
A divergent synthesis of 3,10-dialkylpicenes†‡
Xi-Chao
Chen
a,
Shuhei
Nishinaga
a,
Yasuhiro
Okuda
a,
Jia-Ji
Zhao
a,
Jie
Xu
a,
Hiroki
Mori
a and
Yasushi
Nishihara
*ab
aDivision of Earth, Life, and Molecular Sciences, Graduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushimanaka, Kita-ku, Okayama 700-8530, Japan. E-mail: ynishiha@okayama-u.ac.jp
bACT-C, Japan Science and Technology Agency, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 16th March 2015
Abstract
A series of 3,10-dialkylated picenes has been synthesized from 3,10-dimethoxypicene through a divergent approach involving Ni-catalyzed alkynylation of a C–O bond, alkylation, and hydrogenation.
Introduction
Fused aromatic compounds containing π-conjugated systems are notable for their utility in organic semiconductors and organic field-effect transistors (OFETs).1–8 Research in these field emerged following pioneering discoveries of organic conductors in 19549 and OFETs in 1986.10 Advantageous features of fused aromatic compounds for OFET devices include their mechanical flexibility, light weight, and low costs, relative to inorganic-based FET devices. As a result a number of synthetic approaches to these molecules have been developed.The most frequently studied class of organic semiconductors contain [n]acenes, in particular pentacene derivatives, due to their excellent transistor characteristics.11 Nevertheless, the poor solubility of pentacene and higher acenes in common organic solvents hinders their processability. In addition, because of their narrow energy gaps and high HOMO energy levels, [n]acenes are sensitive to light and oxidants, especially in the internal rings.12,13 Due to this reason, recently [n]phenacenes, armchair-structure molecules have received much attention because of high stability even in the presence of O2 and H2O.14
Picene ([5]phenacene) is an isomer of pentacene with notably high stability resulting from its large band gap (Eg = 3.3 eV) and higher ionization potential (IP = 5.5 eV).15 An OFET device with a picene thin film displayed a high μ value, more than 1.1 cm2 V–1 s–1, and, significantly, the μ value as well as the on-off ratio were dramatically improved under air/O2 conditions.16 Very recently, we reported an efficient synthetic route to picene by Suzuki–Miyaura coupling or Wittig reaction and sequential intramolecular cyclization via C–H bond activation.17 With this methodology in hand, various methoxy-substituted picene derivatives were elaborated.18 Results of theoretical calculations of HOMO and LUMO levels in methoxypicenes indicated that methoxy groups in the 3,10-positions can stabilize the HOMO energy level, the crystal structure of 3,10-dimethoxypicene revealed a 3D-herringbone structure, leading to a higher charge mobility (1.4 × 10–2 cm2 V–1 s–1) than other methoxy-substituted picenes. Despite the promising properties of 3,10-dimethoxypicene toward OFETs, its poor solubility in organic solvents limited its utilization for device fabrication by a solution process, which is desired for application in organic electronic devices. A general strategy to improve the solubility and, as a result, processability is to install long alkyl substituents onto the parent core.19,20
In recent years, various synthetic methods for substituted picenes have been reported. In 2010, Nakano et al. reported the synthesis of 3,10-dialkylpicenes through the Pt-catalyzed cycloaromatization reaction of terminal alkynes (Scheme 1A).21 However, a drawback of this protocol is the need of an initial stage for the introduction of alkyl groups: Suzuki–Miyaura coupling of alkyl-substituted arylboronic acids with halobenzene, which limits the diversity derivatization of alkylated picenes that can be obtained. In general, although a divergent synthesis might allow for a more benign and straightforward route for the preparation of alkylated picenes, to the best of our knowledge, there is only one known example on the installation of alkyl groups onto a picene core at later stage.22 We herein report the first general and straightforward synthesis of various 3,10-alkylated picenes from 3,10-dimethoxypicene as the common starting material. This involves introduction of alkyl groups through the Ni-catalyzed alkynylation of a C–O bond as the key step (Scheme 1B).
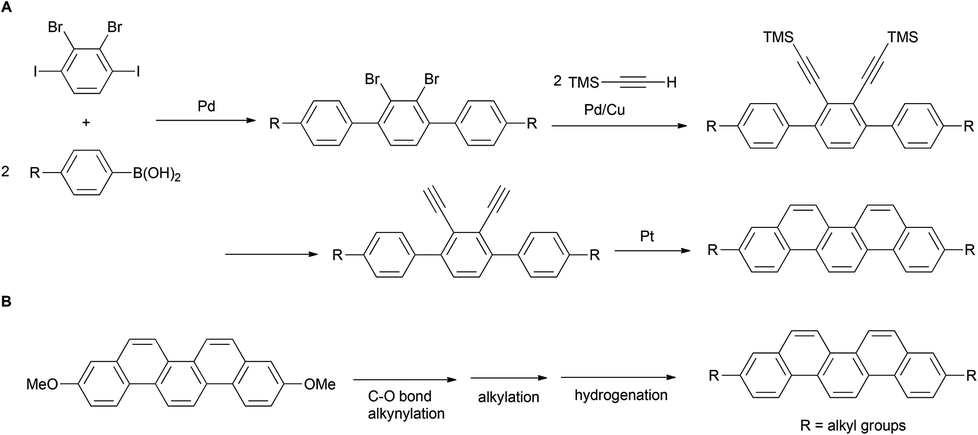 | ||
| Scheme 1 A synthetic strategy for 3,10-alkylated picenes. | ||
Results and discussion
To synthesize 3,10-didecylpicene (8-C10) as a benchmark compound, we started to apply our previously reported procedure.17 A synthetic scheme is shown in Scheme 2. A decyl group was incorporated by the reaction of 1,3-dibromobenzene (1) with an equimolar amount of nBuLi, followed by 1-bromodecane to afford 2 in 90% yield. Preparation of 3 was accomplished by Sonogashira–Hagihara coupling of 2 with trimethylsilylethyne (TMS = SiMe3). After desilylation of 3, Rh-catalyzed hydroboration of the obtained terminal alkyne 4 with HBpin (Bpin = pinacolatoboryl) gave a stereodefined (Z)-alkenylboronate 5 in 60% yield. Precursor 7 was prepared by Pd-catalyzed Suzuki–Miyaura coupling of 5 with 1,4-dichloro-2,3-diiodobenzene (6). Finally, an intramolecular double cyclization of 7via C–H bond activation furnished the target product 8-C10, albeit in 20% yield.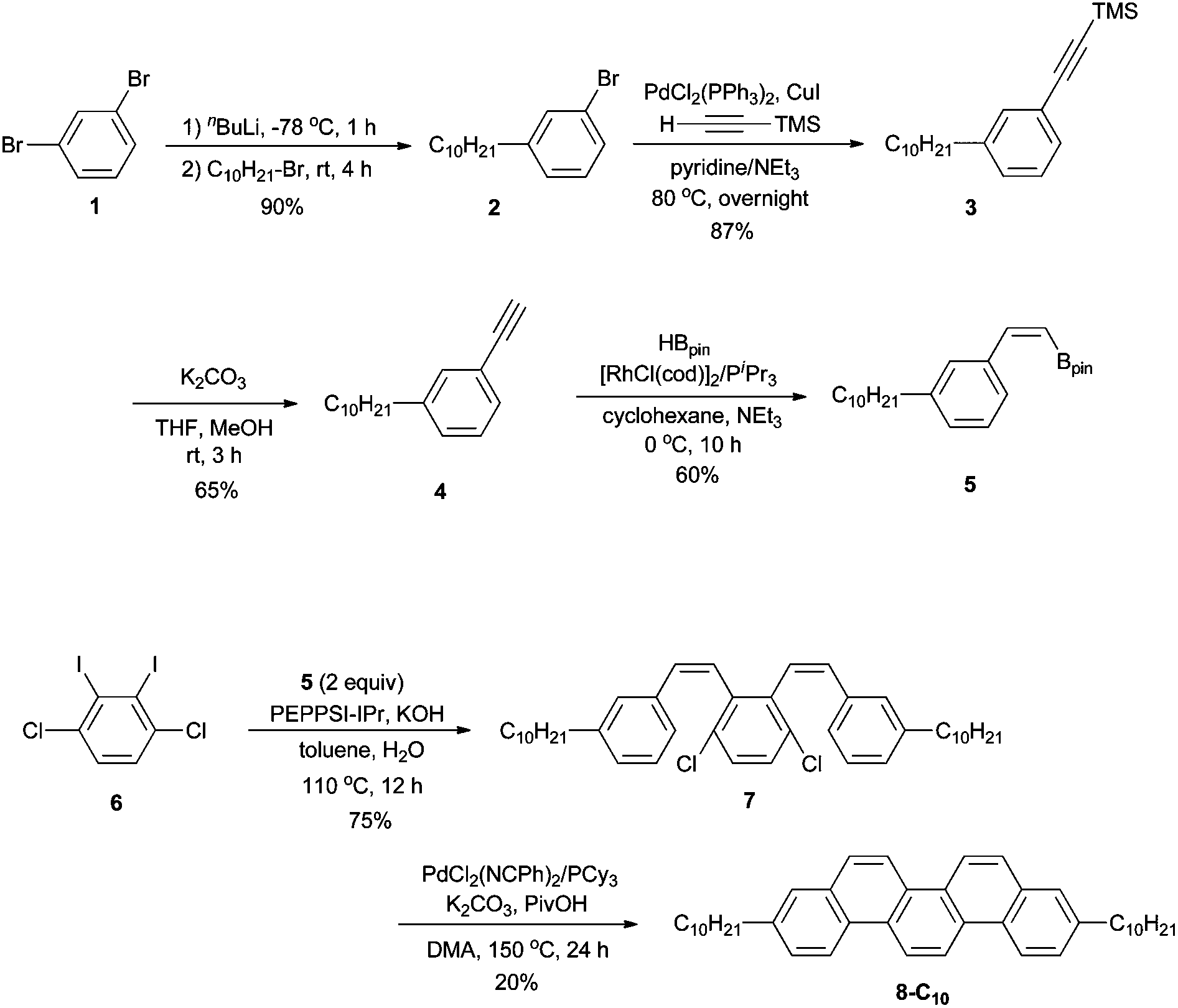 | ||
| Scheme 2 Synthesis of 3,10-didecylpicene (8-C10). | ||
Although alkylated picene 8-C10 could be obtained, the synthetic route shown in Scheme 2 is not ideal for the synthesis of a series of alkylated picenes because the alkyl group has to be introduced at an initial stage. Therefore, we started to investigate more efficient syntheses of 3,10-dialkylpicenes through C–O bond functionalization of 3,10-dimethoxypicene (9)18 as the common starting material. Because of the high stability of the C–O bond, the direct transformations of the C–O bond is a challenging objective.23–36
Very recently, Tobisu and Chatani have reported the direct C–O bond alkynylation reaction of anisole derivatives.37 Inspired by this straightforward transformation of the C–O bond, we optimized the conditions for the double alkynylation of 9 and the results are summarized in Table 1.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (h) | Yieldb (%) | |
| 10 | 10′ | ||||
| a 3,10-Dimethoxypicene (9) (0.05 mmol), the in situ prepared triisopropylsilylethynylmagnesium bromide (0.2 mmol), Ni(cod)2 (0.01 mmol), ICy·HCl (0.02 mmol) in the solvent. b NMR yields based on 1,4-dioxane as an internal standard. c Isolated yields (0.59 mmol for 9). | |||||
| 1 | 1,4-Dioxane | 120 | 18 | 0 | 0 |
| 2 | Triglyme | 160 | 18 | 0 | 0 |
| 3 | NMP | 160 | 18 | 0 | 0 |
| 4 | Diphenyl ether | 160 | 18 | 0 | 0 |
| 5 | Mesitylene | 160 | 18 | 47 | 25 |
| 6 | Mesitylene | 150 | 18 | 70 | 0 |
| 7 | Mesitylene | 140 | 18 | 33 | 17 |
| 8 | Mesitylene | 130 | 72 | 22 | 20 |
| 9 | Mesitylene | 140 | 72 | 76c | 9c |
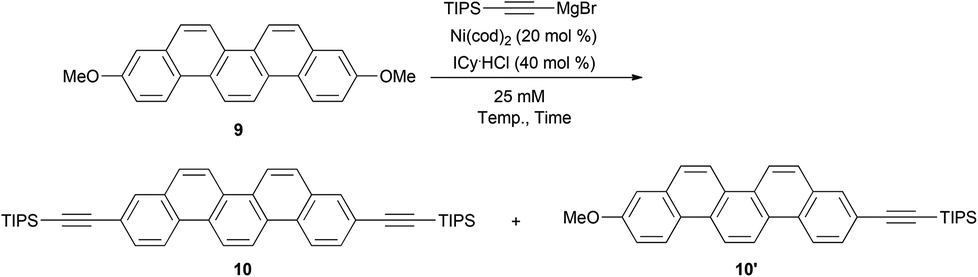
According to the original reaction conditions,37 a mixture of 9 (1.0 equiv.) and triisopropylsilylethynylmagnesium bromide (4.0 equiv.) was heated in 1,4-dioxane (500 mM) at 120 °C for 18 h in the presence of Ni(cod)2 (20 mol%) and ICy·HCl (40 mol%) as the catalyst system (ICy = 1,3-dicyclohexylimidazol-2-ylidene). Unfortunately, however, no desired product 10 was detected and all the substrates were recovered. At a lower concentration (25 mM), the reaction proved similarly unsuccessful (entry 1). In an attempt to identify effective solvents, triglyme, NMP, diphenyl ether, and mesitylene were also evaluated (entries 2–5). To our delight, when mesitylene was used as the solvent, the desired doubly alkynylated product 10 was formed in 47% NMR yield (entry 5). Further optimization of the reaction conditions revealed that reaction at 150 °C gave 10 up to 70% yield (entries 6–8). Consequently, the best result was obtained by the reaction at 140 °C for 72 h; the desired product was obtained in 76% isolated yield (entry 9).
When compound 10 was treated with nBu4NF, diethynylpicene (11) was generated in 91% yield.38
We next screened the reaction conditions for alkyne alkylation by using nBuLi followed by the addition of alkyl bromides in THF.39 When the reaction temperature was increased and 20 mol% of TBAI was added, a wide range of alkylated products 12 were obtained in 46–62% yields (Scheme 3).
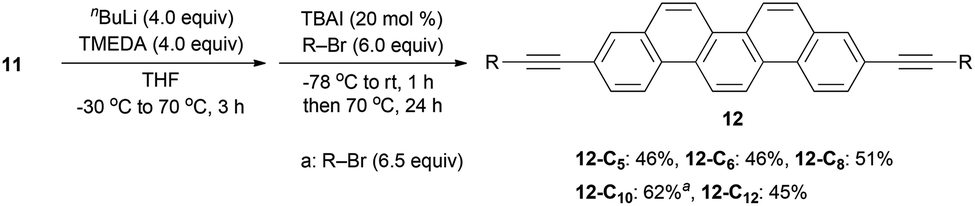 | ||
| Scheme 3 Alkylation of 11. | ||
After obtaining successful alkylation of 11 with different types of alkylating agents, we proceeded to hydrogenate the products to complete the synthesis of alkylpicenes (Scheme 4).40 Interestingly, when using atmospheric pressures of hydrogen gas for hydrogenation we observed low reactivity for substrates bearing longer alkyl chains, and found that a higher reaction temperature (70 °C) was necessary. Gratifyingly, a slight alteration of the reaction conditions (e.g., reaction temperatures and times) afforded full conversion of 12 and reasonable yields of the desired products 8.
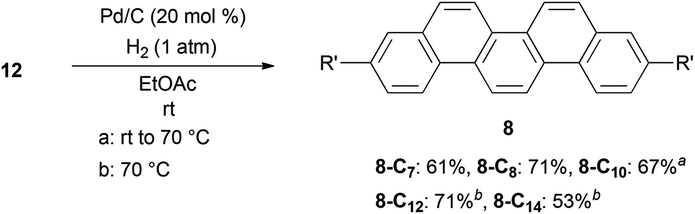 | ||
| Scheme 4 Synthesis of alkylpicenes 8via hydrogenation of 12. | ||
The solubility of a series of alkylpicenes 8 was determined to confirm the solubilizing effect of alkyl substituents. Although the parent picene was almost insoluble in chloroform at room temperature, introduction of the alkyl groups did enhance the solubility (2.9 g L−1 for 8-C7, 3.1 g L−1 for 8-C8, 4.0 g L−1 for 8-C10, 1.1 g L−1 for 8-C12, and 0.4 g L−1 for 8-C14, respectively). We observed that the solubility of picenes with longer alkyl chains (C12 and C14) decreased once the number of carbon atoms in the chain exceeds ten. We believe this is due to the hydrophobic interaction between the alkyl chains, which typically enhance an intermolecular interaction, leading to the lower solubility. This is the so-called “fastener effect”.41–43
Conclusions
In summary, we have developed a new synthetic route to an array of alkyl-substituted picenes via C–O bond activation/alkynylation sequences. This sequence successfully delivered five dialkylated picenes in yields up to 71%. A further elucidation of the physical properties of these compounds, in particular their FET characteristics, is presently underway in our laboratory.Experimental section
General
All reactions were carried out under an Ar atmosphere using standard Schlenk techniques. Glassware was dried in an oven (150 °C) and heated under reduced pressure prior to use. Solvents employed as eluents and other routine operations, as well as anhydrous solvents were purchased from commercial suppliers and employed without any further purification. For thin layer chromatography (TLC) analyses throughout this work, Merck precoated TLC plates (silica gel 60 GF254, 0.25 mm) were used. Silica gel column chromatography was carried out using Silica gel 60 N (spherical, neutral, 40–100 μm) from Kanto Chemicals Co., Ltd. The 1H and 13C{1H} NMR spectra were recorded on Mercury-300 (300 MHz), 400-NMR ASW (400 MHz) and Varian INOVA-600 (600 MHz) spectrometers. Infrared spectra were recorded on a Shimadzu IR Prestige-21 spectrophotometer. GC analyses were performed on a Shimadzu GC-14A equipped with a flame ionization detector using Shimadzu Capillary Column (CBP1-M25-025) and Shimadzu C-R6A-Chromatopac integrator. GC/MS analyses were carried out on a SHIMADZU GC-17A equipped with a SHIMADZU QP-5050 GC-MS system. High-resolution mass spectra (HRMS) analyses were carried out on a JEOL JMS-700 MStation. Elemental analyses were carried out with a Perkin-Elmer 2400 CHN elemental analyser at Okayama University.1,4-Dichloro-2,3-diiodobenzene (6) and 3,10-dimethoxypicene (9) were synthesized according to the literature.18
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), followed by washing with hexane to afford the desired product 10 (287 mg, 0.45 mmol) in 76% yield as a white solid. Mp: 242–243 °C. FT-IR (neat, cm−1): 2943 (s), 2889 (s), 2864 (s), 1593 (w), 1462 (s), 1382 (w), 1244 (m), 997 (m), 951 (s), 883 (s), 804 (s), 748 (s), 685 (s). 1H NMR (CDCl3, 600 MHz, rt): δ 1.20 (s, 42H), 7.78 (dd, J = 8.6 and 1.7 Hz, 2H), 7.95 (d, J = 9.1 Hz, 2H), 8.14 (d, J(long-range) = 1.6 Hz, 2H), 8.73 (d, J = 8.2 Hz, 4H), 8.85 (s, 2H); 13C{1H} NMR (CDCl3, 151 MHz, rt) δ 11.6, 18.9, 91.9, 107.3, 121.9, 122.0, 122.4, 123.3, 127.3, 128.7, 129.1, 130.08, 130.12, 131.7, 132.4; 29Si{1H} NMR (CDCl3, 119 MHz, rt) δ −1.63. Anal. Calcd for C44H54Si2: C, 82.69; H, 8.52%. Found: C, 82.47; H, 8.28%.
:1), followed by washing with hexane to afford the desired product 10 (287 mg, 0.45 mmol) in 76% yield as a white solid. Mp: 242–243 °C. FT-IR (neat, cm−1): 2943 (s), 2889 (s), 2864 (s), 1593 (w), 1462 (s), 1382 (w), 1244 (m), 997 (m), 951 (s), 883 (s), 804 (s), 748 (s), 685 (s). 1H NMR (CDCl3, 600 MHz, rt): δ 1.20 (s, 42H), 7.78 (dd, J = 8.6 and 1.7 Hz, 2H), 7.95 (d, J = 9.1 Hz, 2H), 8.14 (d, J(long-range) = 1.6 Hz, 2H), 8.73 (d, J = 8.2 Hz, 4H), 8.85 (s, 2H); 13C{1H} NMR (CDCl3, 151 MHz, rt) δ 11.6, 18.9, 91.9, 107.3, 121.9, 122.0, 122.4, 123.3, 127.3, 128.7, 129.1, 130.08, 130.12, 131.7, 132.4; 29Si{1H} NMR (CDCl3, 119 MHz, rt) δ −1.63. Anal. Calcd for C44H54Si2: C, 82.69; H, 8.52%. Found: C, 82.47; H, 8.28%.
General procedure for the alkylation of 11
:1) to afford 12-C5 (21.5 mg, 0.046 mmol) in 46% yield as a white solid. Mp: 243–244 °C. FT-IR (neat, cm−1): 2953 (m), 2930 (s), 2860 (m), 1467 (m), 1445 (w), 1274 (m), 887 (m), 810 (s), 750 (m). 1H NMR (CDCl3, 600 MHz, rt): δ 0.97 (t, J = 7.5 Hz, 6H), 1.39–1.45 (m, 4H), 1.49–1.54 (m, 4H), 1.70 (quin, J = 7.2 Hz, 4H), 2.50 (t, J = 6.9 Hz, 4H), 7.70 (dd, J = 8.4 and 1.8 Hz, 2H), 7.91 (d, J = 9.0 Hz, 2H), 8.03 (d, J(long-range) = 1.2 Hz, 2H), 8.69 (d, J = 9.0 Hz, 4H), 8.80 (s, 2H); 13C{1H} NMR (CDCl3, 151 MHz, rt) δ 14.2, 19.7, 22.4, 28.7, 31.4, 80.8, 91.8, 121.9, 122.2, 122.4, 123.2, 127.2, 128.6, 128.8, 129.6, 129.9, 131.6, 131.8. HRMS (EI) Calcd for C36H34: 466.2661. Found: 466.2651.
General procedure for hydrogenation of 8
Acknowledgements
This study was partly supported by the Program for Promoting the Enhancement of Research Universities from MEXT and a Special Project of Okayama University. We gratefully thank Professor Mamoru Tobisu at Osaka University for helpful discussions and Ms. Megumi Kosaka and Mr Motonari Kobayashi at the Department of Instrumental Analysis, Advanced Science Research Center, Okayama University, for the elemental analyses, and the SC-NMR Laboratory of Okayama University for the NMR spectral measurements.Notes and references
- A. Pron, P. Gawrys, M. Zagorska, D. Djurado and R. Demadrille, Chem. Soc. Rev., 2010, 39, 2577–2632 RSC.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- R. A. Street, Adv. Mater., 2009, 21, 2007–2022 CrossRef CAS.
- Y. Kubozono, X. He, S. Hamao, K. Teranishi, H. Goto, R. Eguchi, T. Kambe, S. Gohda and Y. Nishihara, Eur. J. Inorg. Chem., 2014, 3806–3819 CrossRef CAS.
- H. Klauk, Chem. Soc. Rev., 2010, 39, 2643–2666 RSC.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- H. Akamatsu, H. Inokuchi and Y. Matsunaga, Nature, 1954, 173, 168–169 CrossRef CAS.
- A. Tsumura, H. Koezuka and T. Ando, Appl. Phys. Lett., 1986, 49, 1210–1212 CrossRef CAS PubMed.
- M. Bendikov, F. Wudl and D. F. Prepichka, Chem. Rev., 2004, 104, 4891–4945 CrossRef CAS PubMed.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980–4986 CrossRef CAS.
- H. Klauk, U. Zschieschang, R. T. Weitz, H. Meng, F. Sun, G. Nunes, D. E. Keys, C. R. Fincher and Z. Xiang, Adv. Mater., 2007, 19, 3882–3887 CrossRef CAS.
- H. Okamoto, R. Eguchi, S. Hamao, H. Goto, K. Gotoh, Y. Sakai, M. Izumi, Y. Takaguchi, S. Gohda and Y. Kubozono, Sci. Rep., 2014, 4, 5330 CAS.
- H. Okamoto, N. Kawasaki, Y. Kaji, Y. Kubozono, A. Fujiwara and M. Yamaji, J. Am. Chem. Soc., 2008, 130, 10470–10471 CrossRef CAS PubMed.
- N. Kawasaki, Y. Kubozono, H. Okamoto, A. Fujiwara and M. Yamaji, Appl. Phys. Lett., 2009, 94, 043310 CrossRef PubMed.
- N. H. Chang, X. C. Chen, H. Nonobe, Y. Okuda, H. Mori, K. Nakajima and Y. Nishihara, Org. Lett., 2013, 15, 3558–3561 CrossRef CAS PubMed.
- H. Mori, X. C. Chen, N. H. Chang, S. Hamao, Y. Kubozono, K. Nakajima and Y. Nishihara, J. Org. Chem., 2014, 79, 4973–4983 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- K. Hyodo, H. Nonobe, S. Nishinaga and Y. Nishihara, Tetrahedron Lett., 2014, 55, 4002–4005 CrossRef CAS PubMed.
- Y. Nakano, M. Saito and H. Nakamura, PCT/Japan Patent Kokai, WO 2010-016511, 2010 Search PubMed.
- H. Okamoto, S. Hamao, H. Goto, Y. Sakai, M. Izumi, S. Gohda, Y. Kubozono and R. Eguchi, Sci. Rep., 2014, 4, 5048 CAS.
- F. Kakiuchi, M. Usui, S. Ueno, N. Chatani and S. Murai, J. Am. Chem. Soc., 2004, 126, 2706–2707 CrossRef CAS PubMed.
- S. Ueno, E. Mizushima, N. Chatani and F. Kakiuchi, J. Am. Chem. Soc., 2006, 128, 16516–16517 CrossRef CAS PubMed.
- M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866–4869 CrossRef CAS PubMed.
- T. Shimasaki, Y. Konno, M. Tobisu and N. Chatani, Org. Lett., 2009, 11, 4890–4892 CrossRef CAS PubMed.
- M. Tobisu, T. Shimasaki and N. Chatani, Chem. Lett., 2009, 38, 710–711 CrossRef CAS.
- M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946–2948 RSC.
- M. Tobisu, A. Yasutome, K. Yamakawa, T. Shimasaki and N. Chatani, Tetrahedron, 2012, 68, 5157–5161 CrossRef CAS PubMed.
- M. Tobisu, A. Yasutome, H. Kinuta, K. Nakamura and N. Chatani, Org. Lett., 2014, 16, 5572–5575 CrossRef CAS PubMed.
- D. G. Yu, B. J. Li and Z. J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS PubMed.
- B. J. Li, D. G. Yu, C. L. Sun and Z. J. Shi, Chem. – Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed.
- F. Zhao, D.-G. Yu, R.-Y. Zhu, Z. Xi and Z.-J. Shi, Chem. Lett., 2011, 40, 1001–1003 CrossRef CAS.
- F. Zhao, Y.-F. Zhang, J. Wen, D.-G. Yu, J.-B. Wei, Z. Xi and Z.-J. Shi, Org. Lett., 2013, 15, 3230–3233 CrossRef CAS PubMed.
- C. Wang, T. Ozaki, R. Takita and M. Uchiyama, Chem. – Eur. J., 2012, 18, 3482–3485 CrossRef CAS PubMed.
- T. Iwasaki, Y. Miyata, R. Akimoto, Y. Fujii, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2014, 136, 9260–9263 CrossRef CAS PubMed.
- M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Org. Lett., 2015, 17, 680–683 CrossRef CAS PubMed.
- M. Rawat, V. Prutyanov and W. D. Wulff, J. Am. Chem. Soc., 2006, 128, 11044–11053 CrossRef CAS PubMed.
- M. Buck and J. M. Chong, Tetrahedron Lett., 2001, 42, 5825–5827 CrossRef CAS.
- J. E. Bullock, R. Carmieli, S. M. Mickley, J. Vura-Weis and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 11919–11929 CrossRef CAS PubMed.
- H. Inokuchi, G. Saito, P. Wu, K. Seki, T. B. Tang, T. Mori, K. Imaeda, T. Enoki, Y. Higuchi, K. Inaka and N. Tasuoka, Chem. Lett., 1986, 1263–1266 CrossRef CAS.
- I. Doi, E. Miyazaki, K. Takimiya and Y. Kunugi, Chem. Mater., 2007, 19, 5230–5237 CrossRef CAS.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and the characterization data of the new compounds. See DOI: 10.1039/c5qo00039d |
| This journal is © the Partner Organisations 2015 |