
Catalyst-free cross-coupling of N-tosylhydrazones with chromium(0) Fischer carbene complexes: a new approach to diarylethanone†
Fangdong
Hu
ab,
Jinghui
Yang
a,
Ying
Xia
a,
Chen
Ma
b,
Haiping
Xia
c,
Yan
Zhang
a and
Jianbo
Wang
*a
aBeijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wangjb@pku.edu.cn; Fax: (+86)10-6275-7248; Tel: (+86)10-6275-1708
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
cCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China
First published on 28th August 2015
Abstract
Cross-coupling of N-tosylhydrazones with chromium Fischer carbene complexes under catalyst-free conditions has been developed. This protocol employs stable carbene complexes and reactive diazo compounds, which are in situ generated from N-tosylhydrazones. Through hydrolysis of the initially formed methyl enolate products, the diarylethanone derivatives could be obtained in moderate to good yields.
Introduction
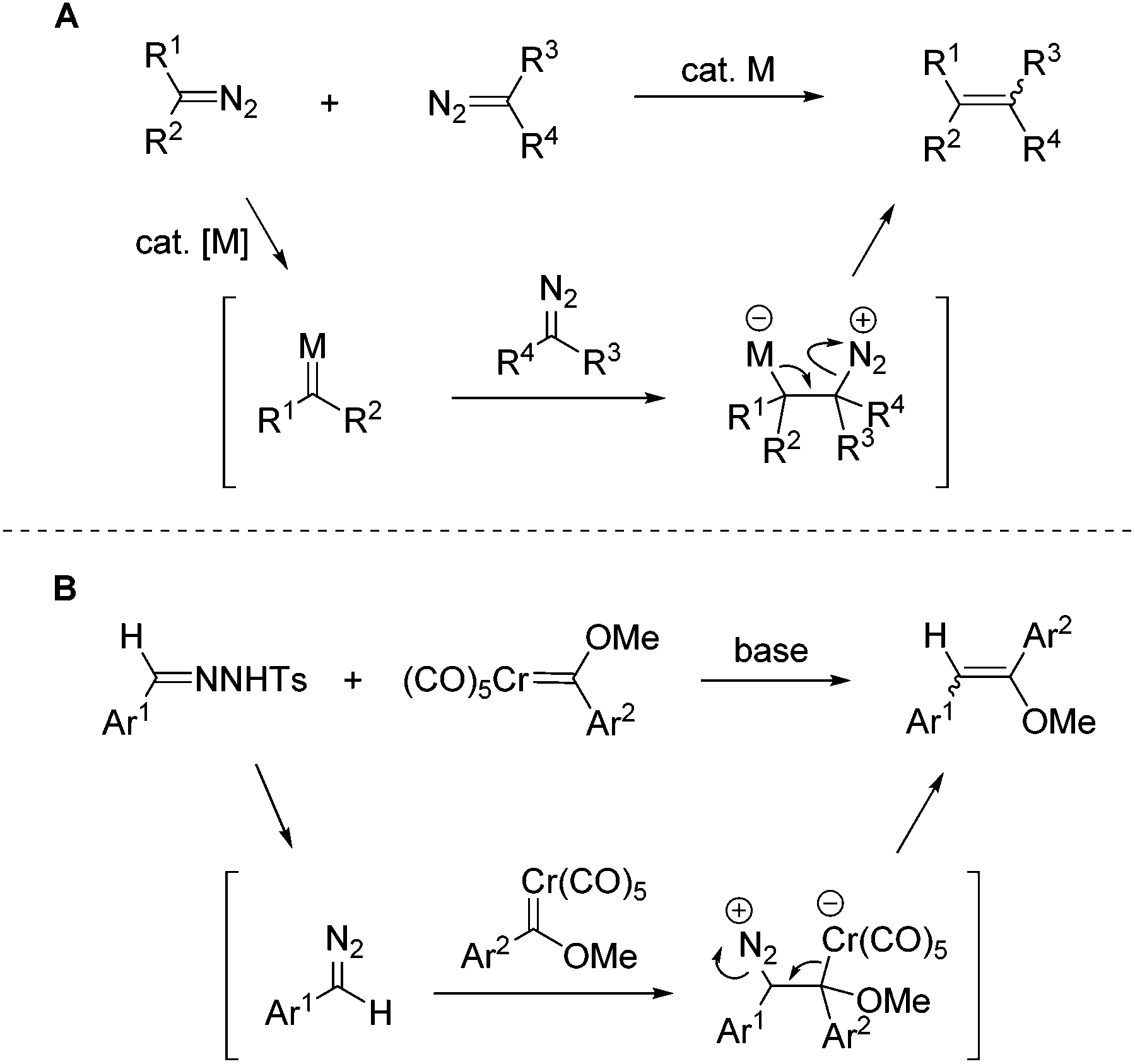
Diazo compounds have been widely employed as carbene sources in transition-metal-catalyzed reactions.1 Through dediazoniation of the diazo substrates in the presence of transition metals, metal carbene species could be generated, which undergo various synthetically useful transformations.2 In this area, carbene dimerization is a ubiquitous process, which is generally regarded as an annoying side reaction pathway. Nevertheless, carbene dimerization provides an alternative method for the synthesis of functionalized alkenes. Recently, efforts have been devoted to this reaction, but only a few successful examples have been reported for intermolecular carbene dimerization of two different diazo substrates.3–5 The apparent problems are the inevitable competition of homo-coupling and the stereoselectivity of the generated double bond. By judiciously adjusting the reactivity of two different diazo substrates, Davies4c and Sun4d have recently realized selective intermolecular carbene dimerization, affording tri-substituted and tetra-substituted alkenes. Although high selectivity has been achieved in these systems, the diazo substrates are largely restricted to specific structures in order to discriminate their reactivities. In contrast, the intramolecular carbene dimerization of diazo compounds or their surrogates N-tosylhydrazones, has been developed into useful methods for the construction of cyclic compounds.6The mechanism of the transition-metal-catalyzed carbene dimerization of diazo compounds is well established.4c,d In this process, the transient reactive metal carbene species is firstly formed and subsequently reacts with another diazo substrate (Scheme 1A). Notably, in the process of metal carbene generation the originally electron-rich carbon to which the diazo group is attached, undergoes reversal of polarity. Thus the electron-deficient carbenic carbon centre of metal carbene can accept the nucleophilic attack by another diazo substrate.
 | ||
| Scheme 1 Olefin formation through carbene dimerization. | ||
On the other hand, chromium Fischer carbene complexes have been studied extensively over the decades and a number of synthetically useful transformations have been developed.7 The carbenic centre in chromium Fischer carbene complexes is electron-deficient in nature, which is comparable to the metal carbene intermediates generated in the transition-metal-catalyzed reactions of diazo compounds. It is thus conceivable that the reaction of nucleophilic diazo compounds with Fischer carbene complexes may generate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. Indeed such a transformation has been reported by Casey and co-workers with tungsten carbene complexes.11a However, this reaction has not been developed into a synthetically useful transformation due to the limited scope of diazoalkane substrates (only the reactions with diazomethane and diazopropane were reported). Besides, Barluenga and co-workers reported the reaction of chromium Fischer carbene complexes with ethyl diazoacetates.11b
C double bonds. Indeed such a transformation has been reported by Casey and co-workers with tungsten carbene complexes.11a However, this reaction has not been developed into a synthetically useful transformation due to the limited scope of diazoalkane substrates (only the reactions with diazomethane and diazopropane were reported). Besides, Barluenga and co-workers reported the reaction of chromium Fischer carbene complexes with ethyl diazoacetates.11b
The N-tosylhydrazones have been well-established as the precursors for diazo compounds, especially for those without the stabilization of electron-withdrawing substituents.8 In this context, a series of synthetically useful transformations have been recently developed using N-tosylhydrazones as the coupling partners.9 We report herein the cross-coupling of N-tosylhydrazones with chromium Fischer carbene complexes under catalyst-free conditions. The reaction affords olefins, which are further hydrolyzed to diarylethanones.10 In comparison with the above-mentioned carbene dimerization of two diazo compounds, this process employs stable chromium Fischer carbene complexes and reactive diazo compounds, which are in situ generated from N-tosylhydrazones under basic conditions (Scheme 1B).
Results and discussion
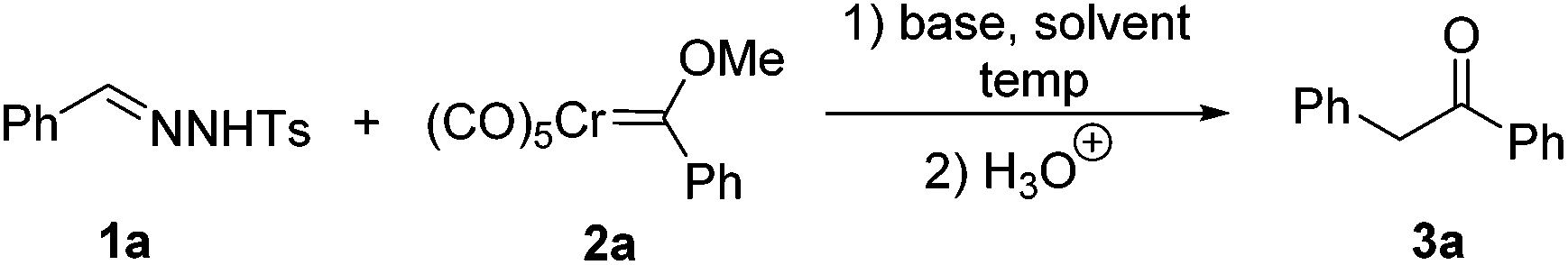
We initiated our investigation with N-tosylhydrazone 1a and chromium Fischer carbene complex 2a as the model substrates. The effects of bases, solvents and temperatures were systematically examined. Among all the bases tested, we found that the best result was obtained with K3PO4 (Table 1) (entries 1–3). Then, the effect of solvents was examined. However, the originally used toluene was proved to be the best solvent for the reaction (entries 4–6). Only trace amounts of the desired product could be detected with MeCN as the solvent (entry 6). Next, we turned our attention to the reaction temperature, the product 3a was obtained in 61% and 70% yields at 80 °C and 100 °C, respectively (entries 9 and 10). As illustrated in entries 7 and 8, increasing the amount of substrate 1a could improve the yield of the corresponding product. Finally, with K3PO4 as the base and toluene as the solvent, the best reaction conditions were established (entry 8). Under the optimized reaction conditions, the desired product 3a was isolated in 78% yield.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | T (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.20 mmol), 2a (0.20 mmol), base (0.30 mmol) in solvent (2 mL) at 90 °C for 1.5 h. The crude product was dissolved in THF (2 mL) and HCl (1 mL, 2 M) at 60 °C for 1 h. b Yield determined by 1H NMR analysis using mesitylene as the internal standard. c 1a (0.24 mmol) was used. d Isolated yield. e 1a (0.30 mmol), K3PO4 (0.40 mmol) and toluene (3 mL) were used. | ||||
| 1 | Toluene | Cs2CO3 | 90 | 54 |
| 2 | Toluene | K2CO3 | 90 | 71 |
| 3 | Toluene | K3PO4 | 90 | 75 |
| 4 | Dioxane | K3PO4 | 90 | 53 |
| 5 | DCE | K3PO4 | 90 | 65 |
| 6 | MeCN | K3PO4 | 90 | <5 |
| 7c | Toluene | K3PO4 | 90 | 80(68)d |
| 8e | Toluene | K3PO4 | 90 | 90(78)d |
| 9 | Toluene | K3PO4 | 80 | 61 |
| 10 | Toluene | K3PO4 | 100 | 70 |
With the optimized reaction conditions established, we then turned our attention to the scope of this reaction. As shown in Scheme 2, a variety of N-tosylhydrazones were subjected to the reaction with the chromium Fischer carbene complex 2a. In all the cases, the reaction afforded the corresponding products in moderate to good yields. Similarly, the desired product 3a was obtained in 74% yield, when pentacarbonyl[(ethoxy)(phenyl)carbene]chromium(0) 2a′ was selected as the reactant. Substituents of different electronic nature at the para-position of N-tosylhydrazones 1a–g were all tolerated under the optimized reaction conditions (3b–g). It should be noted that elevated temperatures were needed for electron-rich substrates (3f–l). The reaction with N-tosylhydrazones bearing multi-substituted aromatic rings also afforded the desired products (3h–j) in moderate yields. Gratifyingly, the naphthyl-substituted substrate also worked in this reaction, giving the product 3k in 52% yield. Hetero-aromatics could equally be accommodated in this protocol, albeit in low yield (3l). Additionally, the process was slightly hampered by ortho substitution (3m). This result indicates that the reaction is influenced by steric hindrance. Our protocol could also be extended to the substrates with meta-substituents (3n–p). It is noteworthy that N-tosylhydrazones bearing electron-withdrawing groups gave the corresponding products in low to moderate yields (3e, 3o, 3p). The decreased nucleophilicity may account for this phenomenon.11a
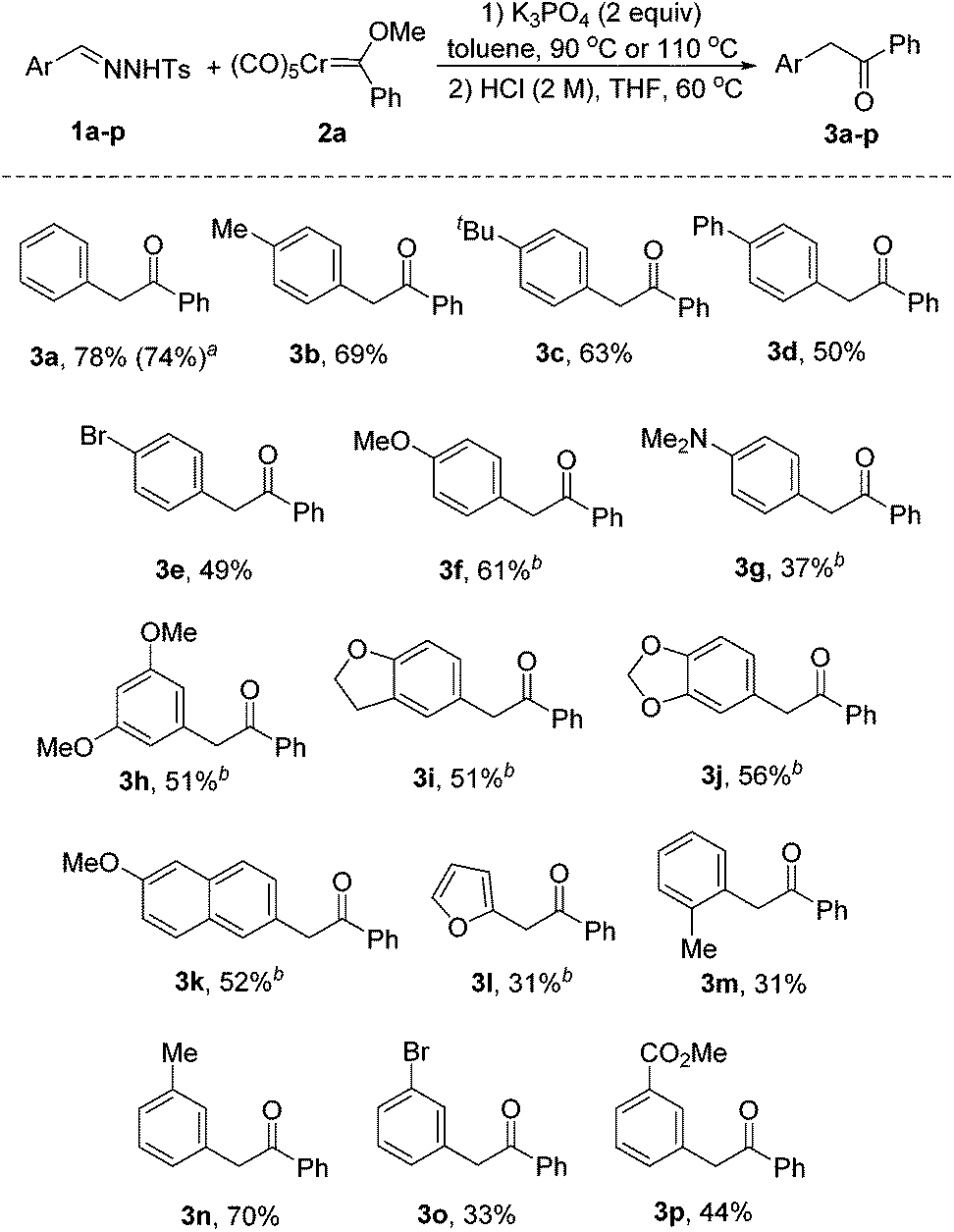 | ||
| Scheme 2 Reaction scope of N-tosylhydrazones. Unless otherwise noted, reaction conditions: 1a–p (0.30 mmol), 2a (0.20 mmol), K3PO4 (0.40 mmol), toluene (3 mL), 90 °C under N2 for 1.5 h. The crude product was dissolved in THF (2 mL), HCl (1 mL, 2 M), 60 °C, 1 h. All the yields refer to isolated yields after silica gel column chromatography. aPentacarbonyl[(ethoxy)(phenyl)carbene]chromium(0) (2a′) was used as a substrate. bThe reaction was carried out at 110 °C. | ||
Encouraged by the results with various N-tosylhydrazones, we next turned our attention to explore the scope of chromium Fischer carbene complexes. Gratifyingly, the substrate 2b with an ortho-substituted group was also reactive in this protocol, affording the product 4a in 69% yield. The reaction proceeded smoothly in the presence of electron-withdrawing or electron-donating substituents at the meta-position of chromium Fischer carbene complexes (4b, 4c). As clearly depicted in 4d–h, the electronic nature of the substituents has no significant impact on the outcome. It is notable that polycyclic aromatic and heteroaryl substrates 2j and 2k were reacted smoothly to deliver the corresponding products 4i and 4j in 63% and 61% yields, respectively (Scheme 3).
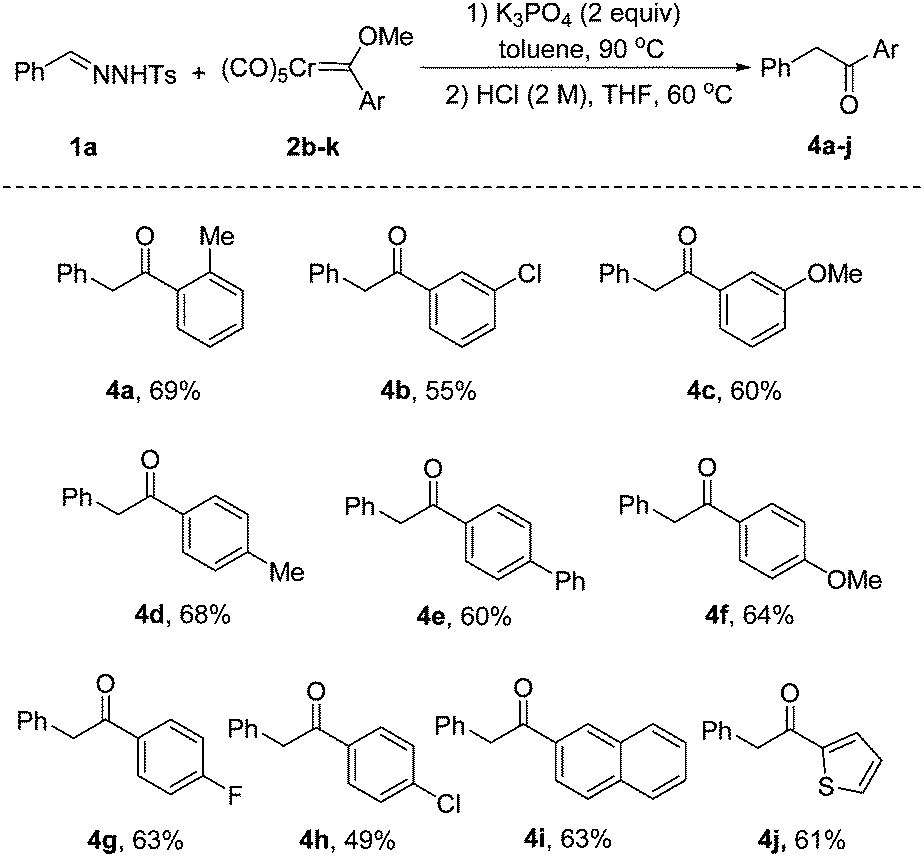 | ||
| Scheme 3 Reaction scope of chromium carbene complexes. Unless otherwise noted, reaction conditions: 1a (0.30 mmol), 2b–k (0.20 mmol), K3PO4 (0.40 mmol), toluene (3 mL), 90 °C under N2 for 1.5 h. The crude product was dissolved in THF (2 mL), HCl (1 mL, 2 M), 60 °C, 1 h. All the yields refer to isolated yields. | ||
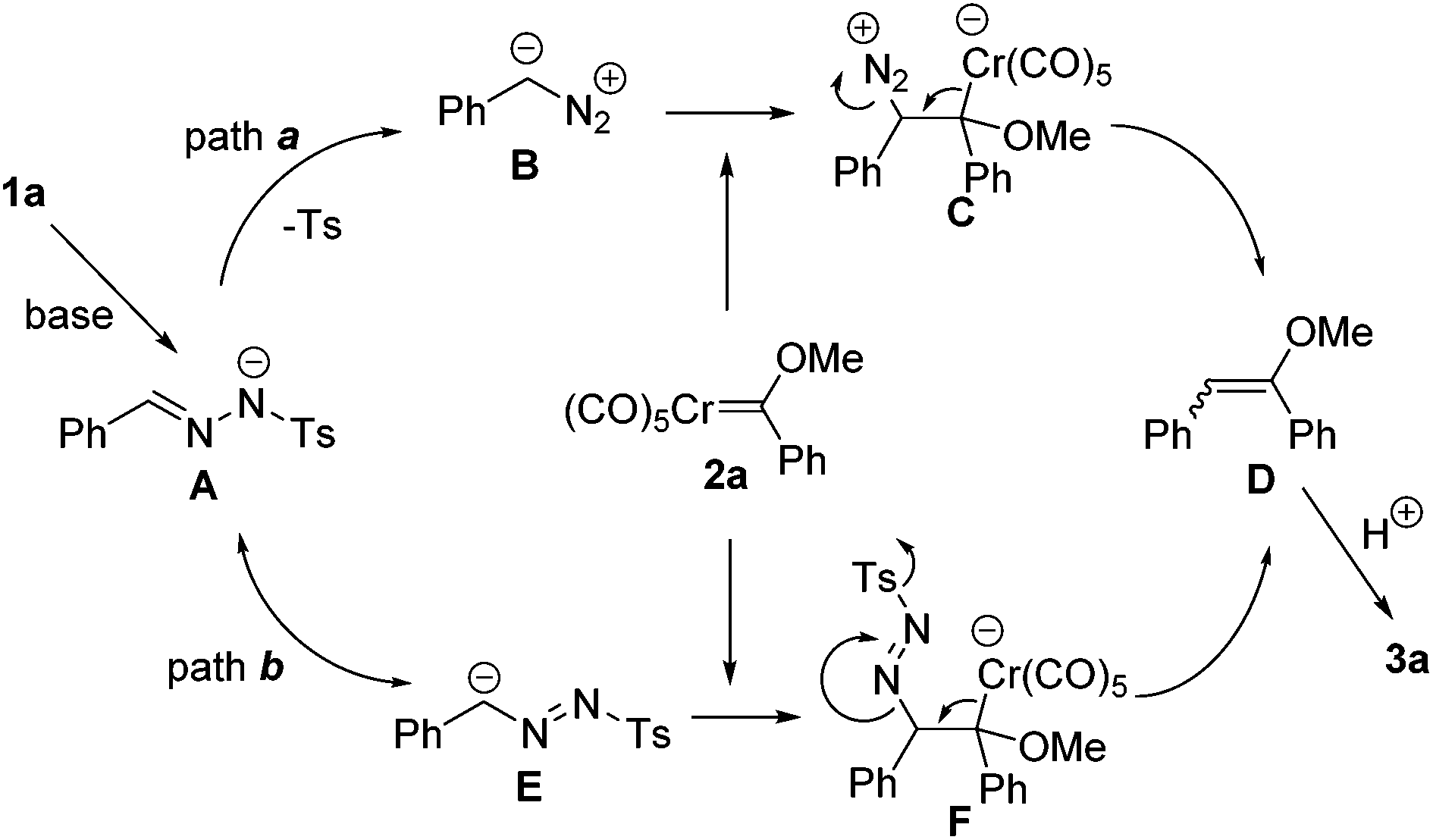
According to our understanding of diazo chemistry and the literature precedents,11 a plausible mechanism was proposed as shown in Scheme 4. Initially, the intermediate A is generated through deprotonation from N-tosylhydrazones. In principle, two reaction pathways are possible from the intermediate A leading to the final product. For path a, the diazo compound intermediate B is formed. Subsequently, the reaction proceeds with the nucleophilic attack of the diazo carbon atom to the electron deficient carbenic carbon center. Then, the olefin D is formed with the release of N2 and the elimination of the chromium carbonyl fragment. In path b, from the anion E, which is the resonance structure of the intermediate A, a similar nucleophilic attack occurs to form F. The release of the Ts group and N2 triggers the elimination of the Cr(CO)5 fragment to afford the product D. Since D is labile in silica gel chromatography, it is converted into arylketone 3a through hydrolysis of the crude product with an acid.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a catalyst-free strategy for the synthesis of diarylethanones from N-tosylhydrazones and chromium Fischer carbene complexes. In contrast to previous carbene dimerization of two diazo substrates, this reaction utilizes stable chromium Fischer carbene complexes as reaction partners; while the highly reactive diazo substrates are generated in situ from N-tosylhydrazones under basic conditions.8 This protocol features simple reaction conditions and wide substrate tolerance. Further exploration of chromium Fischer carbene complexes in the development of novel synthetic methods is currently underway in our laboratory and the results will be reported in due course.Experimental section
General
All reactions were performed under a nitrogen atmosphere in a Schlenk reaction flask. All solvents were distilled under a nitrogen atmosphere prior to use. 1,4-Dioxane and toluene were dried over Na with a benzophenone-ketyl intermediate as an indicator. MeCN and DCE were dried over CaH2. For chromatography, 200–300 mesh silica gel (Qingdao, China) was employed. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz with a Brucker ARX 400 spectrometer. Chemical shifts were reported in ppm using tetramethylsilane as the internal standard when CDCl3 was used as the solvent. IR spectra were recorded with a Thermo Electron Corporation Nicolet AVATAR 330 FT-IR spectrometer. HRMS data were obtained on a VG ZAB-HS mass spectrometer and a Brucker Apex IV FTMS spectrometer. The chromium(0) Fischer carbene complexes were prepared according to the literature procedure: 2a,12a2b,12a2c,12b2d,12a2e,12b2g,12b2h,12b2i,12b2j,12c and 2k.12dTypical procedure for catalyst-free cross-coupling of N-tosylhydrazones with chromium(0) Fischer carbene complexes
N-Tosylhydrazone 1a (82.2 mg, 0.30 mmol), chromium carbene complex 2a (62.4 mg, 0.20 mmol) and potassium phosphate (K3PO4, 85 mg, 0.40 mmol) were successively added to a 25 mL Schlenk reaction flask. The reaction flask was then degassed two times with nitrogen and toluene (3 mL) was added using a syringe. The resulting solution was stirred at the indicated temperatures for 1.5 h. The mixture was then cooled to room temperature and filtered through a short plug of silica gel (washed with petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 10:1). The solvent was removed to obtain a crude mixture. Then, the mixture was transferred to a 25 mL round-bottom flask. After the solvent was removed, 2.0 mL of tetrahydrofuran (THF) and 1.0 mL of aqueous HCl (2 M) were added to the reaction mixture under stirring. The reaction was further stirred for 1 h at 60 °C and then was cooled to room temperature again. Next, the resulting mixture was extracted with diethyl ether (Et2O) three times. After being washed with saturated NaHCO3 and NaCl, the combined organic layers were dried over Na2SO4 and filtered. The solvent was then removed in vacuo to obtain a crude mixture, which was purified by silica gel column chromatography to afford the pure product.
:EtOAc = 10:1). The solvent was removed to obtain a crude mixture. Then, the mixture was transferred to a 25 mL round-bottom flask. After the solvent was removed, 2.0 mL of tetrahydrofuran (THF) and 1.0 mL of aqueous HCl (2 M) were added to the reaction mixture under stirring. The reaction was further stirred for 1 h at 60 °C and then was cooled to room temperature again. Next, the resulting mixture was extracted with diethyl ether (Et2O) three times. After being washed with saturated NaHCO3 and NaCl, the combined organic layers were dried over Na2SO4 and filtered. The solvent was then removed in vacuo to obtain a crude mixture, which was purified by silica gel column chromatography to afford the pure product.
Acknowledgements
The project was supported by the National Basic Research Program (973 Program) (Grant 2012CB821600) and the National Natural Science Foundation of China (Grants 21332002 and 21272010).Notes and references
- For selected reviews, see: (a) T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (b) M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed; (c) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS PubMed; (d) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115 DOI:10.1021/acs.chemrev.5b00121.
- For recent reviews: (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (b) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (c) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC; (d) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (e) H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857 RSC; (f) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC; (g) X. Xu and M. P. Doyle, Acc. Chem. Res., 2014, 47, 1396 CrossRef CAS PubMed.
- For the examples of homo-coupling of diazo substrates: (a) W. Baratta, A. Del Zotto and P. Rigo, Chem. Commun., 1997, 2163 RSC; (b) A. Del Zotto, W. Baratta, G. Verardo and P. Rigo, Eur. J. Org. Chem., 2000, 2795 CrossRef CAS; (c) M. P. Doyle and M. Yan, J. Org. Chem., 2002, 67, 602 CrossRef CAS PubMed; (d) D. M. Hodgson and D. Angrish, Chem. Commun., 2005, 4902 RSC.
- For the examples of cross-coupling of diazo substrates: (a) D. W. Hodgson and D. Angrish, J. Mol. Catal. A: Chem., 2006, 254, 93 CrossRef CAS PubMed; (b) D. M. Hodgson and D. Angrish, Chem. – Eur. J., 2007, 13, 3470 CrossRef CAS PubMed; (c) J. H. Hansen, B. T. Parr, P. Pelphrey, Q. Jin, J. Autschbach and H. M. L. Davies, Angew. Chem., Int. Ed., 2011, 50, 2544 CrossRef CAS PubMed; (d) D. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070 CrossRef CAS PubMed; (e) C. Zhu, G. Xu, D. Ding, L. Qiu and J. Sun, Org. Lett., 2015, 17 DOI:10.1021/acs.orglett.5b02037.
- For a report on carbene dimerization from propargylic esters and diazo compounds: C. Vovard-Le Bray, S. Dérien and P. H. Dixneuf, Angew. Chem., Int. Ed., 2009, 48, 1439 CrossRef CAS PubMed.
- For selected examples: (a) J. Font, F. Serratosa and J. Valls, J. Chem. Soc. D, 1970, 721 RSC; (b) S. Kulkowit and M. A. McKervey, J. Chem. Soc., Chem. Commun., 1978, 1069 RSC; (c) M. P. Doyle, W. Hu and I. M. Phillips, Org. Lett., 2000, 2, 1777 CrossRef CAS PubMed; (d) G.-Y. Li and C.-M. Che, Org. Lett., 2004, 6, 1621 CrossRef CAS PubMed; (e) Y. Xia, Z. Liu, Q. Xiao, P. Qu, R. Ge, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 5714 CrossRef CAS PubMed; (f) C. Zhu, G. Xu, K. Liu, L. Qiu, S. Peng and J. Sun, Chem. Commun., 2015, 51, 12758 Search PubMed.
- For selected reviews: (a) W. D. Wulff, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, U.K., 1995, vol. 12, p. 470 Search PubMed; (b) L. S. Hegedus, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, U.K., 1995, vol. 12, p. 549 Search PubMed; (c) D. F. Harvey and D. M. Sigano, Chem. Rev., 1996, 96, 271 CrossRef CAS PubMed; (d) M. A. Sierra, Chem. Rev., 2000, 100, 3591 CrossRef CAS PubMed; (e) A. De Meijere, H. Schirmer and M. Duetsch, Angew. Chem., Int. Ed., 2000, 39, 3964 CrossRef CAS; (f) J. Barluenga, J. Santamaría and M. Tomás, Chem. Rev., 2004, 104, 2259 CrossRef CAS PubMed; (g) K. H. Dötz and J. Stendel, Chem. Rev., 2009, 109, 3227 CrossRef PubMed; (h) I. Fernández, F. P. Cossío and M. A. Sierra, Acc. Chem. Res., 2011, 44, 479 CrossRef PubMed.
- W. R. Bamford and T. S. Stevens, J. Chem. Soc., 1952, 4735 RSC.
- For reviews, see: (a) J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479 CrossRef CAS PubMed; (b) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS PubMed; (c) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC; (d) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (e) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024 CrossRef CAS PubMed; (f) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS; (g) F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986 RSC.
- (a) M. E. Jung, J. Cordova and M. Murakami, Org. Lett., 2009, 11, 3882 CrossRef CAS PubMed; (b) B. Gao, Y. Zhao and J. Hu, Angew. Chem., Int. Ed., 2015, 54, 638 CAS.
- (a) C. P. Casey, S. H. Bertz and T. J. Burkhardt, Tetrahedron Lett., 1973, 16, 1421 CrossRef; (b) J. Barluenga, L. A. López, O. Löber, M. Tomás, S. García-Granda, C. Alvarez-Rúa and J. Borge, Angew. Chem., Int. Ed., 2001, 40, 3392 CrossRef CAS.
- (a) M. E. Bos, W. D. Wulff, R. A. Miller, S. Chamberlin and T. A. Brandvold, J. Am. Chem. Soc., 1991, 113, 9293 CrossRef CAS; (b) C. F. Bernasconi and L. García-Río, J. Am. Chem. Soc., 2000, 122, 3821 CrossRef CAS; (c) J. Barluenga, I. Pérez-Sánchez, M. G. Suero, E. Rubio and J. Flórez, Chem. – Eur. J., 2006, 12, 7225 CrossRef CAS PubMed; (d) M. G. Suero, R. De la Campa, L. Torre-Fernández, S. García-Granda and J. Flórez, Chem. – Eur. J., 2012, 18, 7287 CrossRef CAS PubMed.
- Y. Su, X. Sun, G. Wu and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 9808 CrossRef CAS PubMed.
- K. Huang, G. Li, W.-P. Huang, D.-G. Yu and Z.-J. Shi, Chem. Commun., 2011, 47, 7224 RSC.
- S.-H. Kim and R. D. Rieke, J. Org. Chem., 2000, 65, 2322 CrossRef CAS.
- C. Cao, L. Wang, Z. Cai, L. Zhang, J. Guo, G. Pang and Y. Shi, Eur. J. Org. Chem., 2011, 1570 CrossRef CAS PubMed.
- J.-C. Hsieh, Y.-C. Chen, A.-Y. Cheng and H.-C. Tseng, Org. Lett., 2012, 14, 1282 CrossRef CAS PubMed.
- E. Bellur, H. Görls and P. Langer, Eur. J. Org. Chem., 2005, 2074 CrossRef CAS PubMed.
- Y.-C. Wong, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2010, 12, 1736 CrossRef CAS PubMed.
- J.-W. Yu, S. Mao and Y.-Q. Wang, Tetrahedron Lett., 2015, 56, 1575 CrossRef CAS PubMed.
- M. Liu, Z. Hyder, Y. Sun, W. Tang, L. Xu and J. Xiao, Org. Biomol. Chem., 2010, 8, 2012 CAS.
- J. Sävmarker, J. Lindh and P. Nilsson, Tetrahedron Lett., 2010, 51, 6886 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. See DOI: 10.1039/c5qo00241a |
| This journal is © the Partner Organisations 2015 |