
Synthetic strategies toward the decalin motif of maklamicin and related spirotetronates†‡
Michelle H.
Lacoske
a,
Jing
Xu
ab,
Noel
Mansour
a,
Chao
Gao
a and
Emmanuel A.
Theodorakis
*a
aDepartment of Chemistry & Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA. E-mail: etheodor@ucsd.edu
bDepartment of Chemistry, South University of Science and Technology of China, Shenzhen, Guangdong 518055, China. E-mail: xuj@sustc.edu.cn
First published on 16th February 2015
Abstract
Herein we describe a scalable approach to the decalin moiety of maklamicin. Key to the synthesis is an intramolecular Diels–Alder (IMDA) reaction that proceeds via an endo-axial transition state to generate the desired stereochemistry. We explored the diastereoselectivity of the IMDA reaction as a function of both chiral catalysis and acyclic precursor stereochemistry.
Introduction
Isolated from various Micromonospora strains, spirotetronate polyketides constitute a peculiar family of natural products characterized by a complex chemical architecture and intriguing bioactivity as antitumor antibiotics.1 Tetrocarcin A, the defining member of this family, contains an aglycon core, referred to as tetronolide (1),2 that is glycosylated at the C9 and C17 centers (Fig. 1). This natural product displays promising anticancer properties in vitro and in animals3 that may stem from its ability to induce cell stress and inhibit Bcl-2 expression.4 Tetrocarcin A also exhibits potent cytotoxicity against several Gram-positive bacteria (e.g. MIC: 0.38 μM against Bacillus subtilis).3b,5 Interestingly, it has been shown that glycosylation of C9 is essential for antimicrobial activity but inconsequential for anticancer activity.5c,6 The influence of glycosylation to the antimicrobial potency has also been observed in other spirotetronates.5c,6,7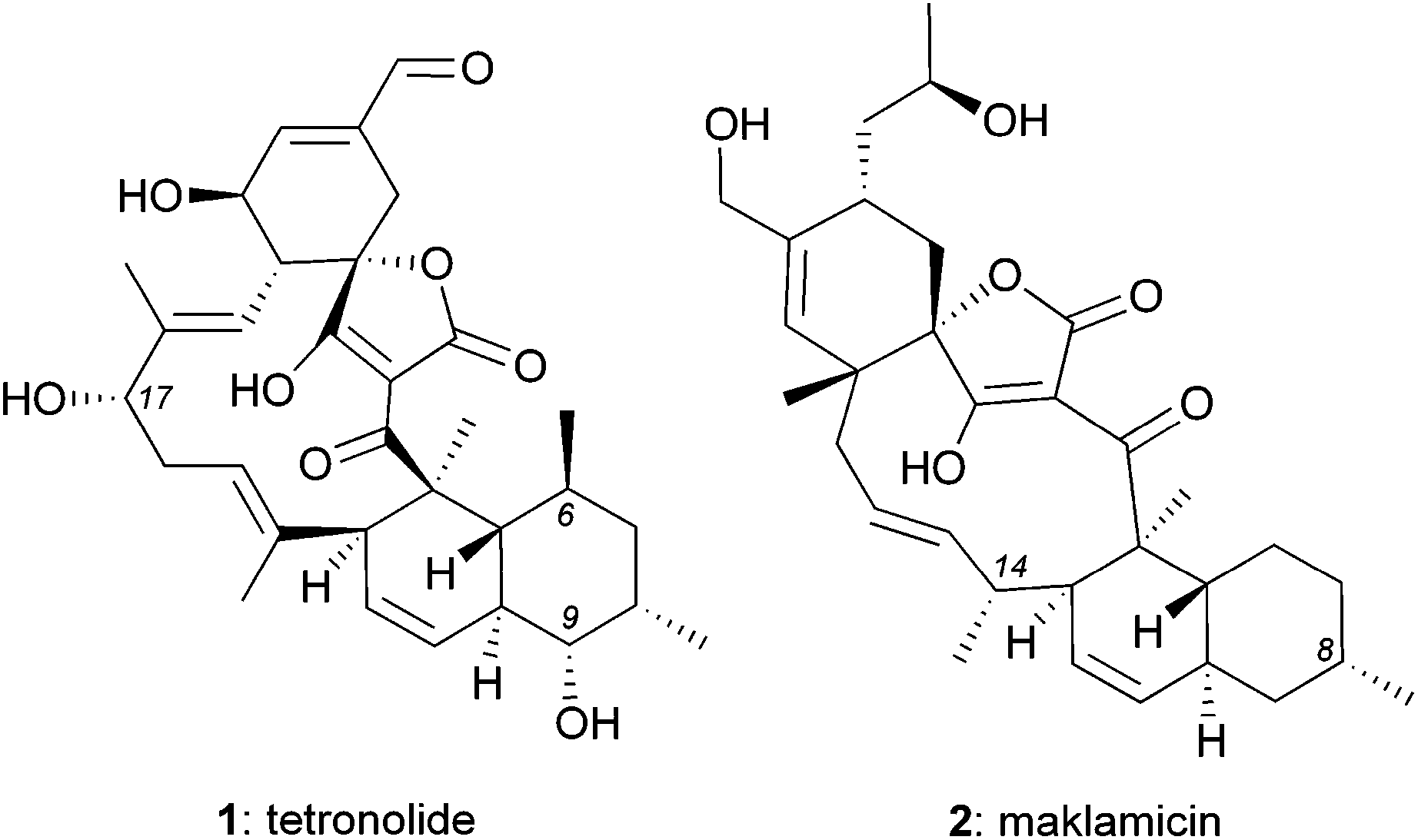 | ||
| Fig. 1 Structures of tetronolide (1) and maklamicin (2). | ||
The biological and pharmacological potential of spirotetronates has fuelled efforts to isolate new family members. Along these lines, Igarashi et al. reported the isolation of maklamicin (2) from Micromonospora sp. GMKU326.8 This novel spirotetronate was shown to exhibit both antimicrobial activities against various Gram-positive bacteria (e.g. MIC: 0.30 μM against Bacillus subtilis) and anticancer activities (IC50: 17–34 μM against various cancer cells lines).8 Intriguingly, maklamicin exhibits potent antimicrobial activity in the absence of C9 glycosylation, which is essential for other spirotetronate polyketides.5c,6,7
Structurally, maklamicin (2) encapsulates a trans-decalin and a spiro-tetronic acid moieties within a strained 11-membered macrocyclic motif.8,9 Herein we describe synthetic approaches towards the decalin moiety of 2.
Inspired by the biosynthetic pathway of spirotetronates, we envisioned that the decalin moiety of maklamicin would arise from an intramolecular Diels–Alder reaction (IMDA).10 This strategy has been successfully applied to the synthesis of related natural products.11,12 Closer analysis of this IMDA reveals four possible transition states that give rise to four distinct decalin diastereomers (Fig. 2). The two endo transition states derive from the relative orientation of the C8 methyl group during the transition state of the IMDA reaction that influences the facial selectivity of this cycloaddition. Based on the C8 methyl group orientation, we define these transition states as endo-equatorial and endo-axial.13 In a similar fashion, depending on the C8 methyl group orientation, we can have the exo-equatorial and exo-axial transition states (Fig. 2). To obtain the desired stereochemistry of the maklamicin decalin unit (i.e. compound 5), the IMDA should proceed via an endo-axial transition state (3: endo-ax. TS). Published reports indicate that the facial selectivity of this type of an IMDA is expected to proceed via an endo-equatorial transition state (3: endo-eq. TS).14 With this in mind, we sought to explore various methods that could alter the facial selectivity of the IMDA in favor of that encountered in the structure of maklamicin.
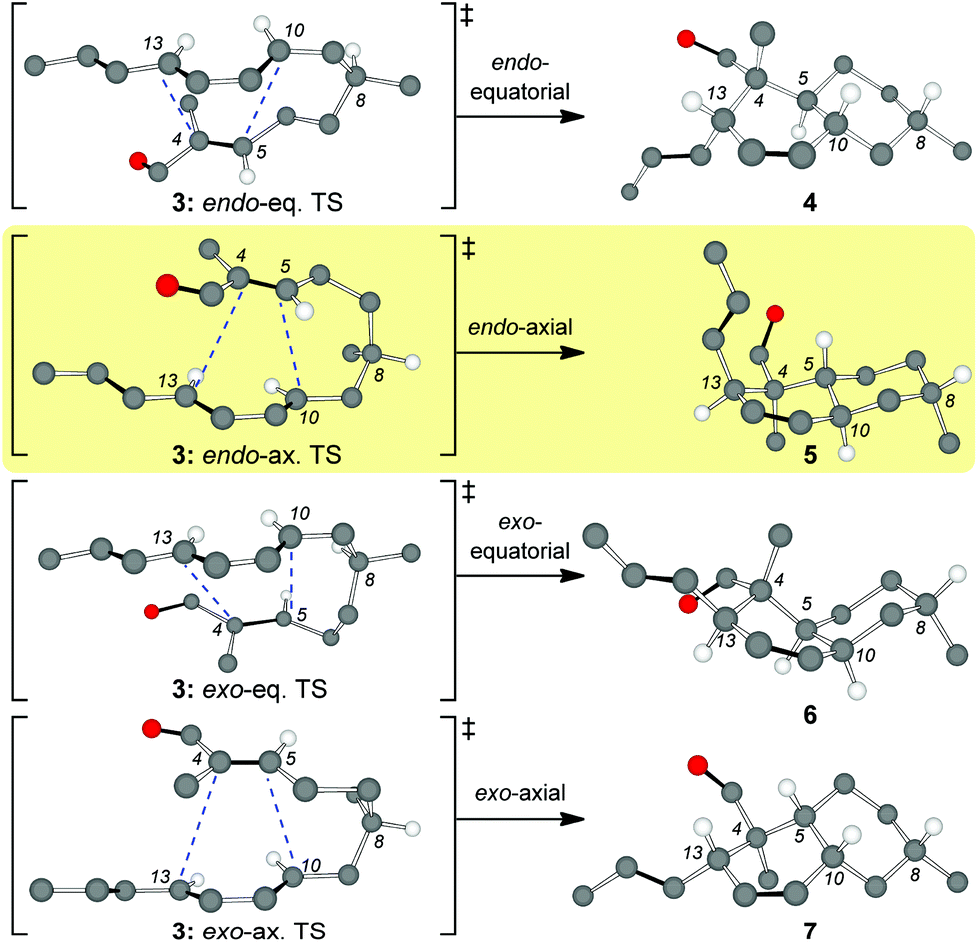 | ||
| Fig. 2 Possible transition states for the IMDA of compound 3 (only selected hydrogens are shown). The terms axial and equatorial refer to the relative orientation of the C8 methyl group. | ||
Our model studies toward the synthesis of the maklamicin decalin core are highlighted in Scheme 1–3. Previously, we demonstrated that triene 3, synthetically available from (–)-citronellal (10) in 3 steps, undergoes an IMDA in the presence of Et2AlCl at −78 °C to produce the endo-equatorial product 4.15 We hypothesized that an organocatalytic process could overcome the inherent substrate selectivity of this system and favor construction of cycloadduct 5, the one encountered in the structure of maklamicin. These studies are summarized in Scheme 1. We performed this reaction in the presence of various imidazolidinone catalysts 8 under previously optimized conditions (20 mol% HClO4, 20 mol% catalyst, MeCN or EtOH, −20 °C).12c,16 Under these conditions, the IMDA reaction was slow (3 days for completion) and produced exclusively the endo-equatorial adduct 4.15 The results indicate that the catalysis was not effective in our substrate, presumably due to the presence of the C4 methyl group adjacent to the carbonyl group. This methyl group likely prevents the transiently formed iminium intermediate from assuming the geometry needed for the desired facial selectivity of IMDA, thus diminishing the ability of the catalyst to direct the stereochemical outcome of this reaction.16,17
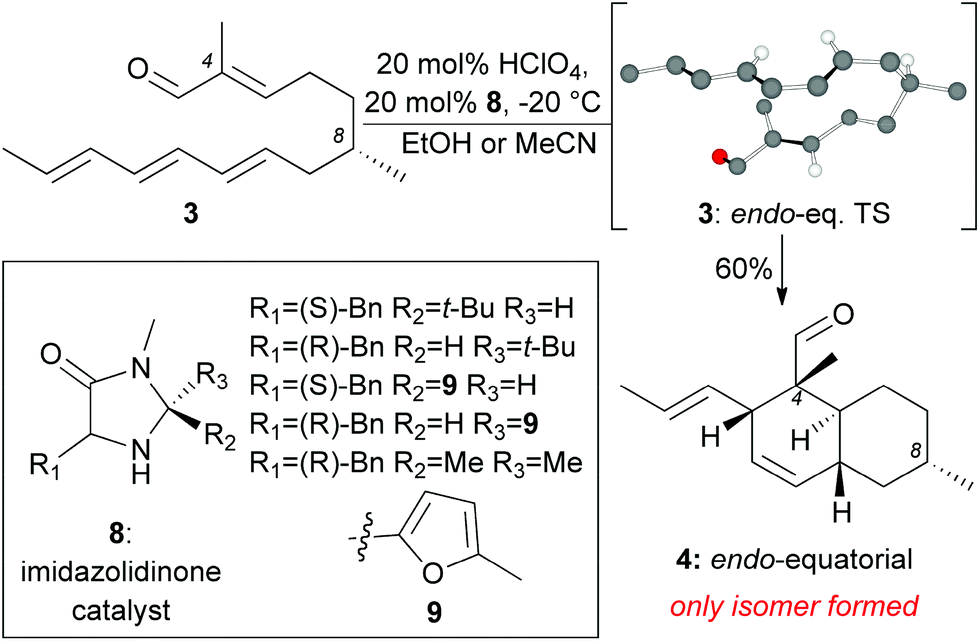 | ||
| Scheme 1 Studies on the IMDA of 3 under imidazolidinone organocatalysis. | ||
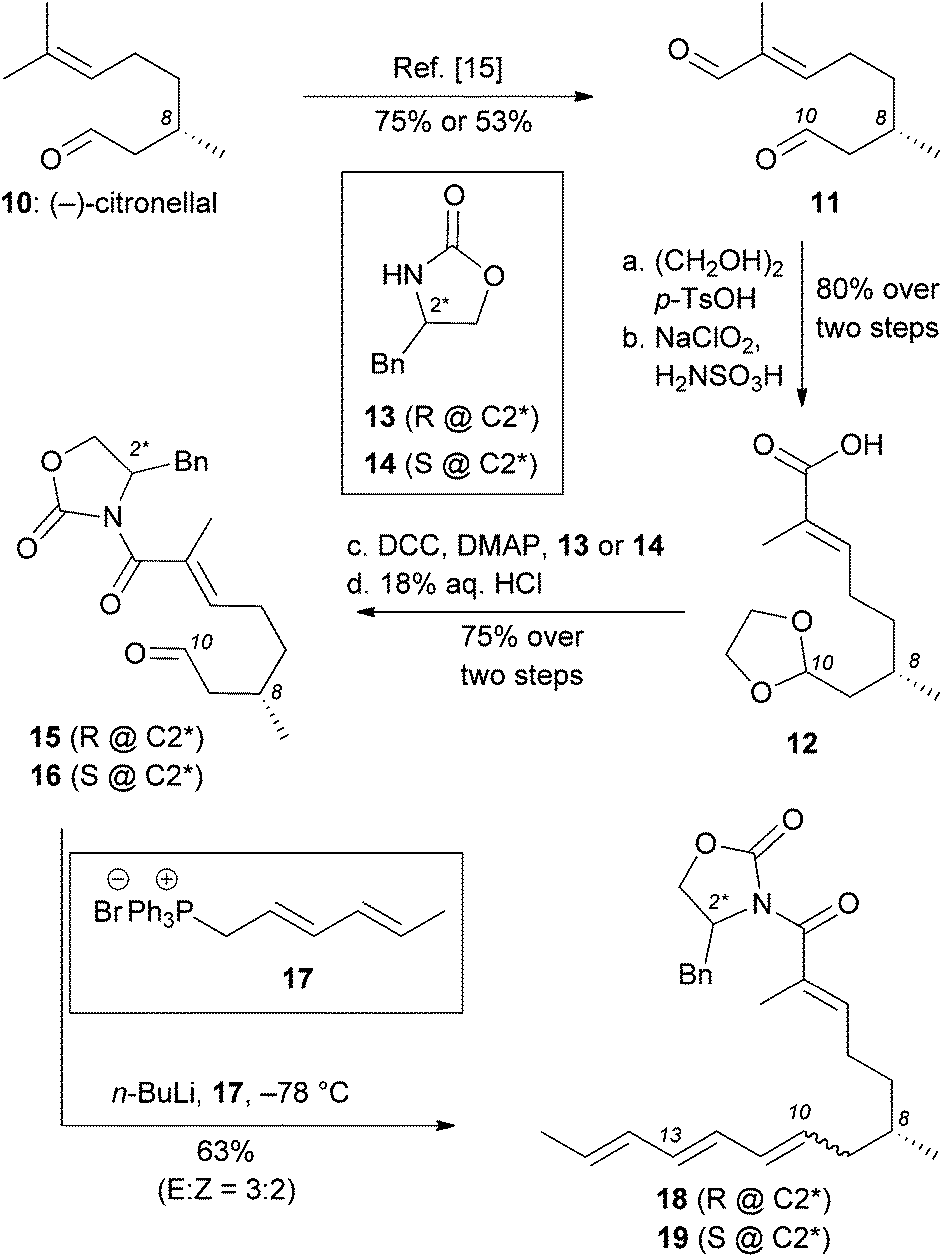 | ||
| Scheme 2 Synthesis of polyenes 18 and 19 containing benzyl oxazolidinones as chiral auxiliaries. | ||
 | ||
| Scheme 3 Products obtained from an IMDA reaction of 18 and 19. | ||
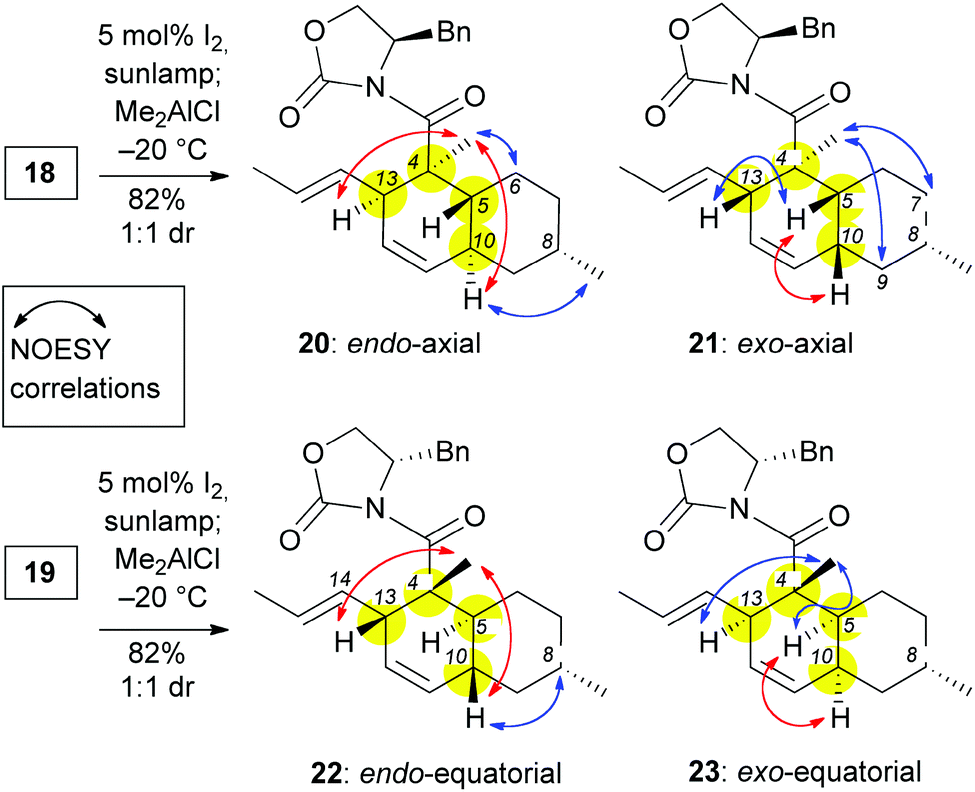
In light of these results, we pursued an alternative strategy, shown in Scheme 2, in which the desired facial stereoselectivity of IMDA could be achieved using Evans oxazolidinones (e.g. compounds 13 and 14) as chiral auxiliaries.18 The modified strategy departed from (S)-(–)-citronellal (10) that, following established protocols, was converted to enal 11.15,19 The difference in the chemoselectivity between the two carbonyl groups of 11, allowed selective protection of aliphatic aldehyde with ethylene glycol and tosylic acid. The resulting acetal underwent Pinnick oxidation to afford the corresponding carboxylic acid 12 (80% yield over two steps). DCC coupling of 12 with (R)-4-benzyl oxazolidinone or (S)-4-benzyl oxazolidinone followed by acetal deprotection (18% aqueous HCl) yielded aldehydes 15 and 16 (75% yield over two steps). Olefination of aldehyde 15 and 16 with a Wittig ylide, produced in situ from phosphonium salt 17 with n-BuLi, afforded trienes 18 and 19 respectively in about 60% yield. The selectivity of this olefination was moderate (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z = ∼3:2) and could not be improved using related olefination techniques.
:Z = ∼3:2) and could not be improved using related olefination techniques.
The (E,E) stereochemistry of the C10 and C13 diene is essential for the desired IMDA. With this in mind, a photoisomerization of 18 and 19 was conducted with 5 mol% of iodine in dichloromethane under sun-lamp photoirradiation,20 to produce almost exclusively the E,E-alkenes (Scheme 3). For characterization purposes, these compounds can be isolated after the photoisomerization reaction (see ESI‡). Nonetheless, these polyenes can undergo the IMDA reaction in one pot, immediately after the photoisomerization. To this end, polyenes 18 and 19 were each treated with 1.1 eq. of Me2AlCl at −78 °C and then the reaction was allowed to warm to −20 °C. Each of these reactions produced two readily separable cycloadducts in a 1:1 isolated yield. NOESY experiments were performed to assign the relative stereochemistry of these compounds (Scheme 3). In compounds 20 and 22 we observed key correlations between the C4 methyl and both protons at C10 and C13 (marked with red arrows in Scheme 3). These correlations, together with the absence of a signal between the C10 and the C5 protons indicate the presence of a trans-decalin ring and thus an endo IMDA reaction. Compound 20 also displayed NOESY correlations between (a) the C10 proton and C8 methyl; and (b) the C4 methyl and C6axial proton (marked with blue arrows in Scheme 3) supporting the notion that the IMDA proceeded through an endo-axial transition state. This assignment is consistent with the NOESY data reported for the decalin moiety of maklamicin.8 In addition to the key correlations of an endo adduct (marked with red arrows in Scheme 3), compound 22 displayed NOESY correlation between the protons at C10 and C8 supporting the assignment of the endo-equatorial adduct. Similar correlations have been observed in related equisetin derivatives.21 In a similar manner, compounds 21 and 23 were identified as exo adducts since they displayed a NOESY signal between the protons at C10 and C5 as expected in a cis-decalin motif (marked with a red arrow in Scheme 3). Compound 21 displayed additional correlations between: (a) the C4 methyl and protons at C7axial and C9axial; and (b) protons at C13 and C5 (marked in blue arrows in Scheme 3). These correlations confirm that adduct 21 was produced via an exo-axial IMDA transition state. Similar correlations have been reported for the cis-decalin motif of ascosalipyrrolidinone.22 On the other hand, compound 23 displayed additional correlations between the C4 methyl and protons at both C5 and C13 in support of this IMDA proceeding through an exo-equatorial transition state. Similar correlations have been observed in pyrrolocin B, a compound that contains a cis-decalin with an equatorial methyl at C8.21a
The above data demonstrate that all stereoisomers of the IMDA reaction can be accessible using the chiral auxiliary approach. In the specific case of maklamicin synthesis, formation of the endo-axial configuration requires use of the (R)-4-benzyl oxazolidinone (13). Having established a strategy for the synthesis of the desired decalin moiety, we then turned our attention toward an appropriately functionalized acyclic precursor of the maklamicin decalin core.
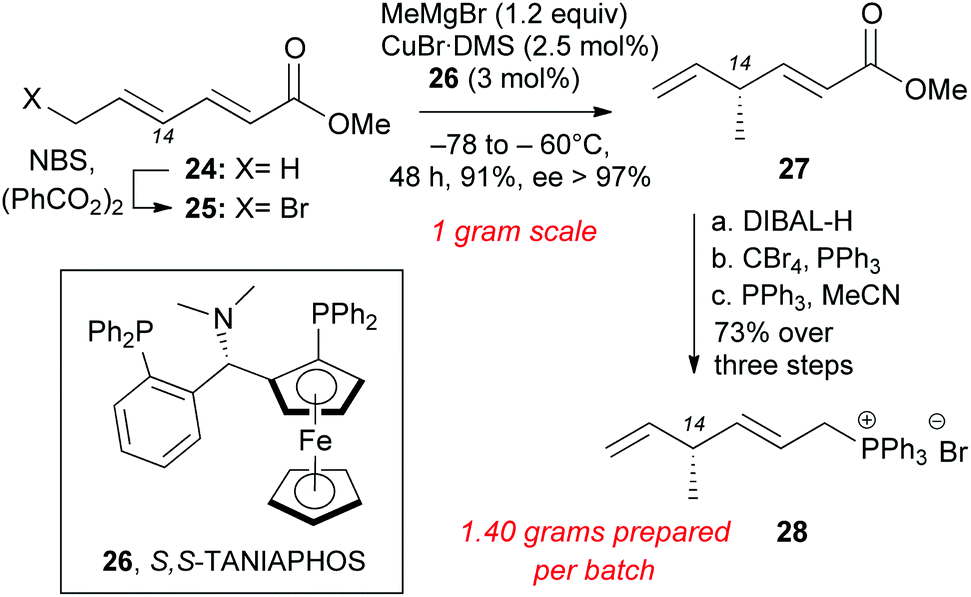
Installation of the C14 methyl group at the polyene precursor of maklamicin was accomplished as highlighted in Scheme 4. Commercially available methyl sorbate (24) underwent allylic bromination to form 25 in 90% yield.23 SN2′ methylation at C14 was accomplished in a stereoselective manner using Feringa's protocol that involves slow addition of 25 to a solution of methyl cuprate and S,S-TANIAPHOS (26).24 Lowering the amounts of CuBr·DMS and 26 to 2.5 mol% and 3 mol% respectively, maintained the same level of enantioselectivity and afforded skipped diene 27 (91% yield, 97% ee). Due to the volatility of product 27, its isolation yield is maximized when the alkylation reaction takes place on one-gram scale.24,25 The slow addition of bromosorbate (25) to a cold solution of the catalyst limits the scalability of this reaction. Reduction of methyl ester 27 with DIBAL-H generated the allylic alcohol, which was converted to the allylic bromide under Appel conditions.26 The allylic bromide was immediately converted to the Wittig salt in MeCN and PPh3 to produce 28 (73% over 3 steps).
 | ||
| Scheme 4 Synthesis of functionalized Wittig salt 28. | ||
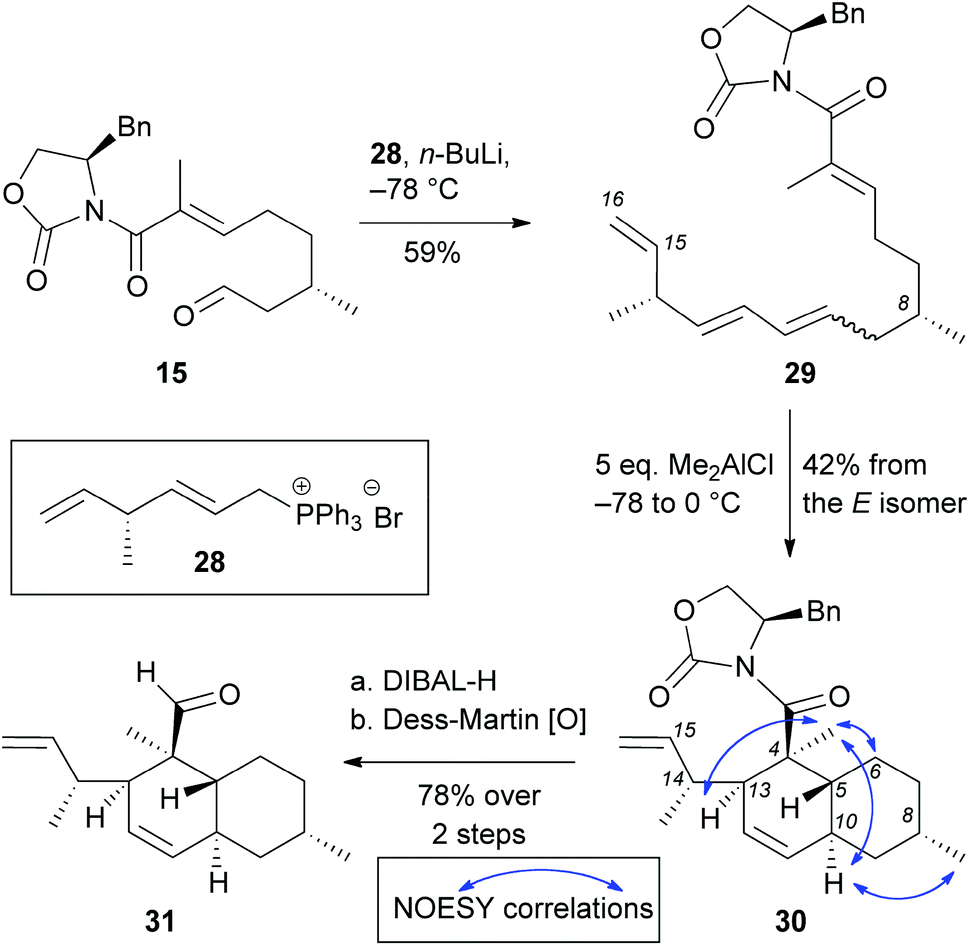
Wittig olefination of aldehyde 15 with phosphonium bromide 28 yielded polyene 29 in 59% yield (E:Z = ∼3:2). Double bond isomerization of this E:Z mixture was attempted but the previously established conditions promoted isomerisation of the terminal C15–C16 olefin to form the conjugated triene. Thus, the E:Z mixture was subjected to the IMDA reaction that proceeded by adding 5 equivalents of Me2AlCl. Under these conditions we isolated cycloadduct 30 in 42% yield (formed from the E-isomer). Not surprisingly, the Z-isomer does not undergo cycloaddition under these conditions.27 NOESY correlation of 30 was compared with compounds 20–23 to confirm that the IMDA proceeded via an endo-axial transition state. Reduction of oxazolidinone 30 with DIBAL-H followed by oxidation of the resulting alcohol with Dess-Martin periodinane yielded decalin aldehyde 31 (78% yield over 2 steps). Albeit short (9 total steps) this strategy has limited scalability due to the following reasons: (a) difficulties in installing the C14 methyl group in large scale due to limitations of the SN2′ alkylation and the cost of the catalyst; (b) inability to isomerize triene 29 to the desired all trans isomer; and (c) low yielding IMDA reaction that only proceeded from E isomer in 42% yield (Scheme 5).
 | ||
| Scheme 5 Synthesis of decalin 31. | ||
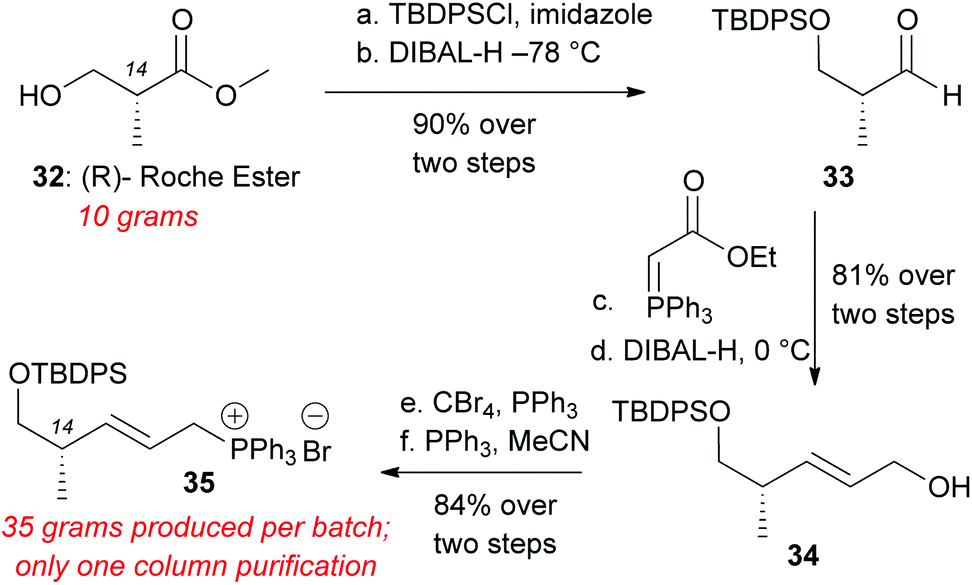
To overcome the above difficulties we sought to develop an alternative route to decalin aldehyde starting from commercially available methyl (R)-(–)-3-hydroxyisobutyrate (Roche's ester) (Scheme 6). This compound was protected with tert-butyldiphenylsilyl chloride (TBDPSCl) and imidazole in DMF in quantitative yield.28 Reduction of the methyl ester to aldehyde 33 was achieved with DIBAL-H administered via syringe pump at −78 °C. Aldehyde 33 was directly subjected to Wittig olefination with (carbethoxymethylene) triphenylphosphorane. The resultant ethyl ester was reduced to the allylic alcohol 34 with DIBAL-H at 0 °C in 81% yield over two steps.28 Appel bromination yielded allylic bromide, which was immediately subjected to PPh3 in MeCN to generate Wittig salt 35 (84% over two steps). The overall preparation of this Wittig salt is high yielding, scalable, and only requires one silica column chromatography after TBDPS protection to produce over 30 grams of Wittig salt 35.
 | ||
| Scheme 6 Synthesis of Wittig salt 35. | ||
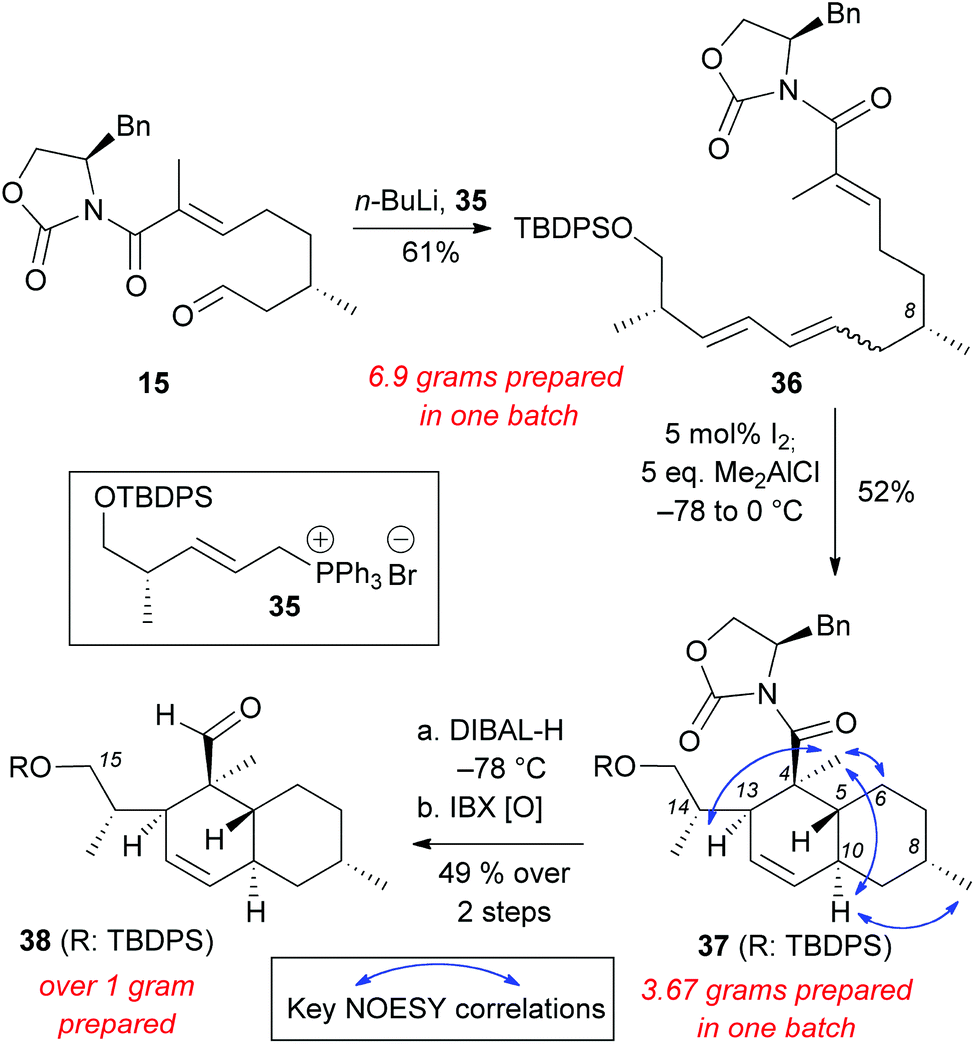
Wittig salt 35 was treated with n-BuLi to generate the ylide in situ and then aldehyde 15 was added to form polyene 36 (61% yield, E:Z = ∼3:2) (Scheme 7). The double bond isomerization and the subsequent IMDA proceeded smoothly to generate decalin 37 (52% yield on more than 3 grams scale). NOESY correlations of 37 were compared to compounds 20–23, 30, and maklamicin to confirm that the desired stereochemistry was achieved.8 Reduction of 37 with DIBAL-H followed by IBX oxidation yielded aldehyde 38 (49% yield, 2 steps). The C15 silylated alcohol in 38 allows further derivatization (e.g. olefination protocols) en route to the chemical synthesis of maklamicin.
 | ||
| Scheme 7 Synthesis of scalable decalin moiety 38. | ||
Conclusions
We have explored the stereochemical outcome of an intramolecular Diels–Alder (IMDA) cycloaddition as a function of the stereochemistry of the acyclic precursor. Initial studies on model systems confirmed the role of the benzyl oxazolidinone on the facial selectivity of the IMDA. We then applied this information to the synthesis of the unusual endo-axial decalin moiety of maklamicin. The chiral methyl groups at C8 and C14 of the IMDA precursor were introduced from enantiomerically pure starting materials. Overall, the strategy is divergent and allows access to the decalin motif 38 in 10 linear steps and good overall yield. Interestingly, decalin 38 is suitably protected for further functionalization towards the synthesis of maklamicin. Importantly, the reported studies pave the way for a general synthetic approach toward the decalin motifs of spirotetronates and related natural products.12b,21,22Acknowledgements
We gratefully acknowledge the National Institutes of Health (NIH) for financial support of this work through Grant Number CA 133002. We thank the National Science Foundation for instrumentation grants CHE9709183 and CHE0741968. We thank Dr Anthony Mrse (UCSD NMR Facility), Dr Yongxuan Su (UCSD MS Facility). We also thank the U.S. Department of Education for a Fellowship to M.H.L. through GAANN grant P200A120223 and the UCSD Academic Senate for partial funding.Notes and references
- For recent reviews on spirotetronate polyketides: (a) M. H. Lacoske and E. A. Theodorakis, J. Nat. Prod., 2014 DOI:10.1021/np500757w; (b) L. Vieweg, S. Reichau, R. Schobert, P. F. Leadlay and R. D. Sussmuth, Nat. Prod. Rep., 2014, 31, 1554–1584 RSC.
- N. Hirayama, M. Kasai, K. Shirahata, Y. Ohashi and Y. Sasada, Tetrahedron Lett., 1980, 21, 2559–2560 CrossRef CAS.
- (a) M. Morimoto, M. Fukui, S. Ohkubo, T. Tamaoki and F. Tomita, J. Antibiot., 1982, 35, 1033–1037 CrossRef CAS; (b) T. Tamaoki, M. Kasai, K. Shirahata, S. Ohkubo, M. Morimoto, K. Mineura, S. Ishii and F. Tomita, J. Antibiot., 1980, 33, 946–950 CrossRef CAS; (c) F. Tomita, T. Tamaoki, K. Shirahata, M. Kasai, M. Morimoto, S. Ohkubo, K. Mineura and S. Ishii, J. Antibiot., 1980, 33, 668–670 CrossRef CAS.
- (a) T. Nakashima, M. Miura and W. Hara, Cancer Res., 2000, 60, 1229–1235 CAS; (b) G. Anether, I. Tinhofer, M. Senfter and R. Greil, Blood, 2003, 101, 4561–4568 CrossRef CAS PubMed; (c) S. Cory, D. C. S. Huang and J. M. Adams, Oncogene, 2003, 22, 8590–8607 CrossRef CAS PubMed; (d) J. Yang, X. S. Liu, K. Bhalla, C. N. Kim, A. M. Ibrado, J. Y. Cai, T. I. Peng, D. P. Jones and X. D. Wang, Science, 1997, 275, 1129–1132 CrossRef CAS.
- (a) K. Namikawa, Y. Sakuma, F. Sunaga and Y. Kanno, Jpn. J. Vet. Sci., 1988, 50, 968–970 CrossRef CAS; (b) M. Ohtomo, K. Yamazaki, S. Ito, K. Shimura, S. Shimizu, T. Minami, T. Fujinaga and K. Shimada, Jpn. J. Vet. Sci., 1985, 47, 581–587 CrossRef CAS; (c) T. Tamaoki, M. Kasai, K. Shirahata and F. Tomita, J. Antibiot., 1982, 35, 979–984 CrossRef CAS; (d) F. Tomita and T. Tamaoki, J. Antibiot., 1980, 33, 940–945 CrossRef CAS.
- M. Kaneko, T. Nakashima, Y. Uosaki, M. Hara, S. Ikeda and Y. Kanda, Bioorg. Med. Chem. Lett., 2001, 11, 887–890 CrossRef CAS.
- (a) L. Cuthbertson, S. K. Ahn and J. R. Nodwell, Chem. Biol., 2013, 20, 232–240 CrossRef CAS PubMed; (b) V. Kren and L. Martinkova, Curr. Med. Chem., 2001, 8, 1303–1328 CrossRef CAS; (c) V. Kren and T. Rezanka, FEMS Microbiol. Rev., 2008, 32, 858–889 CrossRef CAS PubMed.
- Y. Igarashi, H. Ogura, K. Furihata, N. Oku, C. Indananda and A. Thamchaipenet, J. Nat. Prod., 2011, 74, 670–674 CrossRef CAS PubMed.
- Y. Igarashi, T. Iida, N. Oku, H. Watanabe, K. Furihata and K. Miyanouchi, J. Antibiot., 2012, 65, 355–359 CrossRef CAS PubMed.
- (a) J. Fang, Y. P. Zhang, L. J. Huang, X. Y. Jia, Q. Zhang, X. Zhang, G. L. Tang and W. Liu, J. Bacteriol., 2008, 190, 6014–6025 CrossRef CAS PubMed; (b) H. Zhang, J. A. White-Phillip, C. E. Melancon, H. J. Kwon, W. L. Yu and H. W. Liu, J. Am. Chem. Soc., 2007, 129, 14670–14683 CrossRef CAS PubMed.
- Selected syntheses utilizing Diels–Alder reactions: (a) D. Fischer, T. X. Nguyen, L. Trzoss, M. Dakanali and E. A. Theodorakis, Tetrahedron Lett., 2011, 52, 4920–4923 CrossRef CAS PubMed; (b) S. Ghosh, F. Rivas, D. Fischer, M. A. Gonzalez and E. A. Theodorakis, Org. Lett., 2004, 6, 941–944 CrossRef CAS PubMed; (c) T. Ling, B. A. Kramer, M. A. Palladino and E. A. Theodorakis, Org. Lett., 2000, 2, 2073–2076 CrossRef CAS PubMed; (d) T. T. Ling, C. Chowdhury, B. A. Kramer, B. G. Vong, M. A. Palladino and E. A. Theodorakis, J. Org. Chem., 2001, 66, 8843–8853 CrossRef CAS PubMed; (e) J. A. Marshall, J. E. Audia, J. Grote and B. G. Shearer, Tetrahedron, 1986, 42, 2893–2902 CrossRef CAS; (f) J. A. Marshall, J. Grote and J. E. Audia, J. Am. Chem. Soc., 1987, 109, 1186–1194 CrossRef CAS; (g) J. A. Marshall, J. Grote and B. Shearer, J. Org. Chem., 1986, 51, 1633–1635 CrossRef CAS; (h) J. A. Marshall, J. M. Salovich and B. G. Shearer, J. Org. Chem., 1990, 55, 2398–2403 CrossRef CAS; (i) W. R. Roush and B. B. Brown, J. Am. Chem. Soc., 1993, 115, 2268–2278 CrossRef CAS; (j) W. R. Roush, B. B. Brown and S. E. Drozda, Tetrahedron Lett., 1988, 29, 3541–3544 CrossRef CAS; (k) W. R. Roush, M. L. Reilly, K. Koyama and B. B. Brown, J. Org. Chem., 1997, 62, 8708–8721 CrossRef CAS; (l) W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1994, 116, 6457–6458 CrossRef CAS; (m) W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1998, 120, 7411–7419 CrossRef CAS; (n) K. Takeda, E. Kawanishi, H. Nakamura and E. Yoshii, Tetrahedron Lett., 1991, 32, 4925–4928 CrossRef CAS; (o) K. Takeda, T. Kobayashi, K. Saito and E. Yoshii, J. Org. Chem., 1988, 53, 1092–1095 CrossRef CAS; (p) K. Takeda, S. Yano and E. Yoshii, Tetrahedron Lett., 1988, 29, 6951–6954 CrossRef CAS.
- Selected reviews on Diels–Alder reactions: (a) M. Juhl and D. Tanner, Chem. Soc. Rev., 2009, 38, 2983–2992 RSC; (b) G. Li, S. Kusari and M. Spiteller, Nat. Prod. Rep., 2014, 31, 1175–1201 RSC; (c) P. Merino, E. Marques-Lopez, T. Tejero and R. P. Herrera, Synthesis-Stuttgart, 2010, 1–26 CrossRef CAS PubMed; (d) K. C. Nicolaou, S. A. Snyder, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2002, 41, 1668–1698 CrossRef CAS; (e) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
- Note: In similar systems, such transition states are referred to as endo I and endo II. For leading references see: (a) W. R. Roush, Comprehensive Organic Synthesis, Pergamon Press, Oxford, 1991 Search PubMed; (b) D. Craig, Chem. Soc. Rev., 1987, 16, 187–238 RSC; (c) I. Fernandez and F. M. Bickelhaupt, J. Comput. Chem., 2014, 35, 371–376 CrossRef CAS PubMed; (d) H. V. Pham, R. S. Paton, A. G. Ross, S. J. Danishefsky and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 2397–2403 CrossRef CAS PubMed.
- (a) T. K. M. Shing and J. Yang, J. Org. Chem., 1995, 60, 5785–5789 CrossRef CAS; (b) D. R. Williams, M. L. Bremmer, D. L. Brown and J. Dantuono, J. Org. Chem., 1985, 50, 2807–2809 CrossRef CAS.
- (a) J. Xu, E. J. E. Caro-Diaz, L. Trzoss and E. A. Theodorakis, J. Am. Chem. Soc., 2012, 134, 5072–5075 CrossRef CAS PubMed; (b) J. Xu, E. J. E. Caro-Diaz, M. H. Lacoske, C. I. Hung, C. Jamora and E. A. Theodorakis, Chem. Sci., 2012, 3, 3378–3386 RSC.
- (a) R. M. Wilson, W. S. Jen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 11616–11617 CrossRef CAS PubMed; (b) J. B. Brazier, K. M. Jones, J. A. Platts and N. C. O. Tomkinson, Angew. Chem., Int. Ed., 2011, 50, 1613–1616 CrossRef CAS PubMed; (c) A. B. Northrup and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458–2460 CrossRef CAS PubMed.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- (a) D. A. Evans, K. T. Chapman and J. Bisaha, Tetrahedron Lett., 1984, 25, 4071–4074 CrossRef CAS; (b) D. A. Evans, K. T. Chapman and J. Bisaha, J. Am. Chem. Soc., 1984, 106, 4261–4263 CrossRef CAS; (c) D. A. Evans and W. C. Black, J. Am. Chem. Soc., 1993, 115, 4497–4513 CrossRef CAS.
- E. J. E. Caro-Diaz, A. Aung, J. Xu, S. Varghese and E. A. Theodorakis, Org. Chem. Front., 2014, 1, 135–139 RSC.
- C. I. Turner, R. M. Williamson, P. Turner and M. S. Sherburn, Chem. Commun., 2003, 1610–1611 RSC.
- (a) R. C. Jadulco, M. Koch, T. B. Kakule, E. W. Schmidt, A. Orendt, H. He, J. E. Janso, G. T. Carter, E. C. Larson, C. Pond, T. K. Matainaho and L. R. Barrows, J. Nat. Prod., 2014, 77, 2537–2544 CrossRef CAS PubMed; (b) J. H. Jang, Y. Asami, J. P. Jang, S. O. Kim, D. O. Moon, K. S. Shin, D. Hashizume, M. Muroi, T. Saito, H. Oh, B. Y. Kim, H. Osada and J. S. Ahn, J. Am. Chem. Soc., 2011, 133, 6865–6867 CrossRef CAS PubMed; (c) S. B. Singh, D. L. Zink, M. A. Goetz, A. W. Dombrowski, J. D. Polishook and D. J. Hazuda, Tetrahedron Lett., 1998, 39, 2243–2246 CrossRef CAS; (d) R. F. Vesonder, L. W. Tjarks, W. K. Rohwedder, H. R. Burmeister and J. A. Laugal, J. Antibiot., 1979, 32, 759–761 CrossRef CAS.
- C. Osterhage, R. Kaminsky, G. M. Konig and A. D. Wright, J. Org. Chem., 2000, 65, 6412–6417 CrossRef CAS.
- A. Caravano, R. A. Field, J. M. Percy, G. Rinaudo, R. Roig and K. Singh, Org. Biomol. Chem., 2009, 7, 996–1008 CAS.
- Y. G. Huang, M. Fananas-Mastral, A. J. Minnaard and B. L. Feringa, Chem. Commun., 2013, 49, 3309–3311 RSC.
- (a) A. B. Zhang and T. V. RajanBabu, J. Am. Chem. Soc., 2006, 128, 54–55 CrossRef CAS PubMed; (b) A. B. Zhang and T. V. RajanBabu, J. Am. Chem. Soc., 2006, 128, 5620–5621 CrossRef CAS PubMed; (c) M. Fananas-Mastral, B. ter Horst, A. J. Minnaard and B. Feringa, Chem. Commun., 2011, 47, 5843–5845 RSC; (d) M. Perez, M. Fananas-Mastral, P. H. Bos, A. Rudolph, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 377–381 CrossRef CAS PubMed.
- (a) A. Saitman, P. Rulliere, S. D. E. Sullivan and E. A. Theodorakis, Org. Lett., 2011, 13, 5854–5857 CrossRef CAS PubMed; (b) A. Saitman and E. A. Theodorakis, Org. Lett., 2013, 15, 2410–2413 CrossRef CAS PubMed; (c) A. Saitman, S. D. E. Sullivan and E. A. Theodorakis, Tetrahedron Lett., 2013, 54, 1612–1615 CrossRef CAS PubMed; (d) R. Appel, Angew. Chem., Int. Ed., 1975, 14, 801–811 CrossRef.
- (a) F. Fringuelli and A. Taticchi, Dienes in the Diels–Alder Reactions, John Wiley & Sons, New York, 1990 Search PubMed; (b) W. Oppolzer, Comprehensive Organic Synthesis, Pergamon Press, Oxford, 1991 Search PubMed.
- (a) J. S. Yadav, M. Venkatesh, N. Thrimurtulu and A. R. Prasad, Synlett, 2010, 1255–1259 CrossRef CAS PubMed; (b) S. Chandrasekhar, S. R. Yaragorla, L. Sreelakshmi and C. R. Reddy, Tetrahedron, 2008, 64, 5174–5183 CrossRef CAS PubMed; (c) A. E. Pasqua, F. D. Ferrari, J. J. Crawford and R. Marquez, Tetrahedron Lett., 2012, 53, 2114–2116 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Ei-ichi Negishi on the occasion of his 80th Birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00332b |
| This journal is © the Partner Organisations 2015 |