
Reductive decyanation of malononitriles and cyanoacetates using photoactivated neutral organic super-electron-donors†‡
Eswararao
Doni
and
John A.
Murphy
*
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: john.murphy@strath.ac.uk
First published on 25th August 2014
Abstract
A metal-free reductive procedure for decyanation of malononitriles and cyanoacetates, using photoactivated organic electron donors, is described. Decyanation of cyanoacetates is more difficult than for malononitriles and it requires higher loading of the electron donor and extended reaction times. An anionic intermediate is proposed for the observed decyanations and a plausible mechanism is presented.
Introduction
Malononitriles have useful applications in both ionic1 and radical2 reactions. However, they are much less popular tools in organic synthesis than the closely related malonic esters, acetoacetates and sulfonyl acetates. This is partly because of the greater difficulty associated with the removal of the extra functional group in malononitriles i.e. a nitrile group, in comparison to a carboxyl or sulfonyl group in its relatives.3 Accordingly, easier access to decyanation of malononitriles would encourage synthetic applications of malononitriles in organic chemistry. The reductive decyanation of malononitriles to mononitriles using tributyltin hydride/AIBN was discovered by Curran et al.2a during their studies on atom-transfer reactions of iodomalononitriles 1 and later, they made a full study of decyanation reactions.3 Kang et al.4 also reported the decyanation of both malononitriles 4a and cyanoacetates 4b using samarium(II) iodide/HMPA (Scheme 1).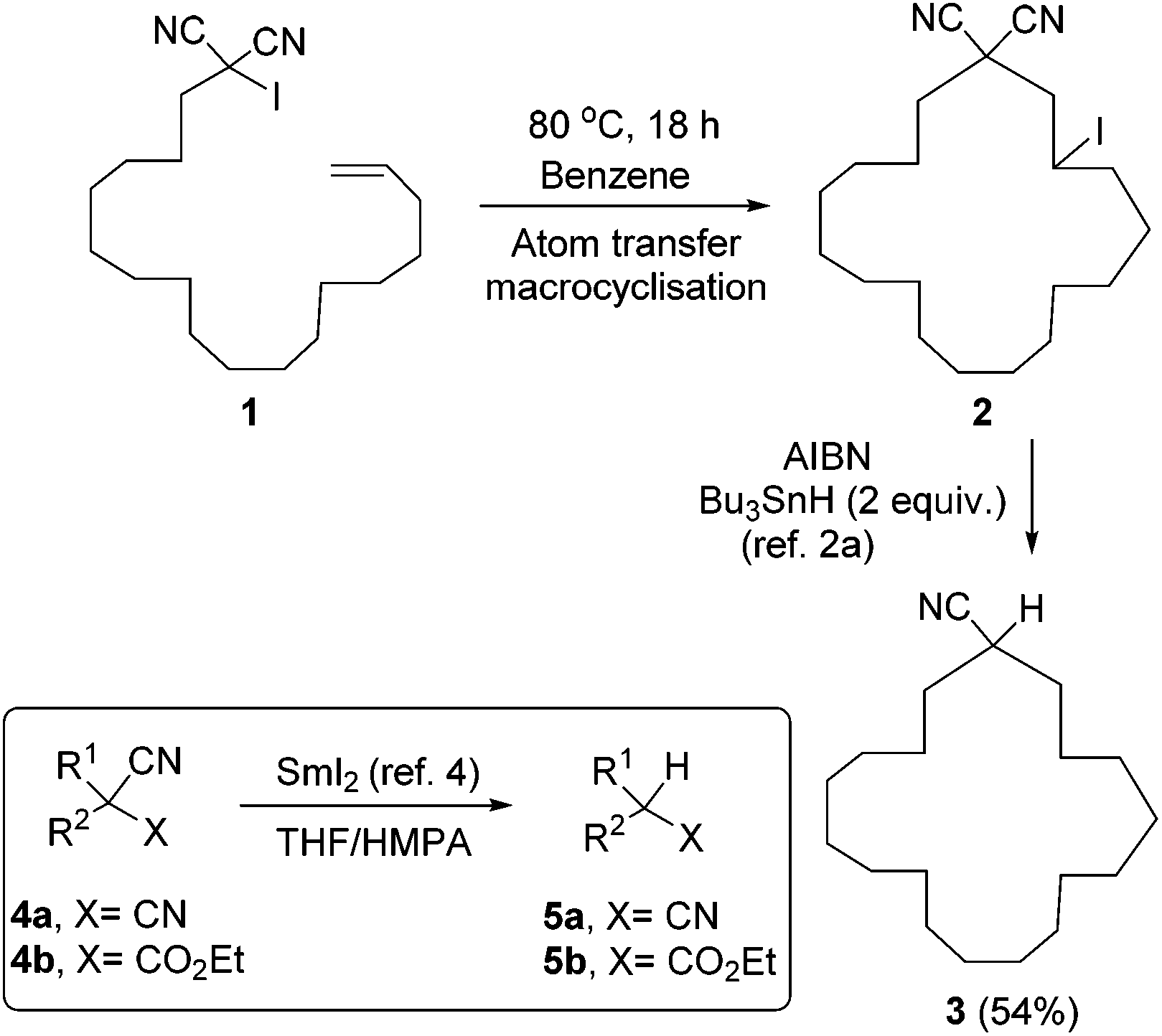 | ||
| Scheme 1 Decyanation of malononitriles and cyanoacetates. | ||
We have recently developed a range of highly reactive neutral, organic super-electron-donors including 6–8 (Scheme 2).5 These molecules donate one or two electrons to suitable substrates and thereby are oxidised to radical cations or dications respectively; the gain in aromaticity in the oxidised products contributes to the driving force for the electron donation. These electron donors perform difficult reduction reactions that are traditionally carried out by metals and metal complexes. For example, efficient single electron transfer (SET) from the donor 6 to unactivated aryl iodides generates the corresponding aryl radicals, while more powerful donors 7 and 8 generate the corresponding aryl anions via double electron transfer. Donors 7 and 8 also reduced arylsulfones,5h arenesulfonamides,5h Weinreb amides,5g acyloin derivatives,5i triflates and triflamides.5l More recently, we showed that photoactivation of the highly coloured donors 7 (vibrant yellow) and 8 (deep purple) enhanced their reducing power and that, under these conditions, they were able to reductively cleave Ar–Cl bonds in chloroarenes in high yields.5k These photoactivated donors were also able to reduce cis-1,2-diphenylcyclopropane 9 to 1,3-diphenylpropane 10 along with epimerised starting material 11.5k
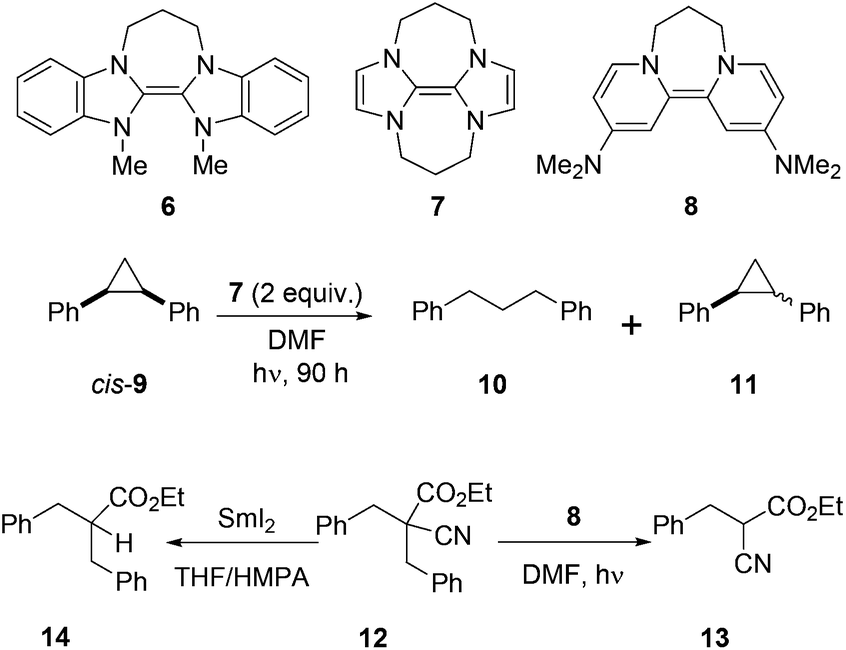 | ||
| Scheme 2 Neutral organic electron donors and their reactivity. | ||
Very recently, we reported selective reduction of arenes over malonate esters and cyanoacetates.5n In that study, substrate 12 selectively provided benzylic C–C bond cleavage product 13. This result was complementary to the earlier work by Kang et al.4 in which the same substrate 12 provided decyanation product 14 upon treatment with SmI2/HMPA. Our interest in decyanation reactions grew as there are very few reports of related chemistry in the literature and also the existing methods featured unfriendly reactions conditions such as using benzene as solvent, HMPA as additive or Bu3SnH as reagent. A new decyanation reaction could be important in extending synthetic applications of malononitriles and, more generally, of nitriles in organic chemistry, and so we now report the results of our detailed study of these reactions.
Results and discussion
We firstly examined the malononitrile substrate 15 (Scheme 3). Surprisingly, the reaction provided both debenzylated product 16 (19%) and decyanation product 17 (75%) (Scheme 2). Decyanation of substrate 15 suggested that an electron donated from a photoactivated donor 8 targeted the nitrile groups instead of the arene ring in the substrate, but it was possible that the arene ring mediated the transfer. Thus, next, it was planned to test dialkyl malononitrile substrates with photoactivated 8, rather than the usual benzyl substrates, to clarify the requirements of the decyanation process. In addition, we needed to establish that the decyanation resulted from photoexcitation of the donor 8, and not from photoactivation of the substrate. We also needed reassurance that potential nucleophilic properties of the donor5i8 played no role in the decyanation reaction. Compound 18 was selected as a test substrate and a set of three parallel reactions (Scheme 3) was carried out simultaneously, to address these points: (a) an original reaction of the substrate with photoactivated 8, (b) a blank reaction under photoactivation conditions, but in the absence of 8 and (c) a blank reaction using 8, but without photoactivation. To our delight, the original reaction (a) provided mononitrile product 19 in excellent yield (94%) while the blank reactions (b) and (c) provided very poor conversion (<2% and <5% respectively) of 18 to 19 but gave excellent recovery of starting material 18 (92% and 90% respectively) (Scheme 3).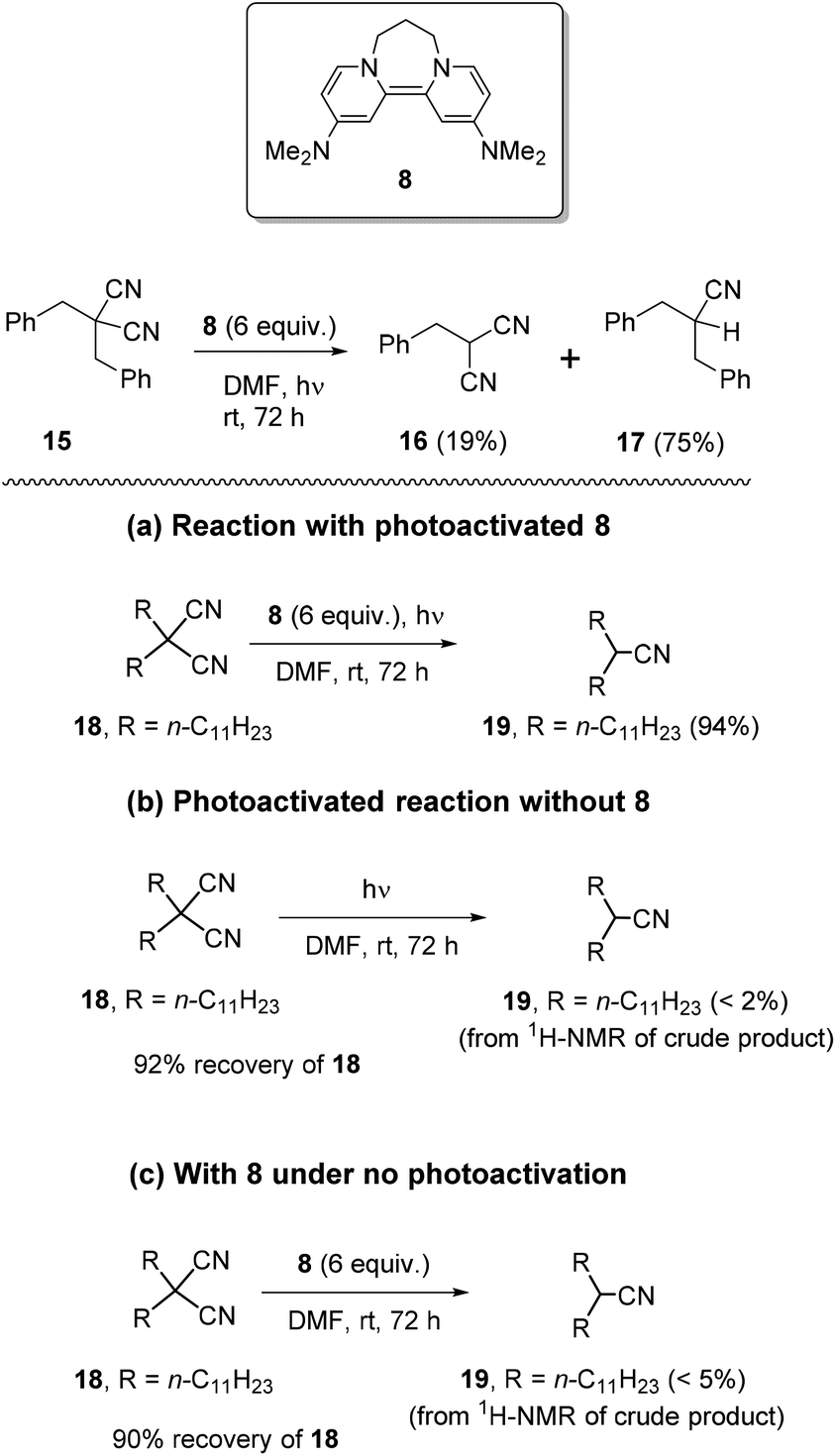 | ||
| Scheme 3 Reactions of photoactivated 8 with malononitriles 15 and 18. | ||
Therefore, the above reactions support electron-transfer as the mechanism for achieving decyanation reactions with photoactivated 8. These results also show that the observed decyanation in 15 can occur without arene rings in the substrate. Encouraged by these results, we now optimised the reaction conditions. Reaction of 18 with photoactivated 8 (4 equiv.) (i) for 48 h provided complete consumption of 18 while (ii) 24 h reaction showed only 89% of conversion of 18 to 19. Later, compound 18 was tested (iii) for 36 h, under the same reaction conditions and showed complete consumption of 18 and provided 19 in excellent isolated yields (92%), following column chromatography (see Table 1 in ESI‡).
Once the optimisation was completed, a series of substrates was prepared and tested with photoactivated 8 and the results are shown in Scheme 4. Substrate 20 showed complete reaction within 24 h, and this faster reaction may be due to mediation of the electron transfer by the two phenyl rings. Substrates 21 and 22, as expected, provided excellent yields of mononitrile products. Substrate 23 with suitably placed alkene groups for cyclisation provided exclusively uncyclised mononitrile product. However, the same compound 23, under Bu3SnH conditions3 had provided both cyclised (24) and uncyclised products and this will be discussed later. Compound 25 afforded exclusive decyanation product and we did not observe any allylic C–C bond cleavage under our reaction conditions and alkene moieties were preserved in the decyanated product.
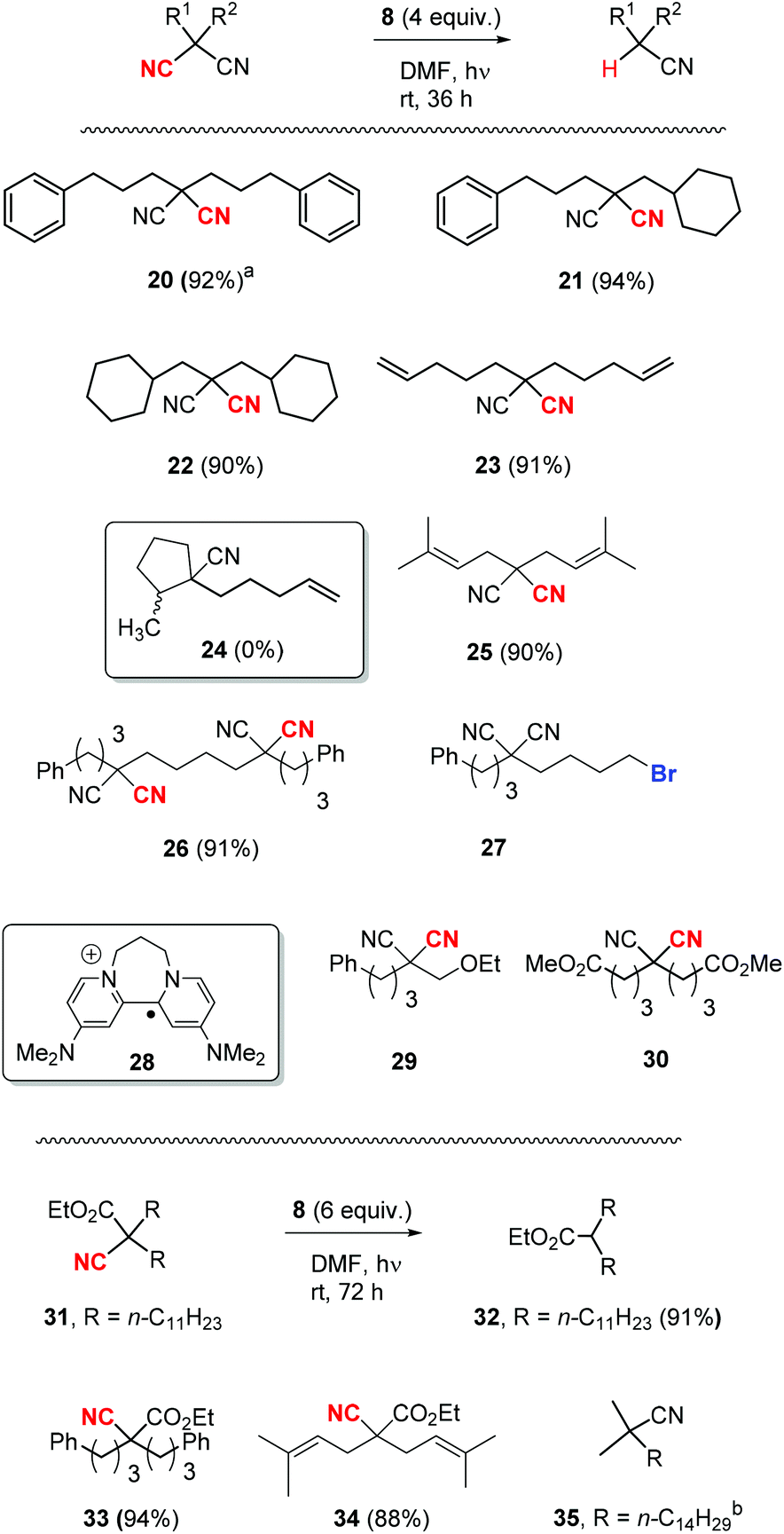 | ||
Scheme 4 Nitrile substrates for decyanation with super-electron-donor 8. Cyano groups depicted in bold red underwent reductive decyanation. The values in parentheses are percent yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was carried out for 24 h. bThis reaction gave recovered 35 (92%). Reaction was carried out for 24 h. bThis reaction gave recovered 35 (92%). | ||
Compound 26 with two malononitrile groups provided decyanation at both sites. Compound 27, featuring two electrophilic sites, (i) a malononitrile moiety and (ii) an alkyl bromide, provided a complete loss of starting material and no products were seen from 1H-NMR analysis of the crude product. This likely results from selective reduction of the alkyl bromide to the corresponding radical intermediate, that is then trapped5p by the persistent radical-cation of the donor 28 to afford water-soluble products. Reactions of mechanistic probes 29 and 30 with photoactivated 8 showed complete consumption of starting materials but yielded complex mixtures of products. This will be discussed later.
After successful decyanation of malononitriles with photoactivated 8, the process was now extended to the closely related cyanoacetates. Decyanation of cyanoacetates is a difficult task6 compared to malononitriles and these reactions had not been successful with tributyltin hydride3 but had worked well using SmI2.4 Firstly, dialkyl cyanoacetate 31 was tested with photoactivated donor 8 and it needed higher amounts of 8 (6 equiv.) along with extended reaction times (72 h) for the complete consumption of starting material but provided an excellent yield (91%) of decyanated product 32. Substrates 33 and 34 were then tested under the same reaction conditions and they provided excellent yields of decyanated products as well. No allylic C–C bond cleavage was observed with 34 under our reaction conditions and the alkene moieties were preserved in the decyanated product (Scheme 4).
Reductive decyanation of simple alkyl mononitriles is highly challenging due to the high C–CN bond dissociation energy (for comparison: 2,2-dimethylmalononitrile vs. isobutyronitrile = 78.9 vs. 126.5 kcal mol−1).6 The mononitrile substrates that underwent successful decyanation reactions by previous methods in the literature were mostly tert-alkyl nitriles, and the decyanation of these substrates was only achieved using solutions of solvated electrons formed from alkali metal.7,8 Intrigued by this, we attempted decyanation of tert-alkyl nitrile 35. However, compound 35 completely resisted attack by our photoactivated 8 and provided recovery of 35 (92%) (Scheme 4).
A plausible mechanism for the above decyanation reactions is shown in Scheme 5. Photoactivated 8 can donate an electron to substrate 36 (X = CN or CO2Et) to form the radical-anion 37 and the radical-cation of the donor, 28. The radical-anion 37 can fragment in two ways i.e. at 37′ to afford cyano radical 38 and stabilised alkyl anion 39 (pathway A, shown in blue) or at 37′′ to afford cyanide anion 40 and alkyl radical 41 (pathway B, shown in red). This alkyl radical 41 can be trapped by the radical cation of the donor, 28, to give water-soluble products, or it can quickly take a second electron from 8 and convert to stabilised alkyl anion 39. The same applies for cyano radical 38 formed in the pathway Ai.e. it can be trapped or further reduced to cyanide anion 40. Finally, stabilised alkyl anion 39, formed from either of pathways A and B, can pick up a proton. From our perspective, both routes are attractive, but what is clear is that under the strongly reductive environment in our reactions, electron-poor radicals are rapidly reduced.5h
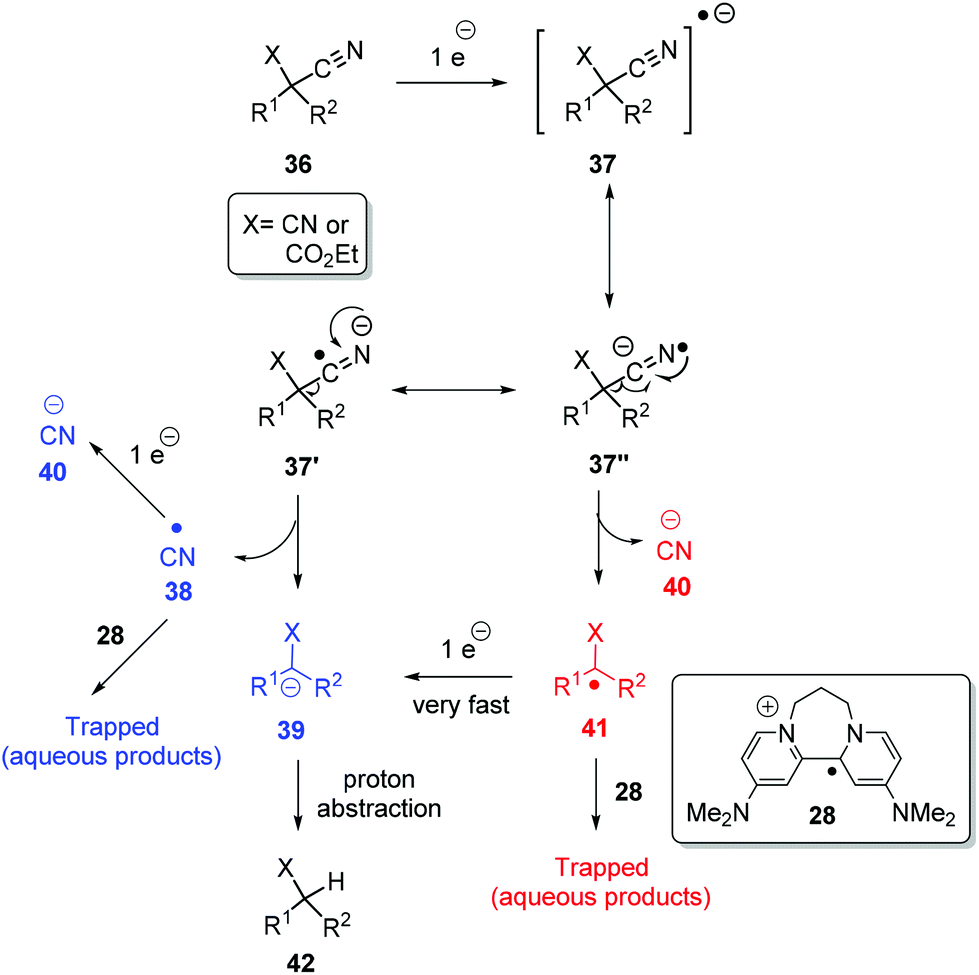 | ||
| Scheme 5 Proposed mechanism for the decyanation reactions. | ||
Returning to discuss substrate 23, it afforded exclusively the uncyclised decyanation product 44 under our reaction conditions. This result differs from Bu3SnH case,3 where the same 23 provided both the cyclised product 24 (major) and uncyclised product 44, arising from radical intermediate 45. So, in our study, we propose stabilised alkyl anion 43 as the key intermediate. As the yields of the reaction are high, trapping of radical intermediates by 28 has very little effect on these reactions and so, second electron transfer in pathway B should be very fast to convert alkyl radical 41 to alkyl anion 39. Substrates 29 and 30 upon reaction with photoactivated 8 provided a complex mixture of products. Again, if an anionic intermediate is the key in these decyanations, these substrates should provide acrylonitrile derivative 47 and β-ketonitrile 49, respectively, and both of these compounds can undergo further reactions or polymerisation under photoactivation conditions (Scheme 6).
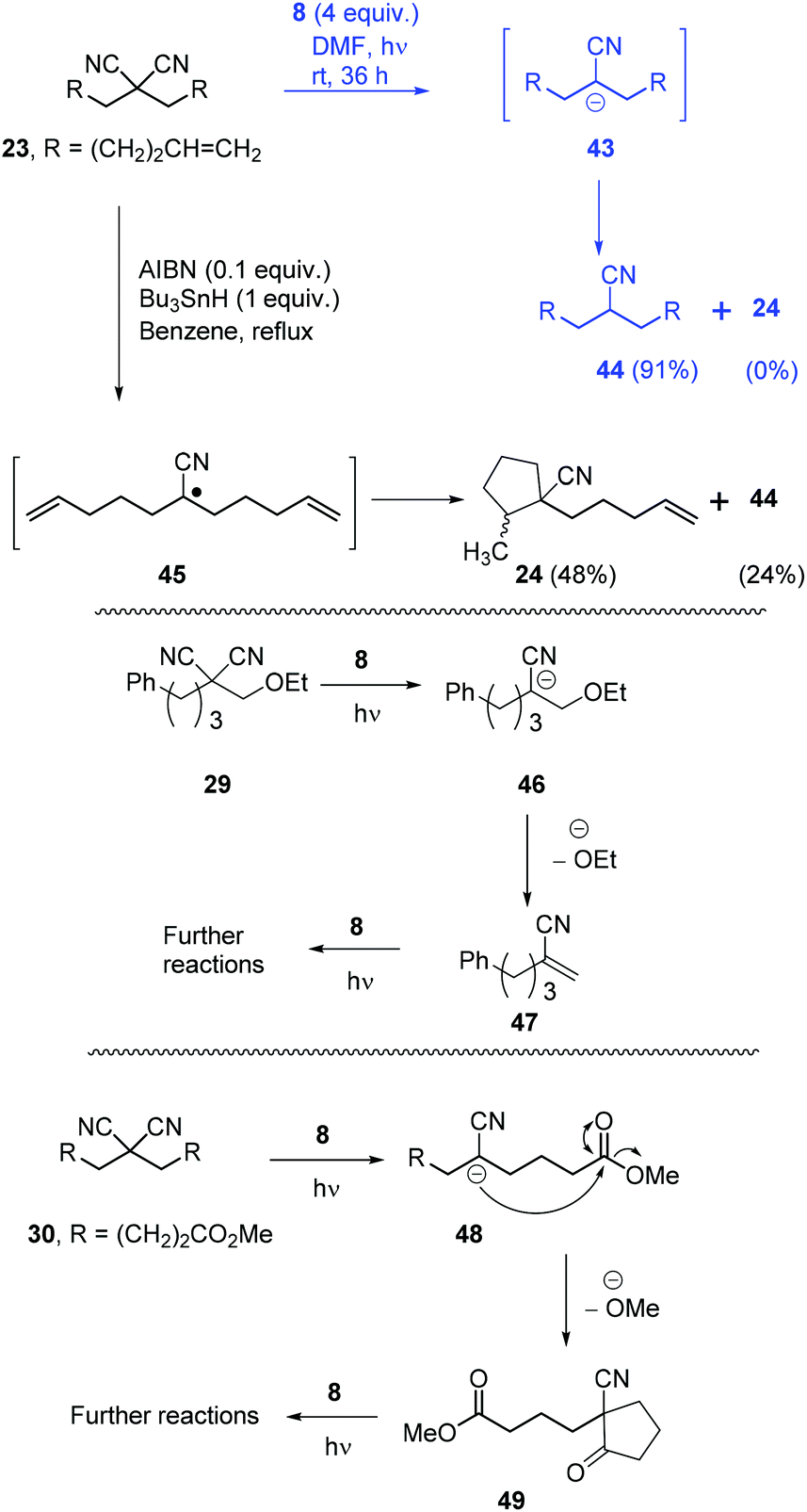 | ||
| Scheme 6 Anionic intermediates are the key in the decyanation reactions. | ||
Conclusions
In conclusion, decyanation of malononitriles and cyanoacetates is effected by neutral organic electron donor 8 under photoactivation. Decyanation of cyanoacetates is more difficult than malononitriles under our reaction conditions, but also proceeds in excellent yields. We propose that stabilised anionic intermediates are the key for these reactions. These reactions have parallels with chemistry induced by SmI2, where decyanation of malononitriles also affords anionic intermediates (no 5-exo cyclisation of intermediates onto alkenes was seen).4 Both types of reaction are conducted at room temperature in the presence of excess reducing agent. However, in the case of SmI2, the presence of HMPA is needed.4 Thus, the current organic electron donors represent an easily prepared and convenient reagent for decyanation of malononitriles and cyanoacetates.Acknowledgements
We thank ORSAS and University of Strathclyde for funding, and the EPSRC national Mass Spectrometry Service for mass spectra.Notes and references
- J. Bloomfield, J. Org. Chem., 1961, 26, 4112 CrossRef CAS.
- (a) D. P. Curran and C. M. Seong, J. Am. Chem. Soc., 1990, 112, 9401 CrossRef CAS; (b) P. Boldt, L. Schulz, U. Klinsmann, H. Köster and W. Thielecke, Tetrahedron, 1970, 26, 3591 CrossRef CAS.
- D. P. Curran and C. M. Seong, Synlett, 1991, 107 CrossRef CAS PubMed.
- H.-Y. Kang, W. S. Hong, Y. S. Cho and H. Y. Koh, Tetrahedron Lett., 1995, 36, 7661 CrossRef CAS.
- (a) E. Doni and J. A. Murphy, Chem. Commun., 2014, 50, 6073 RSC; (b) J. A. Murphy, J. Org. Chem., 2014, 79, 3731 CrossRef CAS PubMed; (c) S. Zhou, H. Farwaha and J. A. Murphy, Chimia, 2012, 66, 418 CrossRef CAS PubMed; (d) J. A. Murphy, T. A. Khan, S. Zhou, D. W. Thomson and M. Mahesh, Angew. Chem., Int. Ed., 2005, 44, 1356 CrossRef CAS PubMed; (e) J. A. Murphy, S. Z. Zhou, D. W. Thomson, F. Schoenebeck, M. Mahesh, S. R. Park, T. Tuttle and L. E. A. Berlouis, Angew. Chem., Int. Ed., 2007, 46, 5178 CrossRef CAS PubMed; (f) J. A. Murphy, J. Garnier, S. R. Park, F. Schoenebeck, S. Z. Zhou and A. T. Turner, Org. Lett., 2008, 10, 1227 CrossRef CAS PubMed; (g) S. P. Y. Cutulic, J. A. Murphy, H. Farwaha, S. Z. Zhou and E. Chrystal, Synlett, 2008, 2132 CAS; (h) F. Schoenebeck, J. A. Murphy, S. Z. Zhou, Y. Uenoyama, Y. Miclo and T. Tuttle, J. Am. Chem. Soc., 2007, 129, 13368 CrossRef CAS PubMed; (i) S. P. Y. Cutulic, N. J. Findlay, S. Z. Zhou, E. J. T. Chrystal and J. A. Murphy, J. Org. Chem., 2009, 74, 8713 CrossRef CAS PubMed; (j) J. A. Murphy, F. Schoenebeck, N. J. Findlay, D. W. Thomson, S. Zhou and J. Garnier, J. Am. Chem. Soc., 2009, 131, 6475 CrossRef CAS PubMed; (k) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673 CrossRef CAS PubMed; (l) P. I. Jolly, N. Fleary-Roberts, S. O'Sullivan, E. Doni, S. Zhou and J. A. Murphy, Org. Biomol. Chem., 2012, 10, 5807 RSC; (m) E. Doni, S. O'Sullivan and J. A. Murphy, Angew. Chem., Int. Ed., 2013, 2239 CrossRef CAS PubMed; (n) E. Doni, B. Mondal, S. O'Sullivan, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2013, 135, 10934 CrossRef CAS PubMed; (o) S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474 CrossRef PubMed; (p) R. Sword, L. A. Baldwin and J. A. Murphy, Org. Biomol. Chem., 2011, 9, 3560 RSC.
- J. C. Lee, H. Y. Koh, Y. S. Lee and H.-Y. Kang, Bull. Korean Chem. Soc., 1997, 18, 783 CAS.
- (a) L. A. Walter and S. M. McElvain, J. Am. Chem. Soc., 1934, 56, 1614 CrossRef CAS; (b) P. G. Arapakos, J. Am. Chem. Soc., 1967, 89, 6794 CrossRef CAS; (c) J. A. Marshall and R. Bierenbaum, J. Org. Chem., 1977, 42, 3309 CrossRef CAS; (d) P. G. Arapakos, M. K. Scott and F. E. Huber Jr., J. Am. Chem. Soc., 1969, 91, 2059 CrossRef CAS; (e) S. D. Rychnovsky, J. P. Bowers and T. J. LePage, J. Am. Chem. Soc., 1992, 114, 8375 CrossRef CAS.
- For electron-transfer induced reduction of mononitriles (a) With SmI2 see: M. M. Szostak, B. Sautier, M. Spain and D. J. Procter, Org. Lett., 2014, 16, 1092 CrossRef CAS PubMed and using other metal reducing agents, see for example: (b) A. R. Doumaux Jr., J. Org. Chem., 1972, 37, 508 CrossRef.
Footnotes |
| † This paper is dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and analyses, together with the NMR spectra of key compounds. See DOI: 10.1039/c4qo00202d |
| This journal is © the Partner Organisations 2014 |