
Copper-catalyzed stereoselective oxytrifluoromethylation of propargyl amides for the construction of oxazolines†
Yanan
Wang
,
Min
Jiang
and
Jin-Tao
Liu
*
Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: jtliu@sioc.ac.cn; Web: http://www.sioc.ac.cn Fax: +(86)-21-64166128
First published on 13th March 2015
Abstract
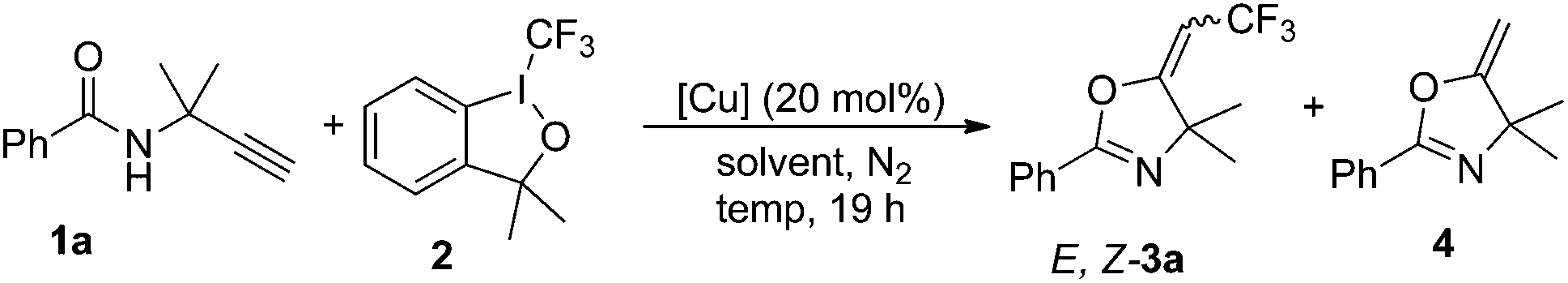
The copper-catalyzed stereoselective oxytrifluoromethylation of terminal propargyl amides took place readily in the presence of Togni's reagent under mild conditions, giving the corresponding trifluoromethylated oxazoline derivatives in moderate to good yields. Using CuBr as catalyst, the E-isomer was obtained as major product. However, the formation of Z-isomer with excellent stereoselectivity was also achieved while Cu(CH3CN)4PF6 was used. Plausible pathways are proposed to account for the excellent stereoselectivity of the reaction.
Introduction
Oxazolines have attracted significant attention due to their abundant presence in pharmaceutically active natural products,1 and have been utilized as useful synthetic intermediates or reagents.2 In addition, they can be used as protecting groups3 and function as valuable catalyst ligands in asymmetric synthesis.4 Nowadays, several versatile methods for the syntheses of oxazolines have been reported, especially the transition metal promoted ones.5 The introduction of a trifluoromethyl group into organic compounds may profoundly change their properties.6 Since the trifluoromethyl group has a significant impact on the electronegativity, hydrophobicity, metabolic activity, and bioavailability of molecule, trifluoromethylated compounds, especially heterocycles, have gained more and more interest in a wide range of applications, such as pharmaceuticals and agrochemicals.7In the last decade, various transition metal-mediated or catalyzed trifluoromethylation reactions have been developed to synthesize trifluoromethyl-containing compounds.8 Very recently, several groups were interested in the trifluoromethylation of alkynes.9–12 For example, the Ding, Fu and Xiao groups have independently developed a copper-catalyzed tandem trifluoromethylarylation/endo-cyclization of internal alkynes [Scheme 1, eqn (1)].10 At the same time, the copper-catalyzed trifluoromethylarylation/exo-cyclization of terminal alkynes was successfully developed in our laboratory [Scheme 1, eqn (2)].11 Hou et al. also reported the syntheses of 4-trifluoromethyl-2,3-dihydropyrroliums through the trifluoromethylation of homopropargyl amines with Umemoto's reagent [Scheme 1, eqn (3)].12 However, to the best of our knowledge, the study on oxytrifluoromethylation of alkynes is quite rare. As part of our continuing interest in trifluoromethylation of unsaturated alkanes,8y,11 we report here a novel copper-catalyzed stereoselective oxytrifluoromethylation of alkynes for the construction of oxazolines [Scheme 1, eqn (4)].
 | ||
| Scheme 1 Previous work and this work. | ||
Results and discussion
On the basis of our previous work, terminal alkyne 1a containing two methyl groups at the propargyl position was chosen as the substrate due to the Thorpe–Ingold effect.13 The initial reaction was carried out with 1.0 equivalent of Togni reagent I (2), 1.5 equivalent of 1a and 20 mol% of CuBr in dichloromethane (DCM) at 45 °C in a sealed tube. Gratifyingly, the corresponding oxazolines E-3a and Z-3a were obtained as a mixture of two isomers in 51% overall yield with a ratio of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table 1, entry 1). The stereochemistry of the newly formed double bond in the major product was established as E-configuration by NOE experiments. Inspired by the result, we screened different conditions to find a suitable protocol for the selective formation of oxazolines. First, we focused on the catalyst effects. In the CuTc or CuBr2-catalyzed reaction, E-3a was also obtained as major product with lower selectivity (entries 2 and 3). However, copper salt Cu(CH3CN)4PF6 was unexpectedly favored to give Z-3a with excellent stereoselectivity (E:Z < 1:20, entry 4). It is worth noting that cyclic compound 4 could be observed in this reaction. Other copper catalysts, such as CuI, Cu(OAc)2 and CuCN, afforded low yield of 3a, albeit with high selectivity for Z-isomer (entries 5–7). Then solvent effect was studied using CuBr and Cu(CH3CN)4PF6 as the catalysts. Various solvents such as CH3CN, DMSO and DMF were screened to examine the feasibility of the reaction with CuBr as catalyst (entries 8–10). It was found that the reaction in DMF proceeded well to give E-3a with high selectivity in good yield (E:Z > 20:1, 77%), and decreasing the temperature to room temperature was beneficial for this reaction (entry 11). While using DMA as solvent, the yield of 3a could be improved to 86% in 3 h (entry 12). A slightly lower yield was obtained with 10 mol% of CuBr (entry 13). Interestingly, only a trace of compound 4 was detected in the reaction systems when the ratio of E,Z-isomers was greater than 20:1. As for the Cu(CH3CN)4PF6-catalyzed reaction, DCM was still the best solvent, affording Z-3a with excellent stereoselectivity (entry 14). It was exciting to find that the yield of 3a was improved when increasing the temperature to 60 °C (entry 15). The catalyst loading could be lowered to 5 mol% without any reduction in yield and stereoselectivity, and the reaction time could also be reduced to 9 h (entry 16). Carrying out the reaction under air atmosphere, 67% of E-3a was obtained in the presence of CuBr, whereas no expected product was detected in the Cu(CH3CN)4PF6-catalyzed reaction (entries 17 and 18). A control experiment showed that copper is essential for the reaction as no reaction was detected in the absence of a copper salt (entry 19).
:1 (Table 1, entry 1). The stereochemistry of the newly formed double bond in the major product was established as E-configuration by NOE experiments. Inspired by the result, we screened different conditions to find a suitable protocol for the selective formation of oxazolines. First, we focused on the catalyst effects. In the CuTc or CuBr2-catalyzed reaction, E-3a was also obtained as major product with lower selectivity (entries 2 and 3). However, copper salt Cu(CH3CN)4PF6 was unexpectedly favored to give Z-3a with excellent stereoselectivity (E:Z < 1:20, entry 4). It is worth noting that cyclic compound 4 could be observed in this reaction. Other copper catalysts, such as CuI, Cu(OAc)2 and CuCN, afforded low yield of 3a, albeit with high selectivity for Z-isomer (entries 5–7). Then solvent effect was studied using CuBr and Cu(CH3CN)4PF6 as the catalysts. Various solvents such as CH3CN, DMSO and DMF were screened to examine the feasibility of the reaction with CuBr as catalyst (entries 8–10). It was found that the reaction in DMF proceeded well to give E-3a with high selectivity in good yield (E:Z > 20:1, 77%), and decreasing the temperature to room temperature was beneficial for this reaction (entry 11). While using DMA as solvent, the yield of 3a could be improved to 86% in 3 h (entry 12). A slightly lower yield was obtained with 10 mol% of CuBr (entry 13). Interestingly, only a trace of compound 4 was detected in the reaction systems when the ratio of E,Z-isomers was greater than 20:1. As for the Cu(CH3CN)4PF6-catalyzed reaction, DCM was still the best solvent, affording Z-3a with excellent stereoselectivity (entry 14). It was exciting to find that the yield of 3a was improved when increasing the temperature to 60 °C (entry 15). The catalyst loading could be lowered to 5 mol% without any reduction in yield and stereoselectivity, and the reaction time could also be reduced to 9 h (entry 16). Carrying out the reaction under air atmosphere, 67% of E-3a was obtained in the presence of CuBr, whereas no expected product was detected in the Cu(CH3CN)4PF6-catalyzed reaction (entries 17 and 18). A control experiment showed that copper is essential for the reaction as no reaction was detected in the absence of a copper salt (entry 19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | Solvent | T (°C) |
3a (E:Z) |
Yieldb (%) |
| a Reaction conditions: 1a (0.15 mmol), 2 (0.10 mmol), copper catalyst (0.02 mmol), solvent (1 mL), 45 °C, 19 h, under N2. b Determined by 19F NMR spectroscopy using hexafluorobenzene as internal standard. c The reaction time was 3 h. d 10 mol% of CuBr was used. e 5 mol% of Cu(CH3CN)4PF6 was used. f The reaction time was 9 h. g The reaction was carried out under air atmosphere. h ND = Not detected. i NR = No reaction. | |||||
| 1 | CuBr | DCM | 45 | 12:1 |
51 |
| 2 | CuTc | DCM | 45 | 7:1 |
41 |
| 3 | CuBr2 | DCM | 45 | 5:1 |
45 |
| 4 | Cu(CH3CN)4PF6 | DCM | 45 | <1:20 |
50 |
| 5 | CuI | DCM | 45 | <1:20 |
23 |
| 6 | Cu(OAc)2 | DCM | 45 | <1:20 |
33 |
| 7 | CuCN | DCM | 45 | — | 6 |
| 8 | CuBr | CH3CN | 45 | 5:1 |
37 |
| 9 | CuBr | DMSO | 45 | >20:1 |
43 |
| 10 | CuBr | DMF | 45 | >20:1 |
77 |
| 11 | CuBr | DMF | rt | >20:1 |
80 |
| 12c | CuBr | DMA | rt | >20:1 |
86 |
| 13d | CuBr | DMA | rt | >20:1 |
77 |
| 14 | Cu(CH3CN)4PF6 | DMF | 45 | 8:1 |
27 |
| 15 | Cu(CH3CN)4PF6 | DCM | 60 | <1:20 |
67 |
| 16e,f | Cu(CH3CN)4PF6 | DCM | 60 | <1:20 |
69 |
| 17g | CuBr | DMA | rt | >20:1 |
67 |
| 18g | Cu(CH3CN)4PF6 | DCM | 60 | — | NDh |
| 19 | — | DCM | 45 | — | NRi |
Next, we studied the scope of propargyl amides for the CuBr-catalyzed oxytrifluoromethylation reaction (Condition A). As shown in Table 2, various amides 1 including aromatic amides and aliphatic amides could react with 2 to give products E-3 with excellent stereoselectivities. Both electron-rich and electron-deficient amides are compatible with the reaction conditions. The substrates bearing methyl or methoxy substituents gave the expected products 3b–3f in high yields. However, the reaction of 2,4,6-trimethyl substituted propargyl amide afforded only 25% yield of 3g due to steric hindrance. Halide-substituted substrates also reacted well to give the corresponding products in good yields (3h–3k). In the case of an iodo-substituted substrate, the iodo substituent remained intact under the reaction conditions, and product 3k was obtained in 71% yield. Furthermore, some important functional groups such as ester, amine and nitro group were tolerated in this oxytrifluoromethylation reaction. Propargyl amides containing other substituents, such as naphthyl, (E)-styryl, heteroaromatic ring and alkyl, could also react with 2 under the standard conditions, giving the corresponding products 3o–3u in moderate yields. Lower yield was obtained when the two methyl substituents at the propargylic position were replaced by a cyclohexyl group.
| a Reaction condition A: 1 (0.18 mmol), 2 (0.12 mmol), CuBr (0.024 mmol), DMA (1.2 mL), rt, 3 h, under N2, isolated yield. b Reaction condition B: 1 (0.18 mmol), 2 (0.12 mmol), Cu(CN3CN)4PF6 (0.006 mmol), DCM (1.2 mL), 60 °C, 9 h, under N2, isolated yield. c The reaction was conducted at 80 °C for 19 h. d Reaction time was 25 h. e Reaction time was 19 h. f Reaction time was 30 h. g The reaction was conducted at 80 °C. |
|---|

|
Propargyl amides 1 were also evaluated in the Cu(CH3CN)4PF6-catalyzed reaction and the results are summarized in Table 2. Gratifyingly, the reaction was compatible with a variety of commonly encountered functional groups such as methyl (3b–3d, 3g), methoxyl (3e–3f), halide (3h–3k), ester (3l), and amine (3m), giving the corresponding products in generally moderate-to-good yields. In each reaction, excellent stereoselectivity was achieved with the Z-isomer as the major product (E:Z < 1:20). The electronic nature of substituents had some influence on the yield of the reaction. Compared with halide and ether-substituted compounds, the yields of 3 containing methyl and methoxy groups were slightly higher. Compound 3m with a nitro-substituent was not obtained in the Cu(CH3CN)4PF6-catalyzed reaction. High stereoselectivities for naphthyl, (E)-styryl, heteroaromatic ring or alkyl-containing products 3o–3v could also be achieved, albeit in lower yield along with the formation of hydrotrifluoromethylation byproducts.
We further examined the stereoselective oxytrifluoromethylation of estrone derivative 1w. As expected, E- and Z-3w were obtained as major products with excellent stereoselectivities in 75% and 66% yield when CuBr and Cu(CH3CN)4PF6 was used as catalyst, respectively (Scheme 2).
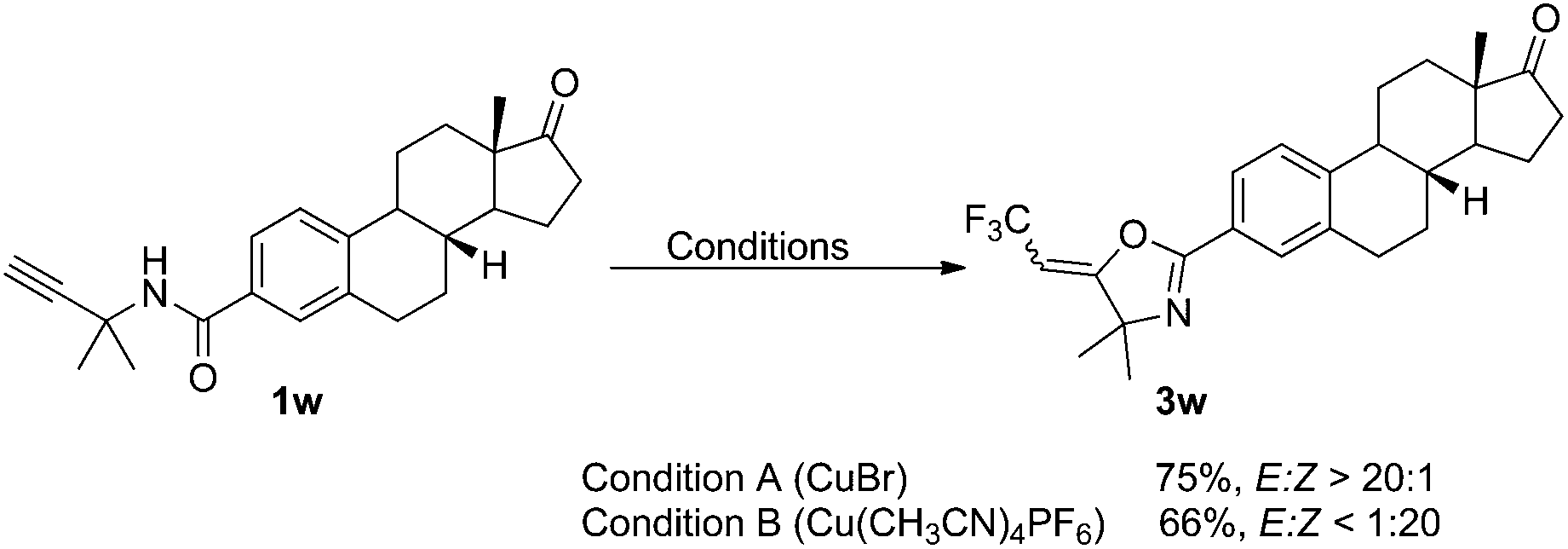 | ||
| Scheme 2 Oxytrifluoromethylation of estrone derivative. | ||
To further explore the scope of this reaction, we also tried the reactions of 2 and alkynes 1x–1z (Scheme 3). Unfortunately, 1x only afforded trace amounts of the hydrotrifluoromethylation product under condition B, and the reactions did not occur in the cases of alkyne 1y and 1z with full recovery of the starting materials under both reaction conditions.
 | ||
| Scheme 3 Other alkyne substrates investigated. | ||
To gain insight into the reaction mechanism, we performed the reaction of 1a and 2 in the presence of 1.0 equivalent of 2,6-di-tert-butyl-4-methylphenol (BHT). The CuBr-catalyzed reaction gave 3a in 78% overall yield, whereas the Cu(CH3CN)4PF6-catalyzed trifluoromethylation reaction was inhibited and 73% of oxazoline 4 was obtained (Scheme 4).
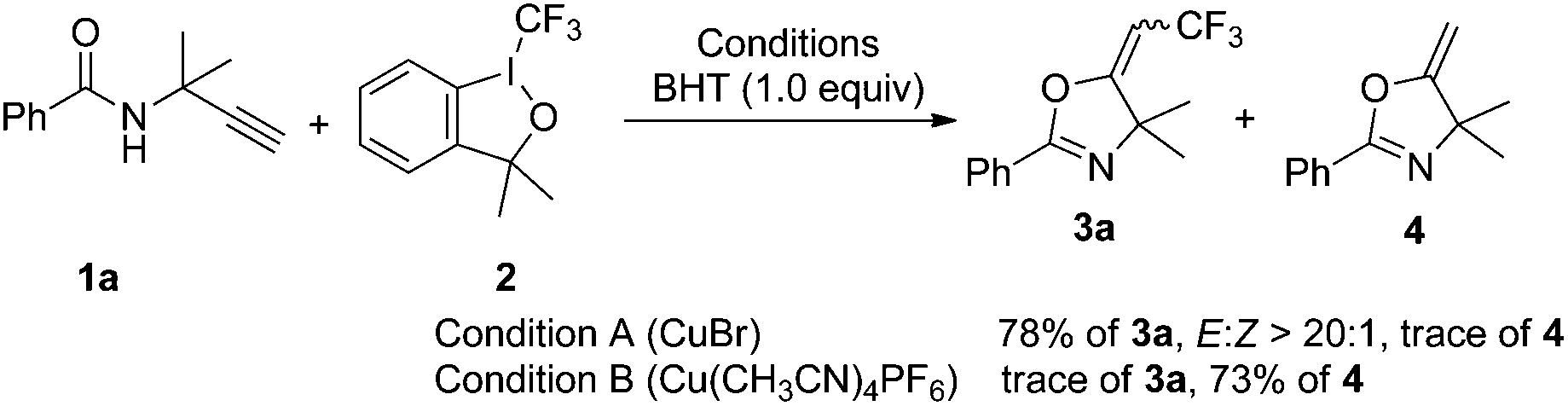 | ||
| Scheme 4 Reactions of 1a with 2 in the presence of BHT. | ||
We then monitored the Cu(CH3CN)4PF6-catalyzed reaction by NMR spectroscopy. It was found that a large amount of cyclic compound 4 was formed after 2 h, and then the yield of desired product 3 increased gradually along with a decrease of amount of 4 (see Table S-1 in ESI†).
Moreover, the trifluoromethylation of compound 4 was investigated under the two standard conditions. To our surprise, 19F NMR spectroscopy showed that product Z-3a was obtained in 56% and 68% yield, respectively, and only a trace amount of E-3a was detected in the presence of CuBr or Cu(CH3CN)4PF6, partially due to the steric hindrance of the two methyl groups (Scheme 5). To further study the reaction of 4 and Togni I, 1.0 equivalent of BHT was added in the CuBr-catalyzed reaction of 4 and 20% of Z-3a was produced. A single-electron-transfer (SET) mechanism for the direct C–H trifluoromethylation of alkene has already been proposed in the literatures.14 However, according to our experimental results, the copper-catalyzed trifluoromethylation of 4 could also take place through cationic process.
 | ||
| Scheme 5 Reactions of 4 under the standard conditions. | ||
We also performed deuterium labeling studies with deuterated alkyne 1a-d (D = 93%) (Scheme 6). Under condition A, the reaction of 1a-d with 2 afforded product E-3a-d with 92% D content at the alkenyl position based on the corresponding 1H NMR spectroscopic data. However, only 50% D content of Z-3a-d was isolated from the reaction under condition B (see the ESI†).
 | ||
| Scheme 6 Deuterium labeling experiments. | ||
Based on the above control experiments, deuterium labeling experiments and the reported work about the copper-catalyzed cyclization of 1a,15 plausible pathways for the formation of oxazoline derivatives 3 are proposed as outlined in Scheme 7. In the CuBr-catalyzed reactions, the interaction of 2 and Cu-catalyst gives iodine(III) species A, which coordinates with carbon–carbon triple bond in alkyne 1 to produce intermediate B (path A).16 Then the carbonyl-oxygen attacks stereoselectively the π-coordinated alkyne from the backside to give the E-product. On the other hand, hypervalent iodine reagents are known to oxidize transition metals easily to their higher oxidation states. Therefore, we cannot rule out the possibility that E-alkene copper intermediate C is formed and further oxidized by Togni I to give a Cu(III)–CF3 complex, which undergoes reductive elimination to afford E-product with the regeneration of copper(I). In the presence of Cu(CH3CN)4PF6, the interaction of 2 and Cu catalyst gives trifluoromethyl radical as evidenced by the inhibiting experiment (path B), and generates the copper(II) species at the same time. The trifluoromethyl radical reacts with the carbon–carbon triple bond in propargyl amide to give intermediate D, which may be quickly converted into its cationic counterpart E by the oxidation of a Cu(II) species. In this step, intermediate D with E-configuration is more favored11 and formed predominantly due to steric effects. Thus, a tandem trifluoromethylation/cyclization process would occur to give Z-3 as the major product. Meanwhile, compound 4 may be formed through intermediate F. In Moran's report,15 the procedure for the formation of 4 includes the proton transfer and protonation. Then the trifluoromethylation of 4 occurs to afford Z-3 stereoselectively under the reaction conditions (path C).
 | ||
| Scheme 7 Proposed pathways for the oxytrifluoromethylation. | ||
Condition A may disfavor the production of cyclic compound 4 and favor the formation of intermediate B to give Z-3 as major isomer, although the reasons are not clear at present. In the Cu(CH3CN)4PF6-catalyzed system, both path B and path C may be involved at the same time and path C is the main process on the basis of the result of the control experiments in Table S-1† and deuterium labeling experiments.
Conclusions
In summary, we have developed a highly stereoselective copper-catalyzed oxytrifluoromethylation of propargyl amides. A series of trifluoromethylated oxazolines with different configuration could be obtained with excellent stereoselectivities under mild conditions by choosing an appropriate copper catalyst. We believe that these fluorinated compounds are useful structural motifs and will find application in the synthesis of bioactive compounds. Plausible mechanisms are also proposed on the basis of experimental results.Acknowledgements
We thank the National Natural Science Foundation of China for financial support (no. 21172243 and 21421002).Notes and references
- (a) J. P. Genet, S. Thorimbert and A. M. Touzin, Tetrahedron Lett., 1993, 34, 1159 CrossRef CAS; (b) P. Wipf and S. Venkatraman, Synlett, 1997, 1 CrossRef CAS PubMed; (c) Q. Li, K. W. Woods, A. Claiborne, S. L. Gwaltney, K. J. Barr, G. Liu, L. Gehrke, R. B. Credo, Y. Hua Hui, J. Lee, R. B. Warner, P. Kovar, M. A. Nukkala, N. A. Zielinski, S. K. Tahir, M. Fitzgerald, K. H. Kim, K. Marsh, D. Frost, S.-C. Ng, S. Rosenberg and H. L. Sham, Bioorg. Med. Chem. Lett., 2002, 12, 465 CrossRef CAS; (d) I. Mohammadpoor-Baltork, A. R. Khosropour and S. F. Hojati, Synlett, 2005, 2747 CrossRef CAS PubMed.
- (a) T. G. Gant and A. I. Meyers, Tetrahedron, 1994, 50, 2297 CrossRef CAS; (b) D. Yang, Y.-C. Yip and X.-C. Wang, Tetrahedron Lett., 1997, 38, 7083 CrossRef CAS; (c) B. H. Lipshutz, Chem. Rev., 1986, 86, 795 CrossRef CAS; (d) H. H. Wasserman, K. E. McCarthy and K. S. Prowse, Chem. Rev., 1986, 86, 845 CrossRef CAS.
- (a) H. Vorbruggen and K. Krolikiewicz, Tetrahedron, 1993, 49, 9353 CrossRef; (b) T. W. Greene and P. G. M. Wutz, in Protecting Groups in Organic Synthesis, John Wiley and Sons, New York, 2nd edn, 1991 Search PubMed.
- (a) H. Nishiyama, H. Sakaguchi, T. Nakamura, M. Horihata, M. Kondo and K. Itoh, Organometallics, 1989, 8, 846 CrossRef CAS; (b) Y. Jiang, Q. Jiang, G. Zhu and X. Zhang, Tetrahedron Lett., 1997, 38, 215 CrossRef CAS; (c) H. A. McManus, S. M. Barry, P. G. Andersson and P. J. Guiry, Tetrahedron, 2004, 60, 3405 CrossRef CAS PubMed; (d) A. Lee, W. Kim, J. Lee, T. Hyeon and B. M. Kim, Tetrahedron: Asymmetry, 2004, 15, 2595 CrossRef CAS PubMed; (e) H. Nishiyama, S. B. Park and K. Itoh, Tetrahedron: Asymmetry, 1992, 3, 1029 CrossRef CAS; (f) A. Gissibl, M. G. Finn and O. Reiser, Org. Lett., 2005, 7, 2325 CrossRef CAS PubMed.
- (a) A. S. K. Hashmi, K. Stephen, A. M. Schuster and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8247 ( Angew. Chem. , 2009 , 121 , 8396 ) CrossRef CAS PubMed; (b) M. Harmata and C.-F. Huang, Synlett, 2008, 1399 CrossRef CAS PubMed; (c) G. C. Senadi, W. P. Hu, J. S. Hsiao, J. K. Vandavasi, C. Y. Chen and J. J. Wang, Org. Lett., 2012, 14, 4478 CrossRef CAS PubMed.
- (a) T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS PubMed; (b) G. K. S. Prakash and A. K. Yudin, Chem. Rev., 1997, 97, 757 CrossRef CAS PubMed; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS PubMed; (d) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (e) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS; (f) Biomedical Frontiers of Fluorine Chemistry, ed. I. Ojima, J. R. McCarthy and J. T. Welch, American Chemical Society, Washington, 1996 Search PubMed; (g) Organofluorine Compounds in Medicinal Chemistry and Biochemical Applications, ed. R. Filler, Y. Kobayashi and L. Agupolskii, Elsevier, Amsterdam, 1993 Search PubMed; (h) H.-J. Bohm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed; (i) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 ( Angew. Chem. , 2012 , 124 , 5134 ) CrossRef CAS PubMed; (j) A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 ( Angew. Chem. , 2012 , 124 , 9082 ) CrossRef CAS PubMed; (k) X. Wang, Y. Zhang and J. Wang, Sci. Sinica Chim., 2012, 42, 1417 CrossRef CAS PubMed; (l) B. E. Smart, Chem. Rev., 1996, 96, 1555 CrossRef CAS PubMed; (m) M. Hird, Chem. Soc. Rev., 2007, 36, 2070 RSC.
- (a) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, Wiley-Blackwell, Chichester, U.K., 2009 Search PubMed; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis Reactivity Applications, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) M. Schlosser, Angew. Chem., Int. Ed., 2006, 45, 5432 ( Angew. Chem. , 2006 , 118 , 5558 ) CrossRef CAS PubMed; (d) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (e) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed; (f) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (g) B. E. Smart, J. Fluorine Chem., 2001, 109, 3 CrossRef CAS.
- (a) L. P. Liu, B. Xu, M. S. Mashuta and G. B. Hammond, J. Am. Chem. Soc., 2008, 130, 17642 CrossRef CAS PubMed; (b) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312 ( Angew. Chem. , 2011 , 123 , 7450 ) CrossRef CAS PubMed; (c) Y. Ye, S. H. Lee and M. S. Sanford, Org. Lett., 2011, 13, 5464 CrossRef CAS PubMed; (d) Z.-L. Zhang, C.-P. Wang, Q.-Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Angew. Chem., Int. Ed., 2011, 50, 1896 ( Angew. Chem. , 2011 , 123 , 1936 ) CrossRef PubMed; (e) A. Hafner and S. Bräse, Angew. Chem., Int. Ed., 2012, 51, 3713 ( Angew. Chem. , 2012 , 124 , 3773 ) CrossRef CAS PubMed; (f) T.-F. Liu, X.-X. Shao, Y.-M. Wu and Q.-L. Shen, Angew. Chem., Int. Ed., 2012, 51, 540 ( Angew. Chem. , 2012 , 124 , 555 ) CrossRef CAS PubMed; (g) Z.-B. He, T. Luo, M.-Y. Hu, Y.-W. Cao and J.-B. Hu, Angew. Chem., Int. Ed., 2012, 51, 3944 ( Angew. Chem. , 2012 , 124 , 4010 ) CrossRef CAS PubMed; (h) C. Feng and T. P. Loh, Chem. Sci., 2012, 3, 3458 RSC; (i) A. T. Parsons, T. D. Senecal and S. L. Buchwald, Angew. Chem., Int. Ed., 2012, 51, 2947 ( Angew. Chem. , 2012 , 124 , 3001 ) CrossRef CAS PubMed; (j) G. Danoun, B. Bayarmagnai, M. F. Grünberg and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 7972 ( Angew. Chem. , 2013 , 125 , 8130 ) CrossRef CAS PubMed; (k) F. Wang, X.-Y. Qi, Z.-L. Liang, P.-H. Chen and G.-S. Liu, Angew. Chem., Int. Ed., 2014, 53, 1881 ( Angew. Chem. , 2014 , 126 , 1912 ) CrossRef CAS PubMed; (l) A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120 ( Angew. Chem. , 2011 , 123 , 9286 ) CrossRef CAS PubMed; (m) J. Xu, Y. Fu, D.-F. Luo, Y.-Y. Jiang, B. Xiao, Z.-J. Liu, T.-J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300 CrossRef CAS PubMed; (n) N. O. IIchenko, P. G. Janson and K. J. Szabó, Chem. Commun., 2013, 49, 6614 RSC; (o) P. G. Janson, I. Ghoneim, N. O. IIchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef CAS PubMed; (p) X. Mu, T. Wu, H.-Y. Wang, Y.-L. Guo and G.-S. Liu, J. Am. Chem. Soc., 2012, 134, 878 CrossRef CAS PubMed; (q) W. Kong, M. Casimiro, E. Merino and C. Nevado, J. Am. Chem. Soc., 2013, 135, 14480 CrossRef CAS PubMed; (r) E. Kim, S. Choi, H. Kim and E. J. Cho, Chem. – Eur. J., 2013, 19, 6209 CrossRef CAS PubMed; (s) L.-L. Chu and F.-L. Qing, J. Am. Chem. Soc., 2010, 132, 7262 CrossRef CAS PubMed; (t) T. D. Senecal, A. T. Parsons and S. L. Buchwald, J. Org. Chem., 2011, 76, 1174 CrossRef CAS PubMed; (u) A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901 CrossRef CAS PubMed; (v) X.-Y. Jiang and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 14177 ( Angew. Chem. , 2013 , 125 , 14427 ) CrossRef CAS PubMed; (w) F. Wang, D.-H. Wang, X. Mu, P.-H. Chen and G.-S. Liu, J. Am. Chem. Soc., 2014, 136, 10202 CrossRef CAS PubMed; (x) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC; (y) Y.-N. Wang, M. Jiang and J.-T. Liu, Adv. Synth. Catal., 2014, 356, 2907 CrossRef CAS.
- (a) Y.-P. Xiong, M.-Y. Wu, X.-Y. Zhang, C.-L. Ma, L. Huang, L.-J. Zhao, B. Tan and X.-Y. Liu, Org. Lett., 2014, 16, 1000 CrossRef CAS PubMed; (b) P. Gao, Y.-W. Shen, R. Fang, X.-H. Hao, Z.-H. Qiu, F. Yang, X.-B. Yan, Q. Wang, X.-J. Gong, X.-Y. Liu and Y.-M. Liang, Angew. Chem., Int. Ed., 2014, 53, 7629 ( Angew. Chem. , 2014 , 126 , 7759 ) CrossRef CAS PubMed.
- (a) Y.-W. Li, Y. Liu, G.-S. Qiu and Q.-P. Ding, Org. Lett., 2014, 16, 4240 CrossRef CAS PubMed; (b) J. Xu, Y.-L. Wang, T.-J. Gong, B. Xin and Y. Fu, Chem. Commun., 2014, 50, 12915 RSC; (c) Y.-L. Ji, J.-H. Lin, J.-C. Xiao and Y.-C. Gu, Org. Chem. Front., 2014, 1, 1280 RSC.
- Y.-N. Wang, M. Jiang and J.-T. Liu, Chem. – Eur. J., 2014, 20, 15315 CrossRef CAS PubMed.
- G.-C. Ge, X.-J. Huang, C.-H. Ding, S.-L. Wan, L.-X. Dai and X.-L. Hou, Chem. Commun., 2014, 50, 3048 RSC.
- (a) R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc. Trans., 1915, 107, 1080 RSC; (b) M. N. Levine and R. T. Raines, Chem. Sci., 2012, 3, 2412 RSC.
- (a) X. Wang, Y.-X. Ye, G.-J. Ji, S.-N. Zhang, J.-J. Feng, Y. Zhang and J.-B. Wang, Org. Lett., 2013, 15, 3730 CrossRef CAS PubMed; (b) Z.-X. Fang, Y.-Q. Ning, P.-B. Mi, P.-Q. Liao and X.-H. Bi, Org. Lett., 2014, 16, 1522 CrossRef CAS PubMed.
- A. Alhalib and W. J. Moran, Org. Biomol. Chem., 2014, 12, 795 CAS.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectral data and copies of spectra. See DOI: 10.1039/c5qo00044k |
| This journal is © the Partner Organisations 2015 |