entry 4). Whilst alkyl groups could be incorporated at R2 yields were generally lower than their benzylic congeners (Table 2, entries 3, 9 and 17). Once again diastereomer ratios were not improved greatly, the best diasteromer ratio came at the expense of yield (Table 2, entry 11) where R1 was 2-bromophenyl (55% yield, 67:33 d.r.).
After further column chromatography purification steps and recrystallization, of crystals of single diastereoisomers suitable for study by single crystal X-ray diffraction study were obtained, in eight cases (Fig. 2), permitting unambiguous relative stereochemical assignment for 6a-cis, 6a-trans, 6d-trans, 6e-cis, 6h-cis, 6m-cis, 6p-cis and 6q-cis. We chose to name cis- and trans- to describe the relationship of the group at R1 relative to the methyl group at C3 (Scheme 3). Combination of crystallography data and nOe studies allowed most compounds to be assigned (see ESI†).
 | ||
| Fig. 2 X-Ray crystal structures of 60a-cis, 6a-trans, 6d-trans, 6e-cis, 6h-cis, 6m-cis, 6p-cis and 6q-cis. One molecule of each plot using Ortep III for windows at 30% probability and PovRay. Certain protons and disorder (in one case) removed for clarity. For full details, collection parameters and analysis see ESI.† | ||
 | ||
| Scheme 3 Example transformations performed on 6 to give O-derivatives 7 and 8 or pyrrolidine 9. | ||
With single diastereoisomers of some examples of 6 in hand some test transformations were attempted. O-Methylation (of 6a-cis) and acylation (of 6p-cis) proceed quantitatively without erosion of stereochemical integrity. Reduction (of 6q-cis) with LiAlH4 gave the desired product (4-hydroxypyrrolidine 9-cis) in 89% isolated yield as a single diastereoisomer.13
We envisaged that increasing the steric bulk of the R3 substituent might improve diastereoselectivity of the cyclisation to γ-lactams. To test this hypothesis compounds 10a–c were prepared with phenyl at the R3 position and benzyl, para-methoxybenzyl and methyl at the R2 positions, respectively, see ESI† for more information.
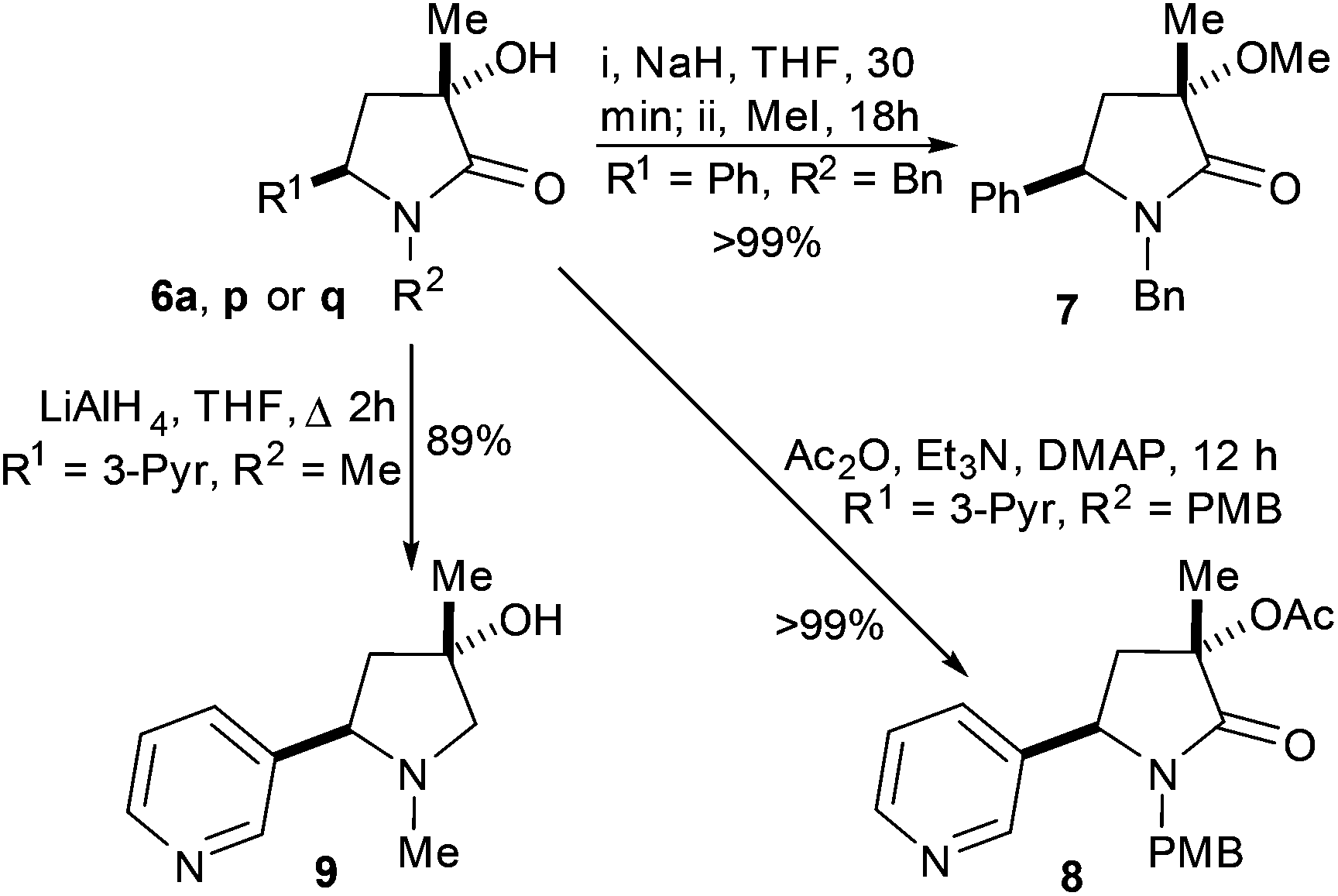
Changing the R3 group to Ph certainly solved the diastereomer ratio problem; single diastereoisomer products were isolated from relatively low conversion reactions (Scheme 4). However, NMR spectra and mass spectrometry data did not match the expected products. Fortunately, XRD studies rapidly identified all three products as intriguing C2-symmetric furan bispyrrolidines (Fig. 3). To the best of our knowledge saturated fused pyrrolidine–furan–pyrrolidine systems like this have not been reported, although we may understand the steps towards formation of our products by contrasting observations made in an authoritative report by Ling et al. relating to oxidative indole coupling.14
 | ||
| Scheme 4 For phenyl substituted homoallyl amines 10a–c exposure to oxidative cyclisation conditions delivers tricyclic compounds 11a–c in 20 to 48% yield. | ||
 | ||
| Fig. 3 X-Ray crystal structures of 11a, 11b and 11c. Plot using Ortep III for windows at 30% probability and PovRay. Certain protons removed for clarity. For full details, collection parameters and analysis see ESI.† | ||
Conclusions
We have shown that changing functionality along the ally group at the β-position of a homoallyl amine, from H to Me and Ph, elicits dramatic structural divergence on reaction outcomes. A versatile route to 1,3,5-trisubstituted γ-lactams with tertiary alcohol functionality at the 3-position was demonstrated, when the homoallyl amine derivative has a methyl group at the β-position. Additionally, a unique reaction manifold to deliver C2-symmetric fused tricyclic pyrrolidine–furan–pyrrolidine systems was accessible, when the starting homoallyl amine bears a phenyl group at the β-position. X-Ray crystallography unambiguously confirmed the identity and relative stereochemistry of each class of product and screening of biological activity of these compounds is on going.Acknowledgements
The authors thank the University of Birmingham and ERDF AWM II for support. JSF is extremely grateful to the Royal Society Industry Fellowship scheme (2012/R1) for on-going support. MHS thanks the HCDP-Kurdistan government for financial support.Notes and references
- R. E. Schwartz, G. L. Helms, E. A. Bolessa, K. E. Wilson, R. A. Giacobbe, J. S. Tkacz, G. F. Bills, J. M. Liesch, D. L. Zink, J. E. Curotto, B. Pramanik and J. C. Onishi, Tetrahedron, 1994, 50, 1675–1686 CrossRef CAS.
- L. P. Dwoskin, L. Teng, S. T. Buxton and P. A. Crooks, J. Pharmacol. Exp. Ther., 1999, 288, 905–911 CAS.
- (a) J. A. Grizzell, A. Iarkov, R. Holmes, T. Mori and V. Echeverria, Behav. Brain Res., 2014, 268, 55–65 CrossRef CAS PubMed; (b) R. Zeitlin, S. Patel, R. Solomon, J. Tran, E. J. Weeber and V. Echeverria, Behav. Brain Res., 2012, 228, 284–293 CrossRef CAS PubMed; (c) V. Echeverria, R. Zeitlin, S. Burgess, S. Patel, A. Barman, G. Thakur, M. Mamcarz, L. Wang, D. B. Sattelle, D. A. Kirschner, T. Mori, R. M. Leblanc, R. Prabhakar and G. W. Arendash, J. Alzheimers Dis., 2011, 24, 817–835 CAS; (d) V. Echeverria, R. Zeitlin, S. Burgess, S. Patel, A. Barman, G. Thakur, M. Mamcarz, L. Wang, D. B. Sattelle, D. A. Kirschner, T. Mori, R. M. Leblanc, R. Prabhakar and G. W. Arendash, J. Alzheimers Dis., 2011, 24, 817–835 CAS.
- (a) J. Isaacson, M. Loo and Y. Kobayashi, Org. Lett., 2008, 10, 1461–1463 CrossRef CAS PubMed; (b) D. J. Wardrop and M. S. Burge, Chem. Commun., 2004, 1230–1231 RSC.
- J. Li, S. C. Liu, S. B. Niu, W. Y. Zhuang and Y. S. Che, J. Nat. Prod., 2009, 72, 2184–2187 CrossRef CAS PubMed.
- W. Sun, L. J. Tian, H. Qi, D. Jiang, Y. Wang, S. Li, J. H. Xiao and X. H. Yang, Chin. J. Chem., 2013, 31, 1144–1152 CrossRef CAS PubMed.
- A. R. Katritzky, S. Mehta, H. Y. He and X. L. Cui, J. Org. Chem., 2000, 65, 4364–4369 CrossRef CAS PubMed.
- (a) A. Feula, L. Male and J. S. Fossey, Org. Lett., 2010, 12, 5044–5047 CrossRef CAS PubMed; (b) A. Feula, S. S. Dhillon, R. Byravan, M. Sangha, R. Ebanks, M. A. Hama Salih, N. Spencer, L. Male, I. Magyary, W.-P. Deng, F. Müller and J. S. Fossey, Org. Biomol. Chem., 2013, 11, 5083–5093 RSC.
- R. Huisgen, H. Hauck, R. Grashey and H. Seidl, Chem. Ber.-Recl., 1968, 101, 2568–2584 CrossRef CAS PubMed.
- L. Yang, D. X. Wang, Z. T. Huang and M. X. Wang, J. Am. Chem. Soc., 2009, 131, 10390–10391 CrossRef CAS PubMed.
- R. Ficarra, M. L. Calabro, S. Alcaro, S. Tommasini, S. Melardi and P. Ficarra, Chromatographia, 2000, 51, 411–416 CAS.
- A. Feula and J. S. Fossey, RSC Adv., 2013, 3, 5370–5373 RSC.
- K. H. Kim, N. H. Lin and D. J. Anderson, Bioorg. Med. Chem., 1996, 4, 2211–2217 CrossRef CAS.
- K. Q. Ling, T. Ren, J. D. Protasiewicz and L. M. Sayre, Tetrahedron Lett., 2002, 43, 6903–6905 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and corresponding spectra. CCDC 1028121–1028130 and 1038737. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00183h |
| This journal is © the Partner Organisations 2015 |