
An iodine-promoted Meyer–Schuster rearrangement for the synthesis of α-iodo unsaturated ketones†
Hai-Tao
Zhu
*a,
Ming-Jin
Fan
a,
De-Suo
Yang
*a,
Xiao-Ling
Wang
a,
Sen
Ke
a,
Chao-Yang
Zhang
a and
Zheng-Hui
Guan
*b
aShannxi Key Laboratory of Phytochemistry, Baoji University of Arts and Sciences, Baoji 721013, China. E-mail: 1888@bjwlxy.edu.cn
bKey Laboratory of Synthesis and Natural Functional Molecule Chemistry of Ministry of Education, Department of Chemistry & Materials Science, Northwest University, Xi'an 710069, China. E-mail: guanzhh@nwu.edu.cn
First published on 13th March 2015
Abstract
A facile and efficient iodine-promoted Meyer–Schuster rearrangement of propargyl alcohols for the synthesis of α-iodo-α,β-unsaturated ketones is presented. The reaction is concisely conducted at ambient temperature and shows good functional group tolerance.
α-Iodo unsaturated ketones are versatile intermediates which have been used for the synthesis of biologically active heterocyclic compounds1 and palladium-catalyzed cross coupling.2 Typically, the direct iodination of unsaturated enones was achieved by 1,4-conjugate addition of a nucleophile to enones sequentially with electrophilic iodination and elimination.3 Lots of α-iodo enones, such as α-iodo cycloalkenones,4,1b,d α-iodo enaminones5 and α-iodo chalcones,6 were synthesized by using the protocol. However, the synthesis of acyclic β-mono/disubstituted α-iodo enones is still a challenging task. Therefore, the development of facile and efficient methods toward these valuable compounds is of great significance.
Recently, Lewis acid7 or Brønsted acid8,9 catalyzed Meyer–Schuster rearrangements of propargyl alcohols have been developed for the synthesis of useful compounds, such as heterocycles, carbocycles, enones and esters. In 2007, an aqueous HI-promoted Meyer–Schuster rearrangement for the synthesis of α-iodo-α,β-unsaturated aldehydes was developed by Wang and coworker (Scheme 1a).9 The reaction was achieved through a stepwise mechanism that included the formation of iodoallene intermediates and their oxygen-mediated oxidation. After that, an Au and Mo co-catalyzed Meyer–Schuster rearrangement for the synthesis of α-iodo-α,β-unsaturated ketones was developed by Zhang and coworker where an iodonium ion was needed for the Au–I exchange (Scheme 1b).10 Recently, Reddy et al. reported an iodine-induced Meyer–Schuster rearrangement of 3-alkoxy propargyl alcohols for the synthesis of α-iodo-α,β-unsaturated esters (Scheme 1c).11 Despite these advances, versatile and efficient methods for the synthesis of α-iodo unsaturated ketones that are easily accessible and the use of readily accessible starting materials remains highly desirable. As a part of our ongoing research on the transformations of propargylic alcohols,12 we herein report a facile iodine-promoted Meyer–Schuster rearrangement of propargylic alcohols for the synthesis of α-iodo unsaturated ketones.
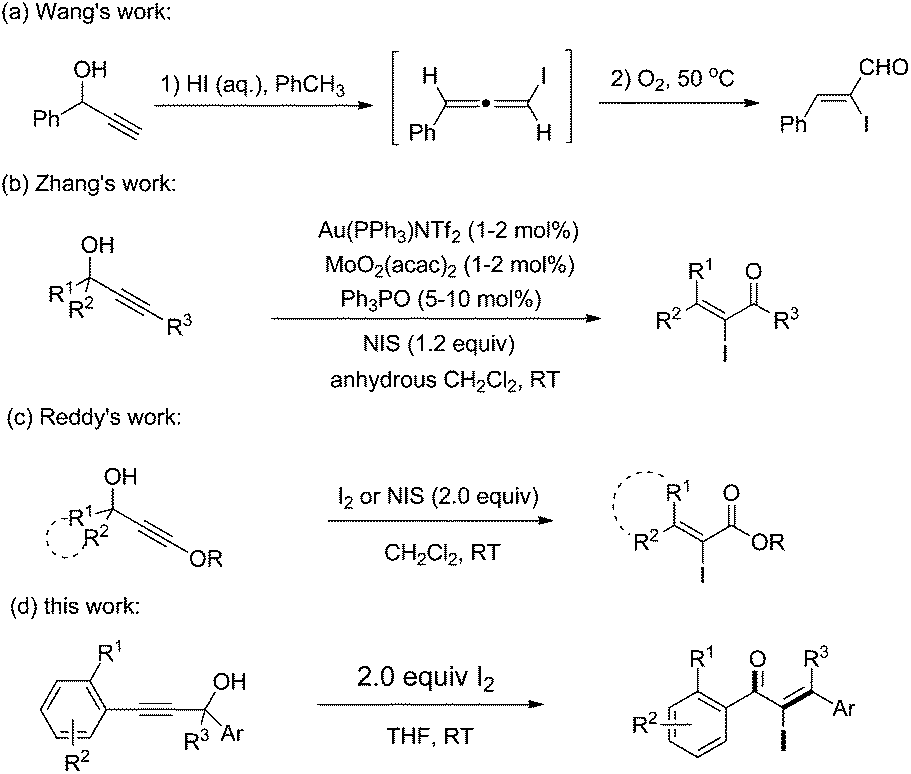 | ||
| Scheme 1 Synthesis of α-iodo unsaturated ketenes. | ||
Initially, the methyl-2-(3-hydroxy-3,3-diphenylprop-1-yn-1-yl) benzoate 1a was selected as the substrate to study the Meyer–Schuster rearrangement in the presence of I2 (1.2 equiv.). To our delight, the desired product methyl-2-(2-iodo-3,3-diphenylacryloyl) benzoate 2a was isolated in 73% yield in THF at room temperature (Table 1, entry 1). The structure of the representative product 2a was determined by X-ray crystallographic analysis (Fig. 1).13 By increasing the loading of I2 to 1.5 equivalents, 76% yield of 2a was obtained (entry 2), and 80% yield of 2a was achieved in the presence of 2.0 equivalents of I2 (entry 3). However, the yield decreased when 3.0 equivalents of I2 were used (entry 4). The screening of different solvents showed that CH3CN and CH3OH were less effective than THF (entries 5 and 6). Furthermore, no better results were obtained when the reaction temperature was varied (entry 7).
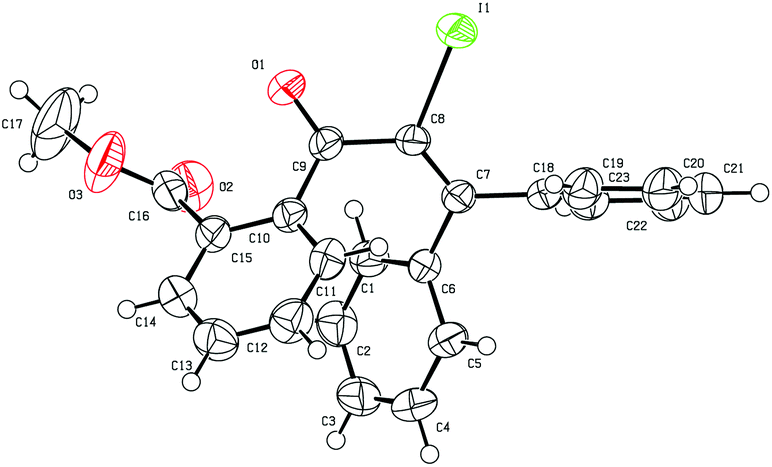 | ||
| Fig. 1 Structure of 2a. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | I2 (equiv.) | Temperature (°C) | Yieldb (%) |
| a All reactions were run under the following conditions, unless otherwise indicated: 0.2 mmol of 1a with I2 in 4 mL of solvent at room temperature. b Isolated yield. | ||||
| 1 | THF | 1.2 | RT | 73 |
| 2 | THF | 1.5 | RT | 76 |
| 3 | THF | 2.0 | RT | 80 |
| 4 | THF | 3.0 | RT | 68 |
| 5 | CH3CN | 1.5 | RT | 68 |
| 6 | CH3OH | 1.5 | RT | 74 |
| 7 | THF | 2.0 | 80 | 75 |
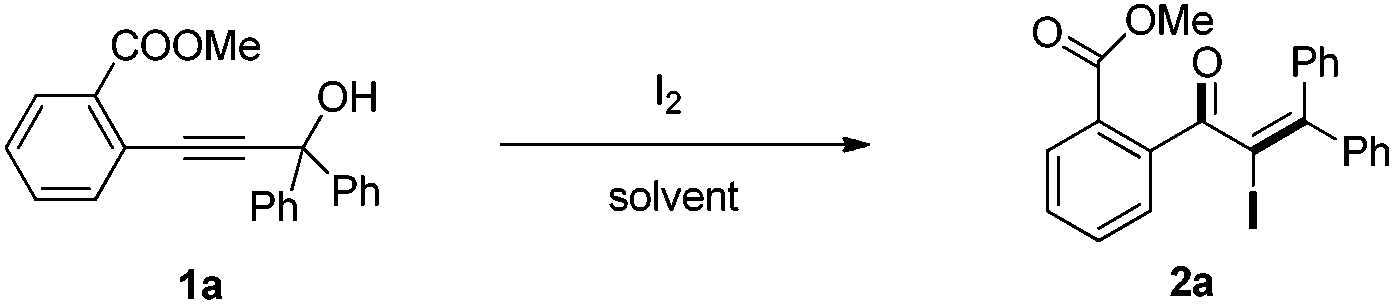
With the optimized reaction conditions established, the scope of the reaction was investigated (Table 2). This iodine-promoted Meyer–Schuster rearrangement of propargylic alcohols14 showed high functional group tolerance and proved to be a concise methodology for the synthesis of α-iodo enones. A variety of substituents, such as carboalkoxyl, formyl, alkyl, alkoxyl, nitro and halo substituents, tolerated the reaction conditions and the corresponding substrates gave α-iodo enones 2a–z in moderate to good yields. The o-carboethoxyl and o-methoxyl phenyl-substituted 2b and 2e, were smoothly obtained in 85% and 82% yields, respectively. These results suggested that the rearrangement was insensitive to the electronic effect of the ortho-substituent on aryl rings (entries 2 and 5). However, substrate 1i with no substituent on the phenyl ring, gave the corresponding product 2i in low yield under the optimized conditions. We also found that m- and p-carbomethoxyl phenyl-substituents α-iodo enones 2j and 2k were not achieved in THF. Fortunately, on changing the solvent from THF to CH3CN, we got better results (entries 9–11). Remarkably, products 2m–q, having m- or p-halo (Cl, F) substituents on a methyl benzoate ring were afforded in excellent yields (entries 13–17). Substrate 1l with an electron-withdrawing nitro group showed a slightly better result than 1r with an electron-rich methyl group (entries 12 and 18). Moreover, we examined the electronic effects of the substituents on R4 and R3 of the aromatic ring. It was found that electron-withdrawing or electron-donating substituents did not affect this transformation (entries 19–22). Interestingly, aliphatic substituted α-iodo enone 2w was also obtained in 62% yield (entry 23).
|
|
|||
|---|---|---|---|
| Entry | Substrate (R1, R2, R3, R4) | Product | Yieldb (%) |
| a All reactions were run under the following conditions, unless otherwise indicated: 0.2 mmol of 1 with I2 in 4 mL of THF at room temperature. b Isolated yield. c The solvent was CH3CN. d The ratio was determined by 1H NMR. | |||
| 1 | R1 = COOMe, R2 = H, R3 = H, R4 = Ph 1a | 2a | 80 |
| 2 | R1 = COOEt, R2 = H, R3 = H, R4 = Ph 1b | 2b | 85 |
| 3 | R1 = COOBn, R2 = H, R3 = H, R4 = Ph 1c | 2c | 81 |
| 4 | R1 = CHO, R2 = H, R3 = H, R4 = Ph 1d | 2d | 73 |
| 5 | R1 = OMe, R2 = H, R3 = H, R4 = Ph 1e | 2e | 82 |
| 6 | R1 = CH2COOMe, R2 = H, R3 = H, R4 = Ph 1f | 2f | 93 |
| 7 | R1 = H, R2 = 4-Et, R3 = H, R4 = Ph 1g | 2g | 86 |
| 8 | R1 = H, R2 = 4-OMe, R3 = H, R4 = Ph 1h | 2h | 76 |
| 9 | R1 = H, R2 = H, R3 = H, R4 = Ph 1i | 2i | 84c |
| 10 | R1 = H, R2 = 3-COOMe, R3 = H, R4 = Ph 1j | 2j | 74c |
| 11 | R1 = H, R2 = 4-COOMe, R3 = H, R4 = Ph 1k | 2k | 56c |
| 12 | R1 = COOMe, R2 = 4-NO2, R3 = H, R4 = Ph 1l | 2l | 86 |
| 13 | R1 = COOMe, R2 = 4-Cl, R3 = H, R4 = Ph 1m | 2m | 92 |
| 14 | R1 = COOMe, R2 = 4-F, R3 = H, R4 = Ph 1n | 2n | 91 |
| 15 | R1 = COOMe, R2 = 3-Cl, R3 = H, R4 = Ph 1o | 2o | 86 |
| 16 | R1 = COOMe, R2 = 5-Cl, R3 = H, R4 = Ph 1p | 2p | 88 |
| 17 | R1 = COOMe, R2 = 5-F, R3 = H, R4 = Ph 1q | 2q | 92 |
| 18 | R1 = COOMe, R2 = 4-Me, R3 = H, R4 = Ph 1r | 2r | 75 |
| 19 | R1 = COOMe, R2 = H, R3 = Me, R4 = 4-MeC6H41s | 2s | 88 |
| 20 | R1 = COOMe, R2 = H, R3 = Cl, R4 = 4-ClC6H41t | 2t | 85 |
| 21 | R1 = COOMe, R2 = H, R3 = F, R4 = 4-FC6H41u | 2u | 81 |
| 22 | R1 = COOMe, R2 = H, R3 = OMe, R4 = 4-OMeC6H41v | 2v | 76 |
| 23 |
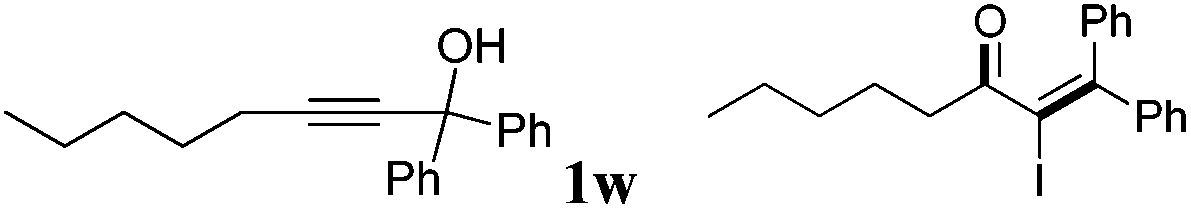
|
2w | 62 |
| 24 | R1 = COOMe, R2 = H, R3 = H, R4 = H 1x | 2x | 73 (>19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)d :1)d |
| 25 | R1 = COOEt, R2 = H, R3 = H, R4 = H 1y | 2y | 60 (17:1)d |
| 26 | R1 = COOMe, R2 = H, R3 = H, R4 = Me 1z | 2z | 46 (15:1)d |
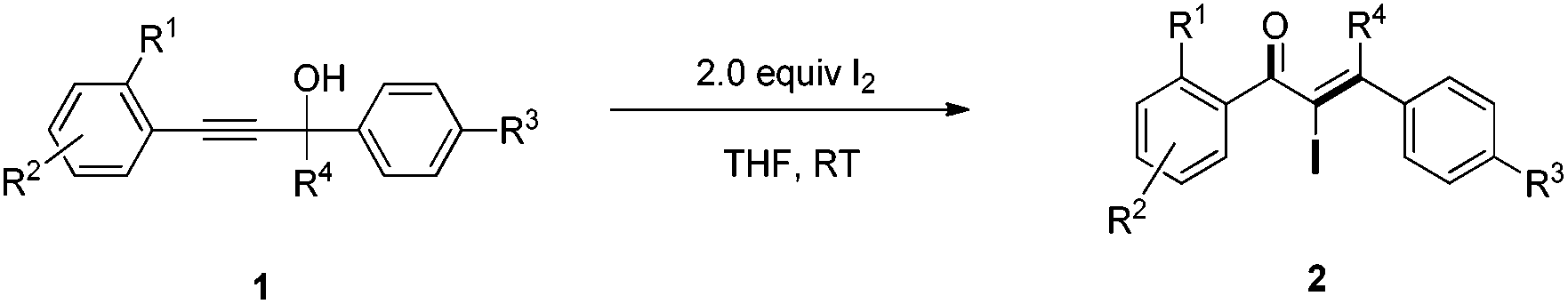
Secondary propargyl alcohols (R4 = H) did not affect this transition. The methyl 2-(3-hydroxy-3-phenylprop-1-yn-1-yl)-benzoate 1x stereo-selectively produced the rearrangement product 2x in good yield (entry 24). However, substrate 1y, bearing an ethoxycarbonyl on R1, formed the inseparable mixture (Z and E) in a 17:1 ratio (entry 25). Compound 1z, bearing a methyl on R4, gave a similar result (entry 26).
Noteworthily, we also investigated the scale-up of this reaction. The 4 mmol of 1a, upon exposure to I2 in THF, afforded the desired product 2a in 79% yield in 1 h. Furthermore, when using IBr as the electrophilic reagent, the desired adduct 2a was also obtained in 90% yield (Scheme 2). The result indicated that the Meyer–Schuster rearrangement is probably induced by the iodonium ion.
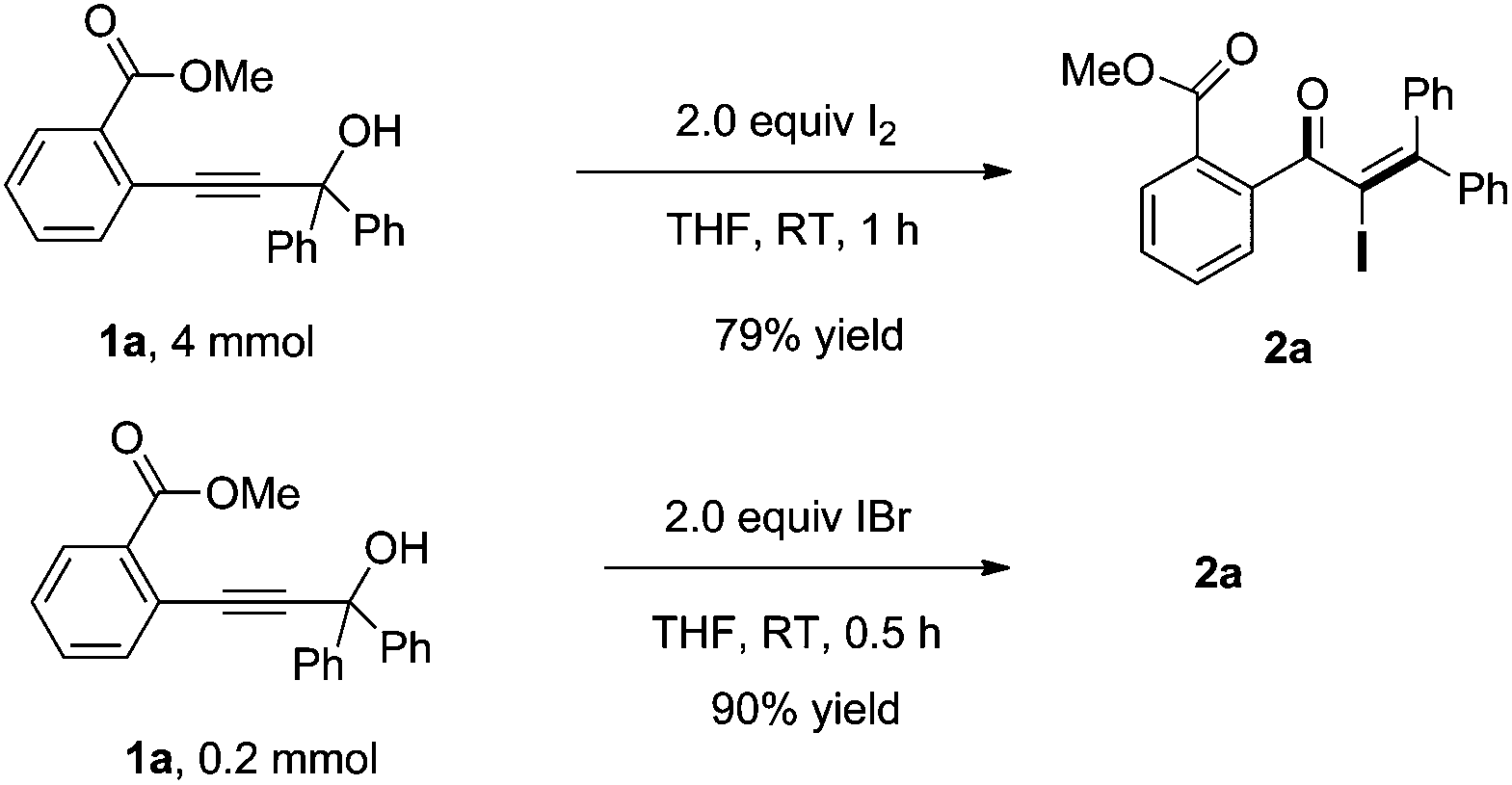 | ||
| Scheme 2 Scale-up of the iodo Meyer–Schuster rearrangement and application of other electrophiles. | ||
As shown in Scheme 3, the α-iodo unsaturated ketone 2a produced by the iodo Meyer–Schuster rearrangement can be further transferred in palladium-catalyzed cross-couplings or reductions. For example, the Suzuki coupling of 2a with p-methoxyl phenyl boronic acid afforded the corresponding product 3a in 45% yield.15 Reductive lactonization and deiodination of 2a and 2x in the presence of NaBH4 produced cyclic compounds 4a and 4x in 79% and 40% yields, respectively.16 The structure of 4 was determined by the 1D NMR, 2D NMR and NOESY spectra (see the ESI†).
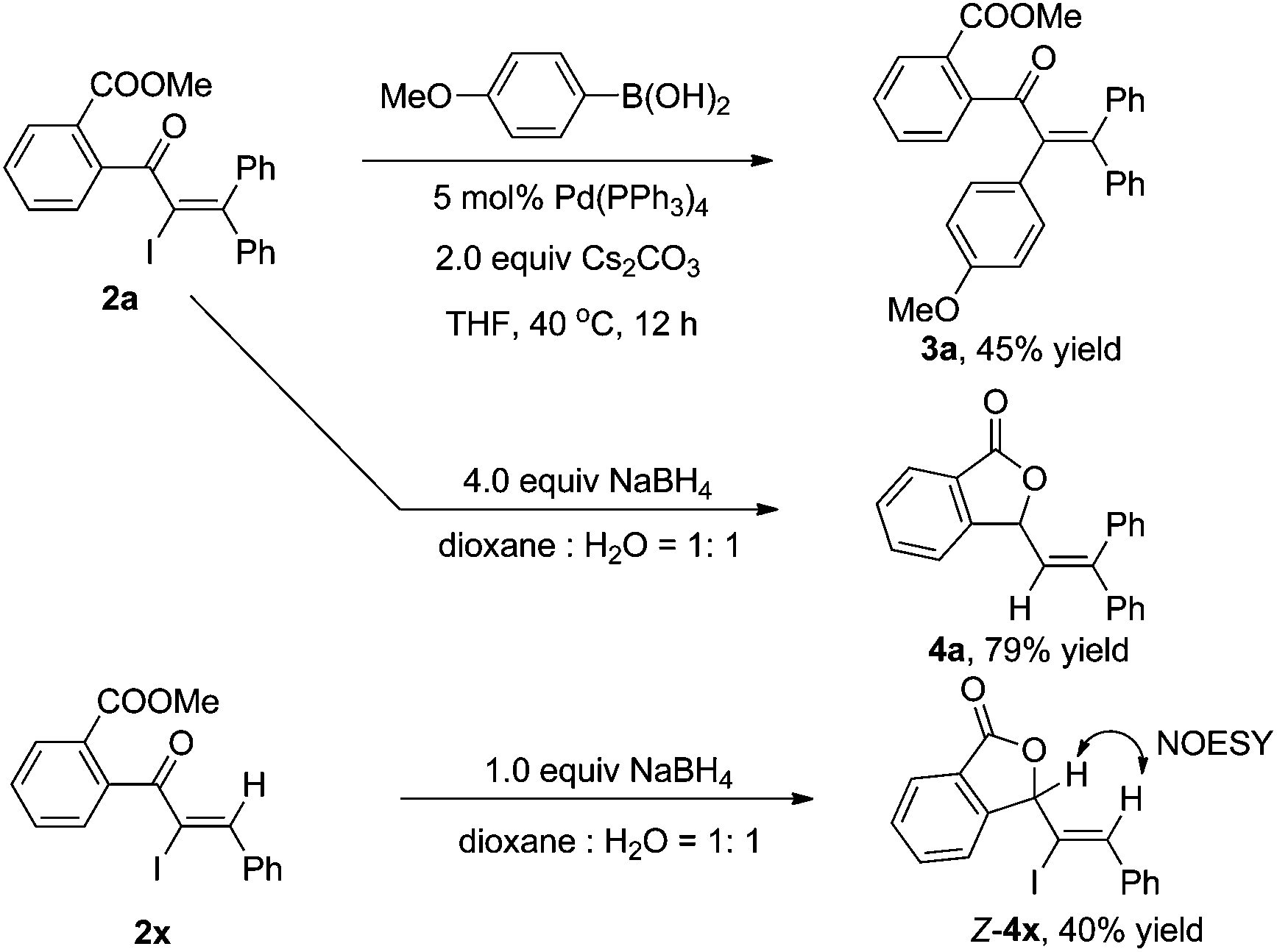 | ||
| Scheme 3 Utilizations of functional groups of α-iodo conjugated enones 2a and 2x. | ||
On the basis of the results obtained above, a tentative mechanism was proposed in Scheme 4. Presumably, in the presence of Lewis acidic iodine, the propargyl hydroxyl group of substrate 1 is activated to afford the intermediate propargyl cation A and hypoiodous acid (HOI).17 Then, A reacts with a hydroxyl anion derived from hypoiodous acid ionization to give allenol intermediate B. Finally, B is induced by an iodide cation to isomerize and produce the major α-iodo unsaturated ketone 2. The E isomer 2′ is unfavorable due to steric hindrance between the two aryl groups.
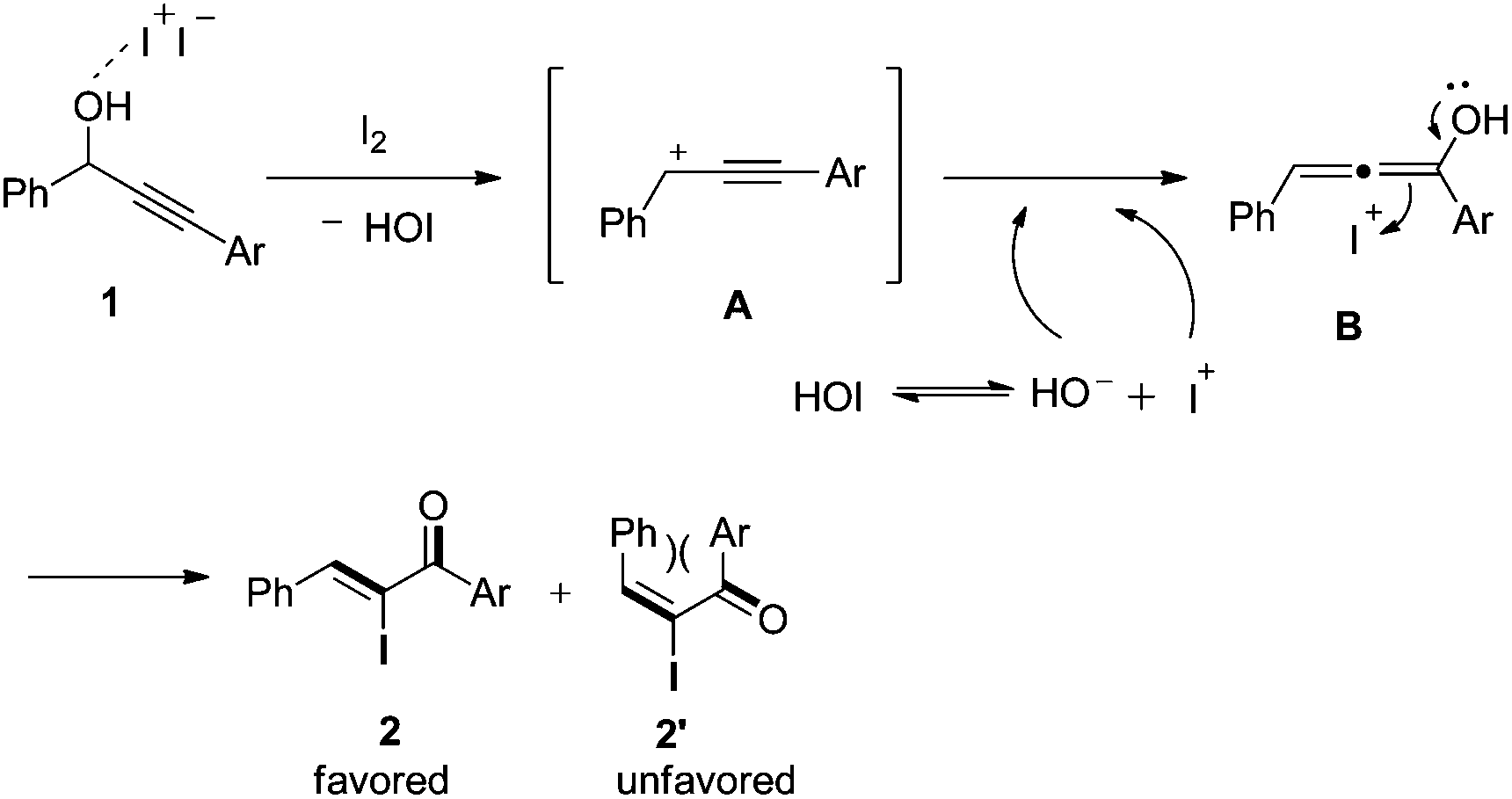 | ||
| Scheme 4 Proposed mechanism for the formation of α-iodo unsaturated ketones. | ||
Conclusions
In conclusion, we have developed a concise and efficient approach to synthesize highly substituted α-iodo-α,β-unsaturated ketones from readily accessible propargylic alcohols under mild reaction conditions. The reaction shows high Z-stereoselectivity and the resulting α-iodo enones can be further exploited by cross-couplings and reductions. The application of α-iodo-α,β-unsaturated ketones for the synthesis of useful polycyclic compounds is in progress.Experimental section
General procedure for synthesis of α-iodo unsaturated ketones
To a solution of propargyl alcohol derivatives 1 (0.20 mmol) in THF (4.0 mL) was added I2 (2.0 equiv., 0.4 mmol) at room temperature. When the reaction was complete, the reaction mixture was quenched by the addition of saturated aqueous sodium thiosulfate, extracted with ethyl acetate (3 × 15 mL), washed with water and saturated brine, dried over Na2SO4 and then evaporated under reduced pressure. The residue was purified by chromatography on silica gel to afford the corresponding α-iodo unsaturated ketenes 2.Methyl-2-(2-iodo-3,3-diphenylacryloyl)benzoate 2a
1H NMR (400M Hz, CDCl3) δ ppm 7.33 (d, J = 7.6 Hz, 2H), 7.32–7.26 (m, 3H), 7.19–7.12 (m, 4H), 6.95–6.91 (m, 5H), 3.90 (s, 3H). 13C NMR (100M Hz,CDCl3) δ ppm 193.3, 168.5, 158.6, 144.8, 139.7, 136.7, 132.1, 131.0, 130.4, 130.1, 129.7, 128.8, 128.7, 128.6, 128.2, 127.9, 101.3, 53.0. IR (neat, cm−1): 2921, 1734, 1654, 1234, 1097, 763. HRMS (ESI) Calcd for C23H17INaO3: M + Na = 491.0115. Found: 491.0121.Acknowledgements
The authors gratefully acknowledge financial support of this work by the Education Department of Shaanxi Province Key Laboratory Project (13JS006). We are also extremely grateful to the Shaanxi Key Laboratory of Phytochemistry, Baoji University of Arts and Sciences for support received for a part of the characterization work.Notes and references
- (a) M. Gartner, J. Qu and G. Helmchen, J. Org. Chem., 2012, 77, 1186 CrossRef PubMed; (b) V. Rauniyar, Z. J. Wang, H. E. Burks and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 8486 CrossRef CAS PubMed; (c) T. J. Greshock and R. L. Funk, J. Am. Chem. Soc., 2006, 128, 4946 CrossRef CAS PubMed; (d) T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164 CrossRef CAS PubMed; (e) M. G. Banwell, B. D. Kelly, O. J. Kokas and D. W. Lupton, Org. Lett., 2003, 5, 2497 CrossRef CAS PubMed; (f) K. C. Nicolaou, D. L. F. Gray, T. Montagnon and S. T. Harrison, Angew. Chem., Int. Ed., 2002, 41, 996 CrossRef CAS; (g) A. D. William and Y. Kobayashi, J. Org. Chem., 2002, 67, 8771 CrossRef CAS PubMed; (h) N. G. Ramesh, E. H. Heijne, A. J. H. Klunder and B. Zwanenburg, Tetrahedron, 2002, 58, 1361 CrossRef CAS; (i) C.-K. Sha, K. C. Santhosh, C.-T. Tseng and C.-T. Lin, Chem. Commun., 1998, 397 CAS.
- (a) G. Pandey and M. Balakrishnan, J. Org. Chem., 2008, 73, 8128 CrossRef CAS PubMed; (b) N. K. Swamy, L. K. Tatini, J. M. Babu, P. Annamalai and M. Pal, Chem. Commun., 2007, 1035 RSC; (c) Q. Yao, E. P. Kinney and Z. Yang, J. Org. Chem., 2003, 68, 7528 CrossRef CAS PubMed; (d) K. C. Nicolaou, M. P. Jennings and P. Dagneau, Chem. Commun., 2002, 2480 RSC; (e) K. Lee, J. Lee and P. H. Lee, J. Org. Chem., 2002, 67, 8265 CrossRef CAS PubMed; (f) L. Timbart and J.-C. Cintrat, Chem. – Eur. J., 2002, 8, 1637 CrossRef CAS; (g) S. T. Handy and X. Zhang, Org. Lett., 2001, 3, 233 CrossRef CAS; (h) M. W. Miller and C. R. Johnson, J. Org. Chem., 1997, 62, 1582 CrossRef CAS; (i) R. C. Larock, M. J. Doty, Q. Tian and J. M. Zenner, J. Org. Chem., 1997, 62, 7536 CrossRef CAS.
- (a) C. R. Johnson, J. P. Adams, M. P. Braun, C. B. W. Senanayake, P. M. Wovkulich and M. R. Uskokovic, Tetrahedron Lett., 1992, 33, 917 CrossRef CAS; (b) D. Elsa, P. Bovonsombat and E. M. Nelis, Synth. Commun., 1997, 27, 2497 CrossRef.
- (a) D. F. Taber and J. F. Berry, J. Org. Chem., 2013, 78, 8437 CrossRef CAS PubMed; (b) K. Li and A. Alexakis, Angew. Chem., Int. Ed., 2006, 45, 7600 CrossRef CAS PubMed; (c) P. Bovonsombat, G. J. Angara and E. Mc Nelis, Tetrahedron Lett., 1994, 35, 6787 CrossRef CAS; (d) C.-K. Sha, C.-Y. Shen, T.-S. Jean, R.-T. Chiu and W.-H. Tseng, Tetrahedron Lett., 1993, 34, 7641 CrossRef CAS; (e) C.-K. Sha and S. J. Huang, Tetrahedron Lett., 1995, 36, 6927 CrossRef CAS.
- (a) J. M. Kim, J. E. Na and J. N. Kim, Tetrahedron Lett., 2003, 44, 6317 CrossRef CAS; (b) P. J. Campos, J. Arranz and M. A. Rodriguez, Tetrahedron Lett., 1997, 38, 8397 CrossRef CAS; (c) I. Papoutsis, S. Spyroudis, A. Varvoglis, J. A. Callies and V. V. Zhdankin, Tetrahedron Lett., 1997, 38, 8401 CrossRef CAS; (d) N. G. Ramesh, N. G. Ramesh, E. H. Heijne, A. J. H. Klunder and B. Zwanenburg, Tetrahedron Lett., 1998, 39, 3295 CrossRef CAS.
- (a) G. Liu, P. D. Wilkerson, C. A. Toth and H. Xu, Org. Lett., 2012, 14, 858 CrossRef CAS PubMed; (b) A. S. K. Hashmi, T. Haffner, M. Rudolph and F. Rominger, Eur. J. Org. Chem., 2011, 667 CrossRef CAS; (c) P. Bovonsombat, R. Rujiwarangkul, T. Bowornkiengkai and J. Leykajarakul, Tetrahedron Lett., 2007, 48, 8607 CrossRef CAS PubMed; (d) M. E. Krafft and J. W. Cran, Syndet, 2005, 1263 CAS; (e) A. Alimardanov and E. Negishi, Tetrahedron Lett., 1999, 40, 3839 CrossRef CAS.
- (a) Y. Zhu, L. Sun, P. Lu and Y. Wang, ACS Catal., 2014, 4, 1911 CrossRef CAS; (b) M. N. Pennell, P. G. Turner and T. D. Sheppard, Chem. – Eur. J., 2012, 18, 4748 CrossRef CAS PubMed; (c) M. Egi, M. Umemura, T. Kawai and S. Akai, Angew. Chem., Int. Ed., 2011, 50, 12197 CrossRef CAS PubMed; (d) D. A. Engel and G. B. Dudley, Org. Lett., 2006, 8, 4027 CrossRef CAS PubMed; (e) E. Jimenez-Nunez and A. M. Echavarren, Chem. Commun., 2007, 333 RSC; (f) A. Furstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410 CrossRef PubMed.
- (a) S. Aiken, K. Booth, C. D. Gabbutt, B. M. Heron, C. R. Rice, A. Charaf-Eddin and D. Jacquemin, Chem. Commun., 2014, 7900 RSC; (b) E. V. Banide, Y. Ortin, B. Chamiot, A. Cassidy, J. Niehaus, A. Moore, C. M. Seward, H. Muller-Bunz and M. J. McGlinchey, Organometallics, 2008, 27, 4173 CrossRef CAS; (c) R. Sanz, A. Martínez, J. M. Alvarez-Gutierrez and F. Rodriguez, Eur. J. Org. Chem., 2006, 2006, 1383 CrossRef; (d) K. H. Meyer and K. Schuster, Chem. Ber., 1922, 55, 819 CrossRef; (e) S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429 CrossRef CAS.
- S. Chen and J. Wang, J. Org. Chem., 2007, 72, 4993 CrossRef CAS PubMed.
- L. Ye and L. Zhang, Org. Lett., 2009, 11, 3646 CrossRef CAS PubMed.
- S. Puri, N. Thirupathi and M. S. Reddy, Org. Lett., 2014, 16, 5246 CrossRef CAS PubMed.
- (a) H.-T. Zhu, K.-G. Ji, F. Yang, L.-J. Wang, S.-C. Zhao, A. Shaukat, X.-Y. Liu and Y.-M. Liang, Org. Lett., 2011, 13, 684 CrossRef CAS PubMed; (b) H.-T. Zhu, L.-J. Wang, K.-G. Ji, X.-Y. Liu and Y.-M. Liang, Chem. – Asian J., 2012, 7, 1862 CrossRef CAS PubMed; (c) H.-T. Zhu, X. Dong, L.-J. Wang, M.-J. Zhong, X.-Y. Liu and Y.-M. Liang, Chem. Commun., 2012, 48, 10748 RSC.
- CCDC 1044075 (2a) contains the supplementary crystallographic data for this paper.
- (a) Y. Dou, P. Xing, Z. Huang and B. Jiang, Chin. J. Chem., 2014, 32, 999 CrossRef CAS; (b) Z. Wang, J. Zheng and P. Huang, Chin. J. Chem., 2012, 30, 23 CrossRef; (c) X. Dong, X. Xu, H. Hou, Q. Wei, D. Xu, N. Jiao, A. Zhang, S. Zhao and S. Ma, Chin. J. Org. Chem., 2001, 21, 1142 CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- (a) W. R. Bowman, C. F. Bridge, P. Brookes, M. O. Cloonan and D. C. Leach, J. Chem. Soc., Perkin Trans. 1, 2002, 58 CAS; (b) A. K. Verma, S. P. Shukla, J. Singh and V. Rustagi, J. Org. Chem., 2011, 76, 5670 CrossRef CAS PubMed.
- F. Yang, T. Jin, M. Bao and Y. Yamamoto, Chem. Commun., 2011, 47, 4013 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1044075. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00048c |
| This journal is © the Partner Organisations 2015 |