
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of polyaniline-wrapped carbon nanotube-supported PtCo catalysts for proton exchange membrane fuel cells: activity and stability tests
Duanghathai Kaewsaia,
Pornpote Piumsomboonab,
Kejvalee Pruksathornab and
Mali Hunsom *ab
*ab
aFuels Research Center, Department of Chemical Technology, Faculty of Science, Chulalongkorn University, 254 Phayathai Road, Bangkok 10330, Thailand. E-mail: mali.h@chula.ac.th; Fax: +66 2 2555831; Tel: +66 2 2187523 5
bCenter of Excellence on Petrochemical and Materials Technology (PETRO-MAT), Chulalongkorn University, 254 Phayathai Road, Bangkok 10330, Thailand
First published on 11th April 2017
Abstract
A series of the polyaniline (PAN)-wrapped carbon nanotube (CNT)-supported PtCo (PtCo/xPAN–CNT) catalysts was prepared for the oxygen reduction reaction (ORR) in a proton exchange membrane (PEM) fuel cell. The effect of the PAN content wrapped around the multiwall CNTs in the range of 0–15 wt% on the activity and stability of the PtCo catalysts was explored. Increasing the PAN content on the CNT surface did not significantly affect the Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co ratio and catalyst loading on the CNT surface, while it positively affected the electrode conductivity, crystallite size, average particle size and the electrochemical surface area (ESA) as well as the hydrophilic property of the PtCo catalyst. Among all the prepared PtCo catalysts, PtCo/10PAN–CNT exhibited the highest catalytic activity towards the ORR, with a kinetic current density of 36.9 mA cm−2 in 0.5 M H2SO4 and a current density of 407 mA cm−2 at 0.6 V (244 mW cm−2) in a PEM fuel cell under a humidified H2/O2 environment at 60 °C and ambient pressure. The presence of PAN on the CNT surface can reconfigure the catalyst-support interaction, resulting in an increased catalyst stability.
:Co ratio and catalyst loading on the CNT surface, while it positively affected the electrode conductivity, crystallite size, average particle size and the electrochemical surface area (ESA) as well as the hydrophilic property of the PtCo catalyst. Among all the prepared PtCo catalysts, PtCo/10PAN–CNT exhibited the highest catalytic activity towards the ORR, with a kinetic current density of 36.9 mA cm−2 in 0.5 M H2SO4 and a current density of 407 mA cm−2 at 0.6 V (244 mW cm−2) in a PEM fuel cell under a humidified H2/O2 environment at 60 °C and ambient pressure. The presence of PAN on the CNT surface can reconfigure the catalyst-support interaction, resulting in an increased catalyst stability.
1. Introduction
The oxygen reduction reaction (ORR) in a proton exchange membrane (PEM) fuel cell controls the fuel cell performance due to its slower kinetics than the hydrogen oxidation reaction (HOR). To increase the rate of the ORR reaction, dispersed platinum (Pt) nanoparticles (NPs) supported on carbonaceous materials are usually used because the physical/chemical properties of the carbon support greatly affect the electrochemical properties of the fuel cell catalyst.1,2 Carbon materials with high surface area and good crystallinity can achieve a high dispersion of Pt nanoparticles (NPs) as well as facilitate electron transfer, resulting in an improved fuel cell performance.3 Actually, various types of carbon materials have been employed as catalyst supports, such as ordered mesoporous carbons,4,5 carbon gels,6–8 carbon nanohorns/nanocoils,9 graphene10,11 and carbon nanofibers.12,13Among all the explored carbon materials, multiwall carbon nanotubes (CNTs) are the most interesting material for use as a catalyst support for low-temperature fuel cells due to their excellent electrical conductivity, high surface area, high chemical stability and high amount of mesopores, which result in a high metal dispersion and a good reactant flux in their tubular structure.14,15 However, the surface of CNTs is accompanied by many carbonaceous impurities. So, prior to use, CNTs are usually treated in a mixture of HNO3/H2SO4.16 In addition, the surface of CNTs lack sufficient binding sites for anchoring some precursor of metal NPs, leading to a poor dispersion and aggregation of metal NPs.15 Thus, the functionalization of CNT surface by the addition of some basic functional groups such as –NH2, –OH, –SH2 or wrapping of CNTs by a Lewis basic functional polymer film was conducted in order to introduce high binding sites and anchoring groups on the surface of CNTs.17 A mixture of 96% H2SO4 and 4-aminobenzenesulfonic acid can change the chemically active and hydrophilic surface of CNTs, which consequently improved the dispersion of the palladium catalyst.18 Grafting of sulfonic acid groups onto the surface of CNTs by in situ radical polymerization of 4-styrenesulfonate also gave a good subsequent dispersion of Pt NPs and accessibility of protons to the active sites, and so achieved a high fuel cell performance.19 Doping of an appropriate amount of nitrogen on the surface of CNTs can help to reduce the agglomeration of Pt NPs,20 control the crystallite size of catalysts21 and serve as the defect sites to enhance nucleation of Pt NPs.22 The chemical reduction of Pt salts on CNTs in ethylene glycol (EG) solution with refluxing in an argon atmosphere initiated the generation of the OH–(CH2)2–O− ion on the CNT surface, which can bind the CNT surface to reduce Pt ions, leading to the formation of a catalyst with a high stability.23 The wrapping of CNTs by poly(benzimidazole) (PBI) allowed a homogeneous Pt immobilization onto the CNT surface, due to the coordination of the Pt ion with the PBI molecules, and resulted in an improved catalyst utilization efficiency.24 The polyaniline (PAN)-wrapped CNTs achieved a high dispersion, narrow particle size distribution and an excellent electrochemical stability of the Pt NPs, as a result of the π–π bonding interaction between the CNTs and PAN.25 The coating of polypyrrole (PPy) on the surface of CNTs induced a more hydrophilic property of the CNT surface, improved the distribution of Pt NPs and preserved the intrinsic properties of the CNTs, such as their chemical resistance, resulting in a simultaneous improvement in the catalytic activity and stability.26 The Pt NPs on CNTs wrapped with pyridine modified PBI (Pt-Py-m-PBI/CNTs) gave a better fuel cell performance than the Pt/C catalyst, where the performance increased with an increased [2,6-pyridinedicarboxylic acid]/[isophthalic acid] molar ratio of the Py-m-PBIs wrapped on CNTs.27
In this work, in order to decrease the fuel cell cost as well as to improve the electronic and geometric parameters of the Pt metal, the Pt was alloyed with cobalt (Co), a cheap and reactive metal,28 and supported on the PAN-wrapped CNTs. The PAN was selected as the wrapping conductive polymer because of its high conductivity, high chemical resistance and large surface area.29 Additionally, it can provide more nitrogen atoms to bond with metal ions, leading to enhanced anchoring effects.30,31 The effect of weight content of PAN wrapped on the CNT surface on the catalytic activity towards the ORR of PtCo catalyst and its stability in an acid environment was explored. The novelty of this work is the use of polyaniline-wrapped carbon nanotube as a support of PtCo catalysts, which had never been studied before.
2. Experimental
2.1 Preparation of the carbon support and PAN
Two types of commercial carbon compounds were used in this work, which were carbon black (Vulcan XC72, Cabot) and a multi-walled CNT (Nano Generation Co. Ltd). The average diameters of the carbon black and CNT were around 50 nm and 20–40 nm, respectively. Prior to use, they were treated in a 7:3 (v/v) ratio of 12 M mixed HNO3 and H2SO4 by dispersing 0.1 g of the respective carbon black and CNT and stirred at 250 rpm for 6 h. The carbon slurry was held afterwards in the fume hood without stirring for 18 h and then separated from the acid solution by vacuum filtration, rinsed several times by deionized (DI) water until the pH of filtrate was constant. The ready-to-use treated carbon support was obtained after drying at 110 °C for 2 h.
The aniline monomer (99.99%, Panreac) was used as the PAN precursor. In brief, it was distilled under a vacuum condition to remove the inhibitor presented in the monomer solution, and subsequently allowed the aniline monomer to polymerize into the PAN.32
2.2 Preparation of the PAN-wrapped CNT
To prepare the PAN-wrapped CNT (PAN–CNT), 100 mL of 2 M HCl (37%, QRëC) containing 1 g CNT powder was sonicated at room temperature for 2 h and then mixed with a stoichiometric amount of aniline monomer. Approximately 20 mL of 0.2 M ammonium persulfate ((NH4)2S2O8, Univar) was slowly added dropwise into the mixture with constant stirring rate in an ice bath at 0–5 °C for 3 h.33 Subsequently, the dark suspension was filtered and washed several times with DI water until the filtrate became colorless. The ready-to-use PAN–CNT was obtained after drying in an oven at 80 °C for 24 h. The PAN–CNT supports had a weight percent of PAN to CNT of 1, 5, 10 and 15 and were denoted as 1PAN–CNT, 5PAN–CNT, 10PAN–CNT and 15PAN–CNT, respectively. For the various weight contents of PAN to CNT, they were denoted as xPAN–CNT.2.3 Synthesis of the supported PtCo catalysts
Various carbon substances, including the treated carbon black, CNT and xPAN–CNT, were used as a PtCo support. The deposition of the PtCo catalyst on all supports was performed by the impregnation-seeding method using a mixture of 2.844 mL of 20 g L−1 H2PtCl6·6H2O (Alfa Aesar) and 4.324 mL of 20 g L−1 CoCl2 (98%, Fluka) as the Pt and Co precursors, respectively. At the same time, 0.1 g of acid-treated carbon black (or acid-treated CNT or xPAN–CNT) was dispersed in DI water, sonicated at 70 °C for 30 min and then adjusted the pH to 2 with 6 M HCl (37% wt, QRëC) to obtain the carbon slurry.34 For the seeding step, 5 mL of the carbon slurry was mixed with 10% (v/v) of the PtCo solution, sonicated at 70 °C for 30 min and then reduced slowly by the addition of 20 mL of 0.2 M NaBH4. The solid catalyst was obtained by the filtration of catalyst slurry and rinsed several times with DI water. The obtained catalyst was used as a seeded PtCo catalyst in the impregnation step by dispersing in 5 mL DI water, sonicated for 30 min and then added into the remaining PtCo solution (90%, v/v). Subsequently, the mixture was reduced again by the addition of 20 mL of 0.2 M NaBH4 under the sonication to obtain the catalyst slurry. The ready-to-use supported PtCo catalyst was then obtained after the filtration of catalyst slurry, washed with DI water till neutral in pH, and finally dried overnight at 110 °C.2.4 Preparation of the carbon sublayer, catalyst ink and membrane electrode assembly (MEA)
The carbon sublayer ink was prepared by mixing 0.5 mL of distilled H2O with 1.334 μL of 60% (w/w) polytetrafluoroethylene (PTFE; Aldrich) and sonicated at ∼30 °C for 30 min. Then, 1.0 mL of i-propanol (99.99%, Fisher) was added into mixture and sonicated at room temperature for 30 min. Afterward, 0.018 g of acid-treated carbon (Section 2.1) was added and sonicated at room temperature for 30 min to obtain the carbon ink, which was then applied onto a 2.25 × 2.25 cm2 gas diffusion layer (GDL) (Carbon cloth, ETEK) by brushing and dried at 80 °C for 2 min to remove an excess solvent. The coating of sublayer ink was repeated to get the sublayer loading of ∼2.0 mg cm−2. Finally, the sublayer ink-coated GDL was dried at 300 °C for 1 h at ambient condition.To prepare the catalyst ink, 25 mg of the respective catalysts (PtCo/C, PtCo/CNT or PtCo/xPAN–CNT) was mixed with 1 mL DI water and sonicated for 1 h. Afterward, 1 mL isopropanol was added and sonicated continuously for 1 h. Subsequently, 0.44 mL of Nafion (5% wt., Fluka) solution was added and sonicated for 2 h at room temperature. The obtained catalyst ink was coated on the first surface of a 5 cm2 treated membrane (Nafion 115, Electrochem, Inc.) at 80 °C using a spray gun (Crescendo, Model 175-7TM) to obtain a catalyst loading of 0.15 mg cm−2. The same procedure was repeated for the second membrane surface, but using the commercial Pt/C (20% (w/w), ETEK) catalyst. Finally, two sheets of sublayer ink-coated GDL were put on both sides of the catalyst-coated membrane and pressed together by a compression mold (LP20, Labtech) at 137 °C with a pressure of 65 kgf cm−2 for 2.5 min to obtain the MEA.
2.5 ORR activity test
The ORR activities of all the catalysts were investigated in an acid solution (0.5 M H2SO4) and in a PEM fuel cell with a H2/O2 environment at ambient pressure. For the first test, 1 μL of each catalyst ink was dripped onto a glassy carbon (GC) electrode of 2 mm in diameter, assembled with the rotating disk electrode (RDE). It was then connected with the Potentiostat/Galvanostat (Autolab, PG Stato 30) as the working electrode, while a Pt rod and Ag/AgCl reference electrode were used as the counter and reference electrodes, respectively. The O2 (99.9999%, Praxair) was supplied to the system during the test to ensure the O2 saturation of the 0.5 M H2SO4. The test was performed with linear sweep voltammetry (LSV) using a scan potential in the range of 0.0 to +0.8 V at different rotation rates (500–2000 rpm) and a scan rate of 10 mV s−1. For the activity test in the PEM fuel cell, the MEA obtained from Section 2.4 was mounted on a commercial single-cell hardware (Electrochem, Inc.) and constructed on an in-house test station at 60 °C under ambient pressure. The H2 (99.99%, Praxiar) and O2 (99.9999%, Praxair) gases were humidified at 65 °C and 60 °C, respectively before fed to the fuel cell at constant flow rates of 100 sccm. The current density–potential data curve was recorded after the fuel cell was run-in or operated at a high current density (>700 mA cm−2) for at least 6–12 h.2.6 Stability test
The stability test of all the prepared catalysts was conducted using a three-electrode system. An Ag/AgCl electrode, Pt and the respective prepared catalysts were used as the reference, counter and working electrodes, respectively. Briefly, to prepare the working electrode, the catalyst ink was dripped onto the GC electrode with 2 mm in diameter, assembled with the RDE and connected with the Potentiostat/Galvanostat (Autolab, PG Stato 30). The measurement was performed at room temperature in N2-saturated 0.5 M H2SO4 solution with a scan rate of 10 mV s−1 in the potential range of −0.2 to +0.8 V via the cyclic voltammetry (CV).2.7 Characterizations
The surface functional group of all xPAN–CNT samples was qualitatively explored by Fourier transform infrared spectroscopy (FTIR; PerkinElmer Spectrum One) using the potassium bromide (KBr) pellet method. The morphologies of all prepared catalysts were analyzed using X-ray diffractometry (XRD; D8 Discover-Bruker AXS), transmission electron microscope (TEM; Phillips Tecnai G2) and a scanning electron microscope (SEM) on a J6M 7610F machine. The PtCo content of each sample was determined by thermogravimetric analysis (TGA; TG 209 F3 Tarsus) with a heating rate of 10 °C min−1. The in-plane conductivity of all catalysts was measured using a four point probe instrument (Jandel, RM3-AR). The electrochemical surface area (ESA) was estimated using the H2 stripping method in N2-saturated 0.5 M H2SO4 via the CV experiment. The ESA of each catalyst was calculated according to eqn (1);35
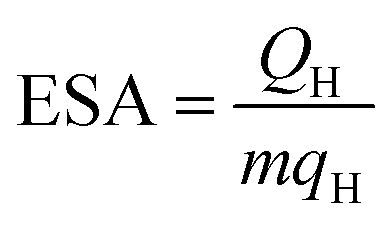 | (1) |
The hydrophobic/hydrophilic property of all prepared PtCo catalysts was measured in terms of contact angle via the Ramé-hart contact angle goniometer (model 200-U1).
3. Results and discussion
3.1 Surface functional group of the PAN–CNT supports
The surface functional groups of the CNT and xPAN–CNT support were explored by FTIR (Fig. 1), where the FTIR spectra of CNT showed a weak peak of CNT backbone stretching mode at a wave number of 1531 cm−1. The FTIR spectra of xPAN–CNT appeared at the wave number of ∼1539 and ∼1460 cm−1, corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC stretch of the quinoid and benzenoid structure of PAN, respectively. The bands at ∼1269 and ∼1207 cm−1 were attributed to the C–N stretching mode of benzenoid units. The absorption peaks at ∼1136 cm−1 were assigned to the C–H in-plane bending, while the peak at ∼808 cm−1 was attributed to the C–H out-of-plane bending. This suggests that the PAN was presented in the form of an emeraldine salt,33,36 which is highly electrically conducting.37
N and CC stretch of the quinoid and benzenoid structure of PAN, respectively. The bands at ∼1269 and ∼1207 cm−1 were attributed to the C–N stretching mode of benzenoid units. The absorption peaks at ∼1136 cm−1 were assigned to the C–H in-plane bending, while the peak at ∼808 cm−1 was attributed to the C–H out-of-plane bending. This suggests that the PAN was presented in the form of an emeraldine salt,33,36 which is highly electrically conducting.37
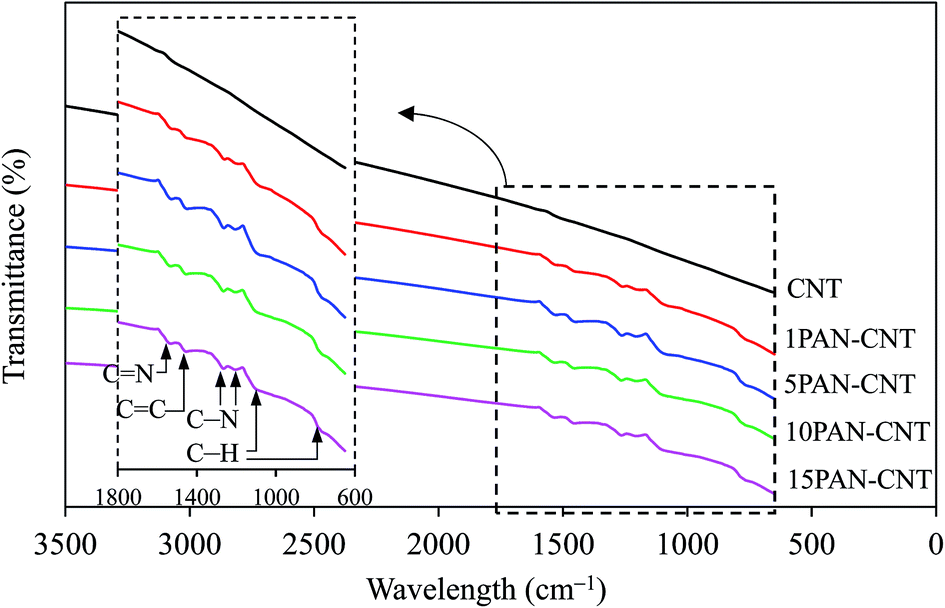 | ||
| Fig. 1 FTIR spectra of the utilized CNT and PAN–CNT supports. | ||
3.2 Catalyst morphology
The morphology of the prepared catalysts was first explored using XRD analysis. As demonstrated in Fig. 2, the XRD of Pt/C showed four obvious peaks. A broad peak at 2θ of 25.01° indicated the graphite structure of the carbon support. The other three peaks appeared at a 2θ of 39.58°, 46.04° and 67.09° represented the characteristic diffraction peaks of Pt(111), Pt(200) and Pt(220), respectively. For the PtCo/C catalyst, no crystalline structure of Co was observed, while all three characteristic diffraction peaks of Pt were still observed with a positive shift in the 2θ value compared with the commercial Pt/C sample. This indicates the formation of the PtCo alloy by the intercalation of the Co particle in the face-centered cubic (FCC) structure of Pt.38,39 With respect to the PtCo/xPAN–CNT catalysts, four XRD peaks of each respective catalyst were also observed. The first peak appeared at a 2θ of 25.66°, ascribed to the diffraction peak of the CNT structure. The three other peaks at a 2θ of 39.78–39.82°, 46.26–46.33° and 67.46–47.54° were the characteristic peaks Pt(111), Pt(200) and Pt(220), respectively. Compared with the peaks of the Pt/C catalyst, the shift in the 2θ of the Pt peaks was still observed, suggesting the formation of the PtCo alloy on the xPAN–CNT support. The lattice parameters of all supported PtCo catalysts were found to be lower than that of the Pt/C catalyst, confirming the formation of the PtCo alloy on the CNT support in both the absence and presence of PAN. | ||
| Fig. 2 Representative XRD patterns of Pt/C and supported PtCo catalysts. | ||
The average crystallite size (D) of all prepared catalysts was then estimated from the half-height of the peak Pt(111) plane using the Scherrer equation,40 as shown in eqn (2);
 | (2) |
As listed in Table 1, the crystallite size of Pt in PtCo/C catalyst was significantly larger than that of the Pt/C catalyst, while the crystallite sizes of PtCo/CNT were slightly greater than that of PtCo/C catalyst, conformable to the average particle size of PtCo catalyst obtained by TEM analysis (Fig. 3 and Table 1). This is probably due to the different quantity of the oxygen surface groups on the surface of CNT compared to that on carbon.41 The high density of oxygen surface groups can facilitate an increased catalyst crystallite size by hindering the reduction of the Pt precursor,42 diminishing the metal-support interaction,43 and reducing the number of surface basic sites, which are the centers for the strong adsorption of PtCl62−.44 The 1PAN–CNT support provided an almost similar crystallite size and average particle size of PtCo catalyst compared with the bare CNT. Further raising the PAN content from 1 to 15 wt% slightly decreased the crystallite size and average particle size of the PtCo catalyst, which is probably due to the presence of high amount of the N-group on the PAN structure that can bond directly with metal ions and consequently allowed a uniform reduction of the metal ions on the CNT surface.30,31
| Catalyst | Lattice parameter (nm) | Crystallite size (nm) | Average particle size (nm) | XRD atomica composition (Pt:Co) |
PtCo loading on xPAN–CNT (wt%) | Electrode conductivity (S cm−1) | ESA (m2 gPt−1) | Contact angle | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated from Vegard's law using (111) data. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pt/C | 0.3938 | 3.84 | 4.02 ± 0.75 | 100:0 |
20.22 | 20.15 | 50.93 | 138.04 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PtCo/C | 0.3918 | 7.04 | 6.55 ± 0.91 | 85.28:14.72 |
27.90 | 18.87 | 58.69 | 144.58 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PtCo/CNT | 0.3920 | 7.72 | 6.87 ± 1.44 | 86.29:13.71 |
27.87 | 22.18 | 58.92 | 151.86 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PtCo/1PAN–CNT | 0.3920 | 7.71 | 6.77 ± 1.25 | 86.29:13.71 |
27.92 | 22.54 | 59.43 | 146.88 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PtCo/5PAN–CNT | 0.3920 | 5.79 | 5.80 ± 0.90 | 86.29:13.71 |
28.09 | 22.97 | 60.07 | 144.34 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PtCo/10PAN–CNT | 0.3920 | 5.49 | 5.73 ± 0.78 | 86.29:13.71 |
28.91 | 25.45 | 63.34 | 143.50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PtCo/15PAN–CNT | 0.3916 | 5.16 | 5.41 ± 0.82 | 84.26:15.74 |
29.99 | 25.54 | 74.58 | 132.96 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
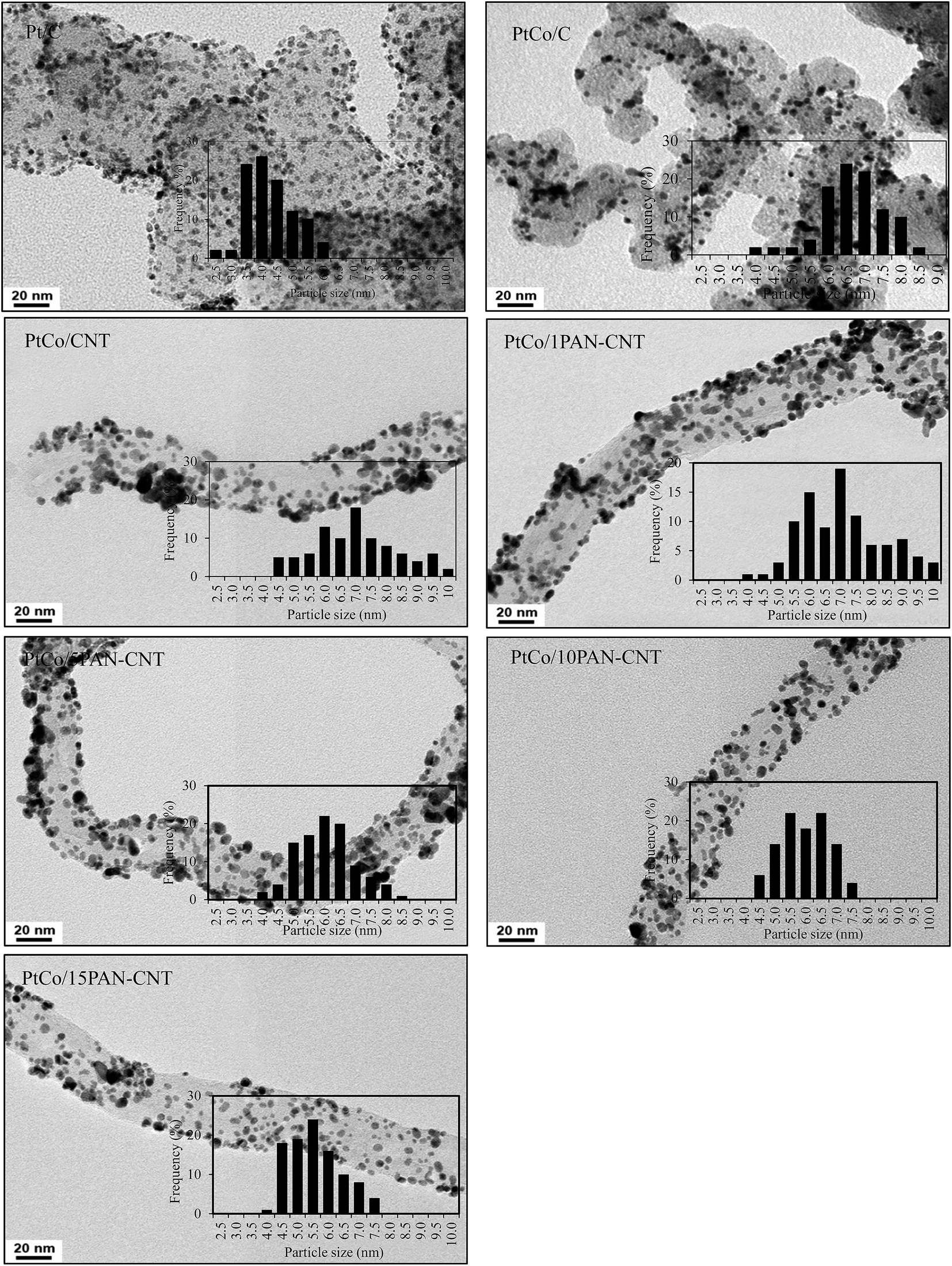 | ||
| Fig. 3 Representative of TEM and particle size distribution of Pt/C and supported PtCo catalysts. | ||
The entire Pt:Co atomic ratio in the bulk chemical composition of the supported PtCo catalysts was calculated using Vegard's law (eqn (3)).45 As summarized in Table 1, it can be seen that the Pt:Co atomic composition in all PtCo catalysts were almost the same (∼85.78:14.22), suggesting that the wrapping of CNT surface by different PAN contents had insignificant effect on the Pt:Co atomic ratio in the bulk chemical composition of the supported PtCo catalysts.
 | (3) |
Variations in the PtCo weight content of the supported PtCo catalysts together with the Pt/C one were determined by TGA analysis (Fig. 4). A complete weight loss was observed for all supports after 600 °C. However, in the presence of the loaded catalyst, the final weight of all catalysts were almost the same at between 27.9–30.0%. Regarding the electrode conductivity, the PtCo/CNT catalyst exhibited a higher electrode conductivity than either the Pt/C or PtCo/C catalysts, attributed to the fact that the CNT has a higher electrical conductivity than the carbon support.46 The wrapping of PAN on the CNT surface increased the electrical conductivity of all the PtCo catalysts, where increasing the PAN content within the investigated range (1–5 wt%) increased the electrical conductivity of the PtCo catalyst from 22.54 to 25.54 S cm−1. The ESA also increased slightly with increased PAN contents, probably due to the optimization of Pt particles by using polar aniline as the dispersant and the polymeric stabilization mechanism of PAN.25 The contact angle of the all PAN-wrapped CNT-supported PtCo catalysts decreased as the increased PAN content (Table 1), indicating an increase of hydrophilic property of the catalyst layer.
 | ||
| Fig. 4 TGA curves of Pt/C and supported PtCo catalysts. | ||
3.3 ORR activity test
The experimental results for the ORR in O2-saturated 0.5 M H2SO4 are summarized in Fig. 5(a). The onset potential for ORR of the PtCo/10PAN–CNT catalyst was shifted to more positive potentials compared with other supported PtCo catalysts as well as that for Pt/C one. In addition, its overpotential at current densities of 1 mA cm−2 was ca. 20 mV lower than that for the pure Pt. These suggests that the PtCo/10PAN–CNT catalyst showed the highest ORR activity compared with the other catalysts. | ||
| Fig. 5 (a) Steady-state polarization curves for ORR at 2000 rpm, and (inset) the Koutecky–Levich plots at 0.6 V (SHE), (b) the kinetic current density at 0.6 V of Pt/C and supported PtCo catalysts in O2-saturated 0.5 M H2SO4, and (c) number of electrons transferred for the oxygen reduction. | ||
From the steady-state polarization curves, the correlation between the current density and the rotation rate can be described by the Koutecky–Levich equation (eqn (4)), which is valid for the first order process with respect to the diffusion species.47
 | (4) |
| B = 0.62neFD2/3ν−1/6C | (5) |
The plot of j−1 vs. ω−1/2 provided a straight line, as demonstrated in the inset of Fig. 5(a), which allowed to determine the kinetic current density (jk), which can be used to demonstrate the catalytic activity towards the ORR in an acid electrolyte. As shown in Fig. 5(b), the PtCo/10PAN–CNT catalyst exhibited a higher ORR activity, measured in terms of the kinetic current density (∼36.9 mA cm−2), than the other supported PtCo catalysts as well as the Pt/C catalyst. This is probably due to a more uniform distribution of ORR active sites on the surface of the catalyst and a high conductivity and ESA. However, although the PtCo/15PAN–CNT catalyst had a higher ESA than the PtCo/10PAN–CNT catalyst, it exhibited a lower ORR activity. This probably due to its high hydrophilic property (Table 1), which allowed the adherence of water layer on catalyst surface, leading to lower active site of catalyst to proceed the reaction, which consequently reduce the ORR activity. Furthermore, number of electrons transferred for the ORR can be estimated using eqn (5). The number of transferred electrons of all samples in the process was ranging from 3.56 to 3.96 (Fig. 5(c)). As a result, it can be said that the ORR on the supported PtCo catalyst involved the 4-electron pathway.
The activity of the prepared supported PtCo catalysts was also explored in a PEM fuel cell under a H2/O2 atmosphere at 60 °C and ambient pressure. As demonstrated in Fig. 6(a), all the prepared catalysts provided an almost similar open circuit potential in the range of 0.935–0.961 V, suggesting that using the different contents of PAN wrapped on the CNT surface did not significantly affect the fuel crossover as well as the internal current formation. Under the activation region (<100 mA cm−2), a difference in the produced current density was observed. In terms of the current density-IR-free cell potential plot (inset of Fig. 6), the current density estimated at 0.9 V of PtCo/C (5.45 mA cm−2) was 1.28-fold smaller than that for the PtCo/CNT (6.98 mA cm−2). The addition of PAN wrapped on the CNT surface at the content of 1–10 wt% enhance an increase of the current density at 0.9 V (j0.9 V,IR-free) of the PtCo catalysts as demonstrated in Table 2. Further raising the PAN content to 15 wt% decreased the j0.9 V,IR-free. This suggests that the PtCo/10PAN–CNT catalyst provided the highest ORR activity, agreeing with the activity tested in acid solution (Fig. 5(b)).
 | ||
| Fig. 6 (a) Current density-potential and (b) power density curves of Pt/C and supported PtCo catalysts in a single PEM fuel cell obtained at 60 °C under the H2/O2 environment and ambient pressure. | ||
| Catalyst | j0.9 V,IR-free (mA cm−2) | j0.6 V (mA cm−2) | Parameters obtained from the NLLS model | R2 | ||||
|---|---|---|---|---|---|---|---|---|
| −b (mV dec−1) | j0 (mA cm−2) | R (Ω cm−2) | m (mV) | n (cm2 mA−1) | ||||
| Pt/C | 3.95 | 228 | 58.0 | 2.48 × 10−5 | 0.8350 | 2.60 | 0.0051 | 0.9997 |
| PtCo/C | 5.45 | 250 | 56.6 | 2.37 × 10−5 | 0.7800 | 2.50 | 0.0050 | 0.9999 |
| PtCo/CNT | 6.98 | 308 | 57.0 | 4.71 × 10−5 | 0.6550 | 2.90 | 0.0053 | 0.9994 |
| PtCo/1PAN–CNT | 8.15 | 319 | 56.5 | 2.68 × 10−5 | 0.6100 | 4.85 | 0.0040 | 0.9997 |
| PtCo/5PAN–CNT | 12.00 | 362 | 59.0 | 1.02 × 10−4 | 0.5200 | 7.25 | 0.0037 | 0.9998 |
| PtCo/10PAN–CNT | 15.95 | 407 | 59.5 | 1.44 × 10−4 | 0.4950 | 7.35 | 0.0033 | 0.9997 |
| PtCo/15PAN–CNT | 11.34 | 332 | 58.0 | 7.10 × 10−5 | 0.6000 | 4.60 | 0.0040 | 0.9997 |
A significant difference in the generated current density among all explored catalysts was observed under a medium-to-high current density (the ohmic region). The PtCo/10PAN–CNT catalyst still provided the highest current density of 407 mA cm−2 at 0.6 V, equivalent to 244 mW cm−2 (Fig. 6(b)).
The kinetic parameters for ORR of the explored catalysts in the PEM fuel cell could be estimated from the current density-potential data using a nonlinear least squares (NLLS) method, as expressed by eqn (6);48
E = E0 − blogj − jR − m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(nj) exp(nj)
| (6) |
|
E0 = Er + blogj0
| (7) |
The ORR parameters obtained from the NLLS method are summarized in Table 2. The coefficient of determination (R2) of the explored catalysts was greater than 0.9994 in all cases, suggesting that the utilized model is adequate to predict the experimental data. The intrinsic Tafel slopes (b) of all the catalysts towards the ORR varied in the range of −56.5 to −59.5 mV dec−1 under similar testing conditions, suggesting the formation of uniform interphase conditions in the presence of the different contents of PAN wrapped on the CNT surface. The variation trend of the exchange current density (j0) was similar to the current density at 0.9 V and 0.6 V, indicating that the PtCo/10PAN–CNT catalyst had the highest ORR activity. The PtCo/10PAN–CNT catalyst also exhibited the lowest total resistance (R), contributed from the charge transfer resistance for ORR and the maximum electronic resistance of the single-cell test and mass-transport resistance than the other investigated catalysts. Interestingly, this catalyst displayed a higher value of m than the other catalysts, suggesting its high mass transport loss compared with the other catalysts. Nonetheless, its high exchange current density, electrical conductivity and ESA and its low resistance and value of n as well as appropriate hydrophilic property led to the higher performance in the PEM fuel cell than the other PtCo catalysts.
Table 3 shows a comparison of the ORR activity in acid solution and in PEM fuel cell between the best catalyst obtained from this work (PtCo/10PAN–CNT) and some previous works. It can be seen that although this catalyst gave the performance in PEM fuel cell at 0.6 V of around 407 mA cm−2, it provided the mass activity higher than the PtCo/C-CeOx,49 Pt3Co1/Ti0.75Ta0.25O250 and PtCo/rG-O51 catalysts both in acid solution and in PEM fuel cell. This suggests that a high catalyst utilization was obtained in this work.
| Author | Type of catalyst | Mass activity in acid solution (mA mg−1) | Activity in PEM fuel cell at 0.6 V |
|---|---|---|---|
| Lee et al.49 | PtCo/C-CeOx | 9.3 | ∼600 mA cm−2 (1.6 mg cm−2 Pt loading, polybenzoxazine-based membrane, at 150 °C) (375 mA mg−1) |
| Stassi et al.50 | Pt3Co1/Ti0.75Ta0.25O2 | — | ∼650 mA cm−2 (0.3 mg cm−2 Pt loading, Nafion 115 membrane, at 80 °C) (2167 mA mg−1) |
| Li et al.51 | PtCo/rG-O | 32 | — |
| This work | PtCo/10PAN–CNT | 246 | ∼407 mA cm−2 (0.15 mg cm−2 Pt loading, Nafion 115 membrane, at 60 °C) (2713 mA mg−1) |
3.4 Stability test
The stability of all the prepared supported PtCo catalysts as well as the Pt/C one was tested by the repetitive CV in N2-saturated 0.5 M H2SO4 at a potential range of −0.2 and +0.8 V at a scan rate of 20 mV s−1. A representative repetitive CV trace is shown in Fig. 7(a), where a well-defined hydrogen adsorption–desorption peak was observed. The peaks with a high current density were observed for the early period of the repetitive CV and their intensity decreased with increasing rounds of the repetitive CV, suggesting an alternation of active surface area of catalyst. The quantitative ESA of all prepared catalysts was then determined via eqn (1) and demonstrated in terms of the normalized ESA, as shown in Fig. 7(b). The PtCo/C catalyst exhibited a slightly higher stability than the Pt/C catalyst, caused by the fact that the Co metal can reconfigure the catalyst-support interaction. As expected, all CNT-supported PtCo catalysts showed a low loss of normalized ESA compared with the PtCo/C catalysts. This loss slightly decreased as the PAN content increased, suggesting that the presence of PAN can help to reconfigure the catalyst-carbon interaction as well as the corrosion of support. | ||
| Fig. 7 (a) Repetitive CV of PtCo/10PAN–CNT and (b) the normalized ESA of Pt/C and supported PtCo catalysts in N2-saturated 0.5 M H2SO4 at a scan rate of 20 mV s−1. | ||
4. Conclusions
The effect of the weight content of PAN wrapped on the CNT surface on the ORR activity and stability in acid environment was explored in this work. It was found that increasing the PAN content wrapped on the CNT surface did not significantly affect the Pt:Co ratio and catalyst loading on the CNT surface. However, the addition of high PAN content decreased the crystallite size and average particle size of PtCo catalyst, but increased the electrode conductivity and hydrophilic property, which consequently effected the ORR activity of the PtCo catalysts. The presence of PAN on the CNT surface can reconfigure the catalyst-support interaction, resulting in an increased catalyst stability. Among all prepared catalysts, the PtCo/10PAN–CNT catalyst exhibited the highest ORR activity and stability compared with other prepared catalysts. That is, it exhibited the highest kinetic current density (36.9 mA cm−2) in 0.5 M H2SO4 and the highest current density (407 mA cm−2 at 0.6 V) in a PEM fuel cell under a humidified H2/O2 environment at 60 °C and ambient pressure. A loss of ESA was around 13% after 500 cycles of repetitive CV test, which was 2.7- and 2.5-fold lower than that of the Pt/C and PtCo/C catalysts, respectively.
Acknowledgements
This research was funded by the Postdoctoral Fellowship under Rachadapisaek Sompote Fund, the Graduate School, Chulalongkorn University to Duanghathai Kaewsai.References
- K. I. Han, J. S. Lee, S. O. Park, S. W. Lee, Y. M. Park and H. Kim, Electrochim. Acta, 2004, 50, 791–794 CrossRef CAS.
- R. V. Hull, L. Li, Y. Xing and C. C. Chusuei, Chem. Mater., 2006, 18, 1780–1788 CrossRef CAS.
- K. W. Park, Y. E. Sung, S. Han, Y. Yun and T. Hyeon, J. Phys. Chem. B, 2004, 108, 939–944 CrossRef CAS.
- V. Raghuveer and A. Manthiram, Electrochem. Solid-State Lett., 2004, 7, A336–A339 CrossRef CAS.
- H. Chang, S. H. Joo and C. Pak, J. Mater. Chem., 2007, 17, 3078–3088 RSC.
- Z. Zhou, W. Zhou, S. Wang, G. Wang, L. Jiang, H. Li, G. Sun and Q. Xin, Catal. Today, 2004, 93, 523–528 CrossRef.
- C. Arbizzani, S. Beninati, E. Manferrari, F. Soavi and M. Mastragostino, J. Power Sources, 2006, 161, 826–830 CrossRef CAS.
- J. Marie, S. Berthon-Fabry, M. Chatenet, E. Chainet, R. Pirard, N. Cornet and P. Achard, J. Appl. Electrochem., 2007, 37, 147–153 CrossRef CAS.
- M. Sevilla, G. Lota and A. B. Fuertes, J. Power Sources, 2007, 171, 546–551 CrossRef CAS.
- M. Sahoo, B. P. Vinayana and S. Ramaprabhu, RSC Adv., 2014, 4, 26140–26148 RSC.
- L. I. Şanlı, V. Bayram, B. Yarar, S. Ghobadi and S. A. Gürsel, Int. J. Hydrogen Energy, 2016, 41, 3414–3427 CrossRef.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- F. Yuan and H. Ryu, Nanotechnology, 2004, 15, S596–S602 CrossRef CAS.
- S. L. Knupp, W. Li, O. Paschos, T. M. Murray, J. Snyder and P. Haldar, Carbon, 2008, 46, 1276–1284 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS.
- W. Z. Li, C. H. Liang, W. J. Zhou, J. S. Qiu, Z. H. Zhou, G. Q. Sun and Q. Xin, J. Phys. Chem. B, 2003, 107, 6292–6299 CrossRef CAS.
- W. Zhang, P. Sherrell, A. I. Minett, J. M. Razal and J. Chen, Energy Environ. Sci., 2010, 3, 1286–1293 CAS.
- S. Yang, X. Zhang, H. Mi and X. Ye, J. Power Sources, 2008, 175, 26–32 CrossRef CAS.
- C. Y. Du, T. S. Zhao and Z. X. Liang, J. Power Sources, 2008, 176, 9–15 CrossRef CAS.
- T. Maiyalagan, B. Viswanathan and U. V. Varadaraju, Electrochem. Commun., 2005, 7, 905–912 CrossRef CAS.
- T. Maiyalagan, Appl. Catal., B, 2008, 80, 286–295 CrossRef CAS.
- H. Y. Du, C. H. Wang, H. C. Hsu, S. T. Chang, U. S. Chen, S. C. Yen, L. C. Chen, H. C. Shih and K. H. Chen, Diamond Relat. Mater., 2008, 17, 535–541 CrossRef CAS.
- C. Gupta, P. H. Maheshwari and S. R. Dhakate, Mater. Renewable Sustainable Energy, 2016, 5, 1–11 CrossRef.
- T. Fujigaya, M. Okamoto and N. Nakashima, Carbon, 2009, 47, 3227–3232 CrossRef CAS.
- D. He, C. Zeng, C. Xu, N. Cheng, H. Li, S. Mu and M. Pan, Langmuir, 2011, 27, 5582–5588 CrossRef CAS PubMed.
- H. S. Oh, K. Kim and H. Kim, Int. J. Hydrogen Energy, 2011, 36, 11564–11571 CrossRef CAS.
- D. Kaewsai, H. L. Lin, Y. C. Liu and T. L. Yu, Int. J. Hydrogen Energy, 2016, 41, 10430–10445 CrossRef CAS.
- T. He, E. Kreidler, L. Xiong, J. Luo and C. J. Zhong, J. Electrochem. Soc., 2006, 153, A1637–A1643 CrossRef CAS.
- D. Prasanna and V. Selvaraj, Korean J. Chem. Eng., 2016, 33, 1489–1499 CrossRef CAS.
- L. Yang, Y. Tang, D. Yan, T. Liu, C. Liu and S. Luo, ACS Appl. Mater. Interfaces, 2016, 8, 169–176 CAS.
- A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef CAS PubMed.
- W. Shi, Environ. Prog. Sustainable Energy, 2016, 35, 840–846 CrossRef CAS.
- H. Y. Lee, W. Vogel and P. P. J. Chu, Langmuir, 2011, 27, 14654–14661 CrossRef CAS PubMed.
- S. Thanasilp and M. Hunsom, Electrochim. Acta, 2011, 56, 1164–1171 CrossRef CAS.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354–2358 CrossRef CAS.
- Y. Qiu and L. Gao, J. Phys. Chem. B, 2005, 109, 19732–19740 CrossRef CAS PubMed.
- A. G. MacDiarmid, Angew. Chem., Int. Ed., 2001, 40, 2581–2590 CrossRef CAS PubMed.
- A. S. Arico, A. K. Shukla, H. Kim, S. Park, M. Min and V. Antonucci, Appl. Surf. Sci., 2001, 172, 33–40 CrossRef CAS.
- T. Lopes, E. Antolini and E. R. Gonzalez, Int. J. Hydrogen Energy, 2008, 33, 5563–5570 CrossRef CAS.
- B. D. Cullity, Elements of X-ray diffraction, Addison-Wesley Publishing, Boston, MA, 1978 Search PubMed.
- S. Limpattayanate and M. Hunsom, J. Solid State Electrochem., 2013, 17, 1221–1231 CrossRef CAS.
- A. Sepulveda-Escribano, F. Coloma and F. Rodriguez-Reinoso, Appl. Catal., A, 1998, 173, 247–257 CrossRef CAS.
- A. Guerrero-Ruiz, P. Badenes and I. Rodriguez-Ramos, Appl. Catal., A, 1998, 173, 313–321 CrossRef CAS.
- M. A. Fraga, E. Jordão, M. J. Mendes, M. M. A. Freitas, J. L. Faria and J. L. Figueiredo, J. Catal., 2002, 209, 355–364 CrossRef CAS.
- K. Endo, K. Nakamura, Y. Katayama and T. Miura, Electrochim. Acta, 2004, 49, 2503–2509 CrossRef CAS.
- P. Serp and J. L. Figueiredo, Carbon Materials for Catalysis, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: Fundamentals and Applications, John Wiley & Sons, New York, 2nd edn, 2000 Search PubMed.
- Y. G. Ryu, S. I. Pyun, C. S. Kim and D. R. Shin, Carbon, 1998, 36, 293–298 CrossRef CAS.
- K. H. Lee, K. Kwon, V. Roev, D. Y. Yoo, H. Chang and D. Seung, J. Power Sources, 2008, 185, 871–875 CrossRef CAS.
- A. Stassi, I. Gatto, V. Baglio, E. Passalacqua and A. S. Aricò, Appl. Catal., B, 2013, 142, 15–24 CrossRef.
- J. Li, X. Fu, Z. Mao, Y. Yang, T. Qiu and Q. Wu, Nanoscale Res. Lett., 2016, 11, 1–8 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |