
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance long chain polyesters via melt copolymerization of cutin-inspired monomers†‡
Zewen
Zhu
a,
Joshua T.
Damron
a,
Jong K.
Keum
 abc,
Logan
Kearney
a,
Vera
Bocharova
*a and
Jeffrey C.
Foster
*a
abc,
Logan
Kearney
a,
Vera
Bocharova
*a and
Jeffrey C.
Foster
*a
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA. E-mail: bocharovav@ornl.gov; fosterjc@ornl.gov
bCenter for Nanophase Materials Science, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
cNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 24th September 2024
Abstract
Biopolymers have exhibited potential as sustainable and circular replacements to existing commodity thermoplastic polymers. However, current biopolymers are limited by poor thermomechanical performance compared with their petroleum-derived counterparts. Herein, we report a simple strategy to achieve good mechanical properties in bio-inspired long-chain polyesters via melt copolymerization. By combining mono- and poly-hydroxyl functionalized long chain fatty acids, we show that tough, semi-crystalline materials can be produced that outperform related biopolymers in terms of their thermomechanical behavior. We envision that long-chain polyesters derived from hydroxylated fatty acids represent an ideal platform to create the next generation of commodity thermoplastics that possess advantaged properties, inherent biodegradability, and feedstock stability.
Sustainability spotlightAchieving a carbon-neutral society and combating the accumulation of plastic waste in the environment requires the development of new bio-based and recyclable plastics. Current bio-based polymers suffer from poor thermomechanical performance that limits their use as replacements for petroleum-based materials. We report our development of a simple procedure to synthesize thermoplastic copolymers using monomers found in various natural sources including plant leaf cuticles and insect shells. Remarkably, these copolymers exhibited similar strength and thermal performance compared with leading consumer plastics such as polyethylene. Our work emphasizes the importance of the following UN sustainable development goals: industry, innovation, and infrastructure (SDG 9), sustainable cities and communities (SDG 11), responsible consumption and production (SDG 12), and climate action (SDG 13). |
Introduction
Consumer plastics such as polyethylene (PE), polypropylene (PP), polyamide (i.e., nylons), and poly(ethylene terephthalate) (PET) are cheap, durable, lightweight, and processable. These petroleum-based materials contribute significantly to human health and safety and enable technologies in fields spanning healthcare, to transportation, to construction. Despite their advantages, these plastics are derived from non-renewable feedstocks and their limited biodegradability drives their accumulation and persistence in the environment. In fact, of the 35.7 million tons of plastic generated in the US in 2018, only 8.7% was recycled, with 5.6 million tons disposed through incineration and the remaining 27 million tons landfilled.1 New materials are needed to address these challenges that combine the low cost and advantaged properties of current plastics with sustainable sourcing and engineered degradability.Biopolymers—produced or derived from living organisms—have emerged as promising alternatives to commodity plastics.2–4 Such materials are synthesized via chemo- or bio-synthetic pathways and possess inherent biodegradability, enabling their circular production and recycling. Poly(lactic acid) (PLA),5,6 poly(butylene succinate) (PBS),7 and polyhydroxyalkanoates (PHAs)8 are exemplar biopolymers, with production capacities approaching >100 tons per year.9 While their production costs are becoming more competitive with their petroleum-based counterparts, their thermomechanical performance falls short.10 As such, there remains a strong demand to produce ever larger quantities of petroleum-based plastics despite the steep resource and environmental costs of their production.
The commodity biopolymers PLA, PBS, and PHA(s) are classified as short-chain polyesters, with ≤6 carbon units between each backbone ester functional group. While their high functionality engenders rapid degradation in the environment, it also tends to limit their thermomechanical performance. Indeed, Mecking and coworkers showed that aliphatic polyesters generally possess properties that diverge from those of HDPE dependent on the spacing of backbone ester functional groups, with closer spacing leading to lower melting temperatures, Tm, and decreased toughness.11 However, biopolymers based on long-chain aliphatic lactone or diester building blocks have achieved good thermomechanical performance and recyclability. For example, Gross and coworkers prepared high molecular weight (MW) poly(ω-pentadecalactone) via lipase catalyzed ring-opening polymerization.12 Excellent tensile behavior was obtained by increasing MW to drive entanglement in this system. More recently, Mecking and coworkers reported the preparation of long-chain aliphatic polyesters from C18 diesters and diols sourced from oleic acid, which exhibited thermomechanical performance similar to LDPE along with good recyclability.13 Based on these inspirations, we view long-chain aliphatic polyesters as potential high-performance alternatives to current, bio-based short-chain polyesters, combining the desirable features of natural and synthetic polymeric materials.
Long-chain polyesters can also be synthesized through condensation polymerization of hydroxylated fatty acids, which are ubiquitous in nature and can be sourced from feedstocks such as plant leaves (cutin), fruit skins, (suberin and cutin), insect resins (shellac), and algae (algaenan).14,15 In biological systems, long-chain polyesters constitute highly branched, amorphous, and hydrophobic materials that serve as waterproof and protective barriers. Current cutin-inspired synthetic materials exhibit limited mechanical strength, low glass transition temperatures, Tg, and possess amorphous microstructures due to their highly branched structures.16–18 In contrast, long-chain polyesters with highly linear structures based on the 16 carbon cutin constituent 16-hydroxyhexadecanoic acid (HHA) are high-strength, semi-crystalline materials.19–21 However, their high crystallinity engenders embrittlement and thus such polyesters typically exhibit very low toughness. Based on these factors, we imagined that biodegradable materials with high strength and toughness could be prepared from cutin-inspired building blocks by leveraging the best aspects of both linear and branched long-chain polyesters. In this contribution, we prepare long-chain polyesters via copolymerization of hydroxylated fatty acid monomers inspired by the constituents of cutin. By combining mono- and poly-hydroxyl functionalized monomers, we obtain copolymers with enhanced toughness, high Young's modulus, and improved energy dissipating capability compared to their homopolymers. Our findings demonstrate the significant potential for long-chain polyesters as alternative materials to current commercial biopolymers. We envision that long-chain polyesters derived from hydroxylated fatty acids represent an ideal platform to create the next generation of LDPE-like plastics that possess advantaged properties, inherent biodegradability, and feedstock stability.
Result and discussion
Synthesis and spectral characterization of long-chain polyesters
We initiated our investigation using the commercially available, cutin-inspired C16 α-hydroxy acids 9,10,16-trihydroxyhexadecanoic acid (aleuritic acid, AA) and HHA. AA is a major component of shellac (insect) resin, and both can be found within the highly branched structure of cutin (plant cuticles), among other natural sources.17,22AA possess a terminal primary hydroxyl group and two pendant secondary hydroxyl groups and forms a highly branched polymer, PAA, when homopolymerized.17PAA exhibits limited mechanical strength and ductility, limiting its applications as a commodity thermoplastic.23 In contrast, HHA possess only a terminal primary hydroxyl group and its polymerization provides a fully linear polymer PHHA that is semi-crystalline and brittle.24 We hypothesized that copolymerization of AA and HHA might afford polymeric materials with properties characteristic of both constituents, with linear HHA segments providing mechanical reinforcement via crystallization and branched, amorphous AA domains affording numerous connections between polymer crystals as well as providing additional strength and toughness via H-bonding.25,26To evaluate our hypothesis, copolymers of AA and HHA were synthesized via self-catalyzed melt condensation polymerization (Fig. 1A). We opted to conduct melt polycondensation in the absence of solvent and heavy metal catalyst to avoid the entrapment of toxic compounds within the synthesized materials. Solid AA and HHA were pre-melted in different weight ratios at 100 °C and mechanically stirred to ensure homogenization. The melted monomer mixtures were cast into Teflon molds and degassed under vacuum at 110 °C. Polymerizations were then carried out by heating the melt to 150 °C for 16 h under dynamic vacuum. Homopolymers of AA and HHA were also prepared for comparison using the same method. After cooling, the polymers were delaminated from the molds for spectral and thermomechanical characterization. PHHA could be dissolved in polar organic solvents, while PAA and the P(AA-co-HHA) copolymers were completely insoluble in any of the tested solvents, including in N,N-dimethylformamide at elevated temperatures (Table S1‡). Condensation reactions with multifunctional monomers (ABf type) or copolymerizations of ABf and AB type monomers are incapable of forming networks and instead afford hyperbranched polymers and copolymers, respectively, that typically exhibit better solubility than analogous linear systems.27,28 However, the insolubility of PAA and P(AA-co-HHA) was indicative of covalent network formation. Similar solubility behavior has been observed for PAA and related condensation polyesters produced from multifunctional hydroxy acids, and these observations have been attributed to covalent crosslinking mediated by oxidative cleavage or dehydrative reactions of 1,2-diols.16,29 We note that no concrete evidence from these covalent crosslinking reactions could be identified in the spectral characterization of polyesters PAA or P(AA-co-HHA), suggesting that their incidence was low but sufficient to induce covalent network formation. However, covalent cross-linking was apparent in the mechanical characterization techniques, as discussed below.
 | ||
| Fig. 1 (A) Chemical structures of monomers AA and HHA and polymers PAA, PHAA, and P(AA-co-HHA) obtained from melt polymerization or copolymerization. (B and C) Overlaid solid-state 13C NMR spectra of monomers and polyesters. (D) Overlaid FT-IR spectra of monomers and copolymers, where transmittance values were normalized to the antisymmetric vibration of methylene functional groups at 2925 cm−1. | ||
The synthesized polyesters were characterized by solid-state 13C NMR and FT-IR spectroscopies to calculate the degree of reaction of their primary and secondary OH and COOH groups and to better understand their solid-state architectures. Monomers AA and HHA were similarly characterized for comparison. Overlaid NMR spectra are shown in Fig. 1B and C and FT-IR spectra in Fig. 1D. Briefly, 13C resonances from C–O carbons in esters and alcohols could be observed in each of the NMR spectra. Signals corresponding with terminal C–OH motifs (ca. 61 ppm) decreased following melt condensation and were completely absent from the spectrum of PHHA, indicative of high polymerization conversion. ω-Hydroxyl conversion was less significant for polymers PAA and P(AA-co-HHA) and was consistent with a competition between primary and secondary alcohols for the formation of ester linkages. Gaussian fitting of these spectra revealed that ca. 56% of the primary alcohols had been converted to ester groups for PAA, while a higher conversion of 79% of terminal alcohols was calculated for P(AA-co-HHA) (see Table S2‡ for details). The latter result was attributed to the relatively lower proportion of secondary alcohols in the copolymer reaction mixture. Also apparent was the decrease in the signal corresponding with carbons of secondary alcohols at ca. 73 ppm for PAA and P(AA-co-HHA) and a coincident increase in intensity at ca. 71 ppm and 75 ppm. Both signals qualitatively exhibited similar integrations, consistent with preferential monoesterification of the 1,2-diol motifs. This neighboring group effect likely originated from steric congestion and electronic deactivation of the α-hydroxyl motifs following monoesterification. Similar conversions of secondary alcohols of 22% and 20% were calculated for PAA and P(AA-co-HHA), respectively, by subtracting the calculated relative conversions of primary alcohols from the total concentration of alcohols in the reaction feed. However, based on the higher proportion of 1,2-diols in PAA compared with P(AA-co-HHA), the former necessarily possessed a higher degree of branching. Ester bond formation was also evident in the upfield regions of the NMR spectra for the various polymers, manifest as new signals at ca. 24 ppm and 31 ppm that were assigned to carbons beta to the ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O linkages, respectively.
O and C–O linkages, respectively.
Fig. 1D shows the infrared spectra of monomers, polymers, and copolymers. The spectral region between 1800 and 1500 cm−1 corresponded to the stretching vibrations of carbonyl groups (CO) in esters, carboxylic acids, and carboxylates.30,31 In this zone, the main band appeared at 1730 cm−1, with shoulders at 1712 and 1687 cm−1. The 1730 cm−1 peak and the 1712 cm−1 shoulder were assigned to free and hydrogen-bonded esters, respectively. The CO vibration of free carboxylic acid was represented by the shoulder at 1687 cm−1 and was most significant for monomers AA and HHA. For the homo- and copolymers, residual COOH signal could not be differentiated from the nearby carbonyl vibrations, indicating high COOH conversion. The spectra were normalized to the antisymmetric vibration of methylene functional groups at 2925 cm−1 to evaluate the degree of esterification. As shown in Fig. 1D, the degree of esterification was comparable across all homo- and copolymers, evidenced by the high intensity of CO bands and the low intensities of the free and hydrogen-bonded COOH bands, and this result was consistent with observations from 1H NMR spectroscopy. Furthermore, the broad band at 3460 cm−1, characteristic of vibration of hydroxyl groups, had comparable intensities and shapes among all copolymers, indicating similar amounts of free and hydrogen-bonded hydroxyls and degrees of association in the copolymers. The frequency value of vibration of hydroxyl groups also indicated different degrees of interaction between them,32 which control polymer association.33 In this respect, the hydroxyls were shifted to a lower wavenumber in the monomers compared to polymers and copolymers, suggesting their stronger association.
The crystalline structure and morphology of the samples were studied with WAXS and SAXS (Fig. 2A). In the high-q WAXS region, crystalline peaks were observed in each sample that co-existed with an amorphous halo, as would be expected for semi-crystalline polymers. The WAXS patterns were characterized by peaks at 1.49 and 1.69 Å−1 which were assigned to 110 and 200 reflections of the orthorhombic cell, in line with structural studies on related polyesters.34 The degree of crystallinity was determined by fitting the high-q region (0.2 Å−1 ≤ q ≤ 2.3 Å−1) using Fityk software as shown in Fig. S1 and summarized in Table S3,‡ and the plot depicting crystallinity vs. weight content of HAA is shown in Fig. 2B. Crystallinity was observed to increase in correspondence with increasing HHA content following a non-monotonic trend. Linear PHHA exhibited the highest crystallinity of 58%, while the highest crystallinity observed for the copolymers was 39% for P(AA0.3-co-HHA0.7). The lowest crystallinities of 24% and 25% were observed for branched PAA and copolymer P(AA0.7-co-HHA0.3), respectively.
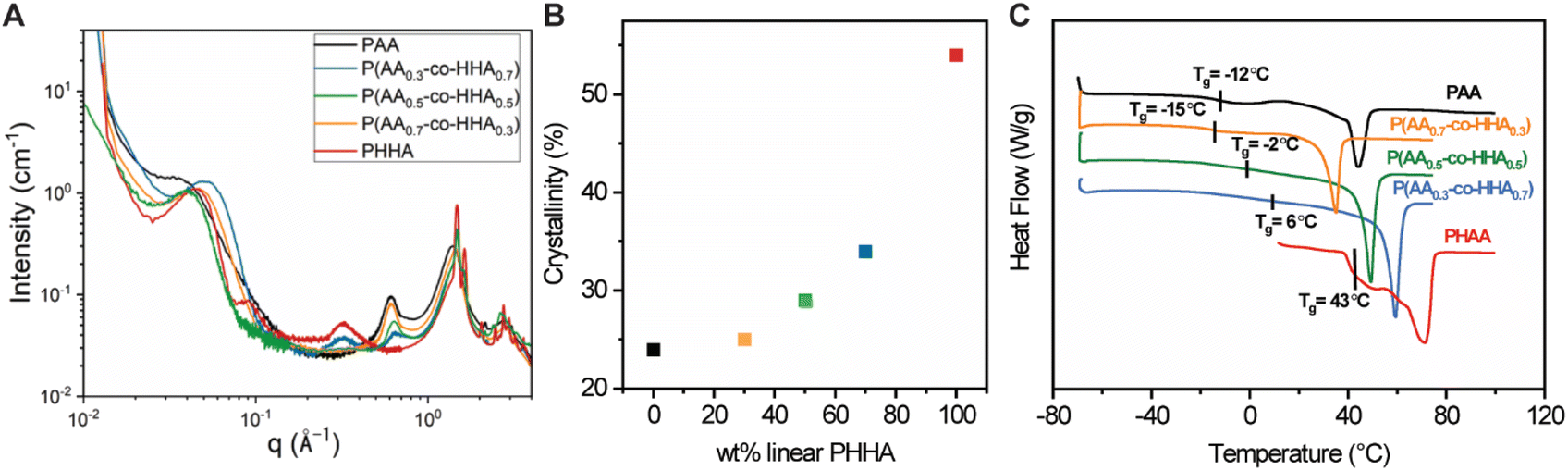 | ||
| Fig. 2 (A) Combined SAXS and WAXS curves of polyesters. (B) Correlation between crystallinity and wt% of added HHA. (C) DSC data obtained for polyesters from second heating cycles. | ||
Peaks with maxima at qmax = 0.31 Å−1 and 0.61 Å−1 showed strong dependencies on the PHHA and PAA content. The peak at q = 0.31 Å−1 was maximized for neat PHHA, while its intensity decayed with increasing PAA. The d-spacing, d = 20.2 Å−1, calculated from the peak maximum by d = 2π/qmax, was assigned to the repeat unit length of PHHA. Hence, we indexed this peak as (001) reflection of the PHHA crystal, in line with the literature.34 The reflection maximum at qmax = 0.61 Å−1, on the other hand, increased in intensity with an increase in the amount of the branched motif. We associated the presence of this peak with the longitudinal organization of the PAA chains, with a repeat distance corresponding to half the length of AA in the crystallized state. The low-q region contained peaks with positions signaling a lamellar mesoscale organization, which is common for semi-crystalline polymers. The characteristic long period for these lamellar is presented in Table S4.‡
The thermal behavior of the various homo- and copolymers was evaluated using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). From TGA (Fig. S2‡), the copolymers showed higher degradation temperatures (5% weight loss) compared to PAA and PHHA. Individual DSC curves in Fig. 2C were characterized by Tg region and, for all samples but PHHA, a melting endotherm, Tm. We note that Tg transitions were broadened in the copolymers relative to their analogous homopolymers. The Tg and Tm of copolymers increased in response to increased loadings of HHA. The Tg of branched PAA was higher than that of P(AA0.7-co-HHA0.3), which could suggest the disruption of the polymer network formed by PAA with the addition of PHHA. A comparison between the first and second heating cycles (Fig. S3A‡) revealed a decrease in melting enthalpy or a complete disappearance of crystalline peaks, as was observed for PAA. This indicated changes in the crystalline structure of the samples and the formation of isomorphs which is in line with literature data on polyesters and polyethylene.34,35 Nevertheless, the observed crystalline organization after recrystallization had a transient nature and tended to return to its initial state if the sample was left at room temperature. We believe that this transition occurred quickly; for example, in the case of P(AA0.3-co-HHA0.7), it took ca. 1 h at room temperature to return to the original structure before thermal treatment, with the most pronounced changes in the sample occurring within the first 10 min (Fig. S3B‡).
To evaluate the degradability of the synthesized homo- and copolymers, the various materials were suspended in solutions of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O/EtOH containing 6 M NaOH and were heated at 80 °C for 16 h. Near complete dissolution of the previously insoluble copolymers was observed in all cases, indicative of their deconstruction to soluble, small molecule byproducts. 1H NMR spectroscopic analysis of these crude reaction mixtures confirmed regeneration of free carboxyl and hydroxyl functionality (Fig. S4‡). Thus, in addition to their good thermomechanical performance (vide infra), these results highlight the potential for P(AA-co-HHA) to be degraded at their end-of-life. Based on the highly crystalline nature of these copolymers, we assume that their hydrolytic degradation would occur slowly relative to short-chain polyesters like PLA.36,37 However, we note that copolymerization could also be leveraged to modulate degradation rates in our systems based on the dependence of crystallinity on copolymer structure.
:1 H2O/EtOH containing 6 M NaOH and were heated at 80 °C for 16 h. Near complete dissolution of the previously insoluble copolymers was observed in all cases, indicative of their deconstruction to soluble, small molecule byproducts. 1H NMR spectroscopic analysis of these crude reaction mixtures confirmed regeneration of free carboxyl and hydroxyl functionality (Fig. S4‡). Thus, in addition to their good thermomechanical performance (vide infra), these results highlight the potential for P(AA-co-HHA) to be degraded at their end-of-life. Based on the highly crystalline nature of these copolymers, we assume that their hydrolytic degradation would occur slowly relative to short-chain polyesters like PLA.36,37 However, we note that copolymerization could also be leveraged to modulate degradation rates in our systems based on the dependence of crystallinity on copolymer structure.
Mechanical properties characterization
To characterize mechanical properties for the various polyesters, dynamic mechanical analysis (DMA) and tensile testing were employed. The storage moduli for all samples, as measured by DMA, are presented in Fig. S5.‡ The presence of a rubbery plateau in the storage moduli of all copolymers and PAA at high temperatures was indicative of covalent network formation. This supposition was further confirmed via isothermal equilibration experiments (1 h at 100 °C), during which the plateau value remained constant. The absence of such a plateau for PHAA highlighted that the crosslinking was initiated by the presence of HAA monomer; however, the degree of crosslinking did not scale proportionally with the amount of branched motif, pointing to a more complex organization. The plateau level was found to change as PAA ∼ P(AA0.3-co-HHA0.7) > P(AA0.5-co-HHA0.5) > P(AA0.7-co-HHA0.3) (Fig. S5‡). The lowest and highest molecular weight between crosslinks, Mc, were found to be 4.3 and 18.0 kg mol−1 for P(AA0.3-co-HHA0.7) and P(AA0.7-co-HHA0.3), respectively (Table S1‡).For the tensile tests, the polyester materials were prepared into dumbbell-shaped specimens. The representative engineering stress–strain curves are shown in Fig. 3a, and the analyses of the data obtained are summarized in Table S1.‡ As observed, PHHA showed the highest Young's modulus and lowest ductility due to its linear semicrystalline structure. On the other hand, PAA exhibited lower Young's modulus and higher elongation at break compared to PHHA. These observations were consistent with previous reports on PAA and PHHA homopolymers.16,22 In stark contrast, the series of P(AA-co-HHA) copolymers exhibited ductile tensile behavior with rather high elongations at break. Compared to homopolymers PAA and PHHA, all three copolymers exhibited significantly increased strain-at-break values of ∼250%, which is similar to commercial low-density polyethylene (LDPE) (Fig. 3b). When increasing the ratio of HHA in the P(AA-co-HHA) copolymers, the yield strength increased in the order P(AA0.7-co-HHA0.3) < P(AA0.5-co-HHA0.5) < P(AA0.3-co-HHA0.7) (Fig. 3b). Young's modulus values increased from 296 MPa for PAA to 493 MPa for P(AA0.7-co-HHA0.3), 892 MPa for P(AA0.5-co-HHA0.5) and 872 MPa for P(AA0.3-co-HHA0.7), in line with the crystallinity trends observed from X-ray scattering (Table S3‡). The toughness of the copolymers also depended on the crystallinity, with increased crystallinity resulting in increased toughness. Specifically, P(AA0.3-co-HHA0.7) had a toughness of 2730 ± 350 J m−3. For P(AA0.5-co-HHA0.5) and P(AA0.7-co-HHA0.3), measured toughness values were 2610 ± 370 J m−3 and 2430 ± 430 J m−3, respectively. The toughness of the homopolymers, 28 ± 8 and 47 ± 7 J m−3 for PAA and PHHA respectively, were much lower than for the copolymers, which could be related to factors like the high crystallinity of PHHA and the high chemical crosslinking of PAA samples that made materials less stretchable.
 | ||
| Fig. 3 Engineering stress–strain curves (a) and summarized tensile testing results (b) for the various polyesters. A LDPE sample is included for comparison. | ||
Cyclic tensile tests were then carried out to investigate the energy dissipation process during deformation. As-prepared samples were incrementally stretched to different strains and interrupted after each step by an unloading-reloading loop. All copolymers exhibited pronounced hysteresis and a large residual strain for each loading and unloading cycle (Fig. S6‡). The hysteresis area, reflecting the dissipation capability of the materials, is plotted as a function of applied strain in Fig. S7.‡ It increased with an increase in strain and leveled off at 50–60% strain for all copolymers. The observed hysteresis area depended on copolymer composition and decreased on the order P(AA0.3-co-HHA0.7) > P(AA0.5-co-HHA0.5) > P(AA0.7-co-HHA0.3), following the degree of chemical crosslinking of copolymers as measured by DMA. It was likely the case that higher degrees of chemical crosslinking provided better dissipating capability due to the stronger contribution from inelastic deformation. These results demonstrate that our bio-inspired, long-chain polyester copolymers exhibit potential as alternative commodity materials with good dampening properties.
Of the tested copolymers, P(AA0.3-co-HHA0.7) possessed an optimal ratio of linear and branched units to achieve a material with high Young's modulus and toughness and good dissipating capability. This material was further studied via uniaxial tensile deformation WAXS and SAXS to understand structural changes occurring during deformation. The 2D WAXS data for P(AA0.3-co-HHA0.7) at stretched and non-stretched states are presented in Fig. 4A. Fig. 4B combines selected 1D SAXS and WAXS curves to demonstrate changes induced by the stretching. For that, the X-ray data of the stretched sample were segregated into equatorial and azimuthal regions. Intensity data integration was performed within angular sectors of 10 degrees around the equatorial and azimuthal axes.
 | ||
| Fig. 4 (A) 2D WAXS images of P(AA0.3-co-HHA0.7) of unstretched, stretched, and, after the heating. (B) 1D SAXS/WAXS curves for P(AA0.3-co-HHA0.7) in the stretched and unstretched states where azimuthal and equatorial curves are reported for stretched sample. | ||
Before the stretching, isotropic scattering rings (scattering peaks) were observed at q = 0.31, 0.61, 1.48, and 1.64 Å−1 for P(AA0.3-co-HHA0.7) (Fig. 4A). As the copolymer was stretched uniaxially, the scattering patterns were characteristic of arc-type scattering, implying an anisotropic crystal orientation in which the chain axes of the stretched copolymers were aligned parallel to the stretching axis. The arc type scattering at q = 0.61 Å−1 and 0.31 Å−1 centered at the meridian confirmed that the scattering peak was related to the atomic correlation along the backbone repeat units of PAA and PHHA, respectively. The arc type scattering at q = 1.48 and 1.64 Å−1, with azimuthal intensity centered at the equator, implied the presence of a lateral interatomic correlation originating from orthorhombic cell (Fig. 4B). Unfortunately, we could not reach the full stretching of the samples (>200%) due to their high toughness. Alternatively, we were able to reach the instances of greater stretching in the sample for P(AA0.5-co-HHA0.5), where the distinct equatorial peaks gradually combined into a singular equatorial peak (Fig. S8‡). This fusion indicated a potential alteration to the hexagonal crystalline structure, with its characteristic reflection from 100 planes.35 Notably, DSC analysis suggested that the observed transitions between subcells could be achieved not only through deformation but also via heating and cooling. As stretching progressed, the amorphous phase reached its maximum stretching, causing deformation in the lamellar structures—such as chain extraction from the crystalline phase. This state of fibril deformation in the samples associated with a high-q shift in the lamella peak signified a reduction in the characteristic lamella distance during deformation (Fig. 4B). We imagine that the crystalline lamellae fragmented to smaller crystallites, resulting in a decrease in the average distance between lamellae which could promote a transition between different subcells. The stretching of the amorphous phase contributed to the extended plateau region in the stress–strain curves, where a transition to the hexagonal crystal phase ultimately led to an upturn in the plateau region and eventual breakage in the stress–strain curve. Photographs of the unstretched and stretched samples are provided in Fig. 4A. After annealing, the sample recovered back to its initial state. 2D WAXS indicated isotropic orientation and complete morphology recovery after annealing. Complete retraction to the original dimensions after heating above melting is also observed in other semicrystalline polymers like PE.38
Conclusions
We demonstrated that the thermomechanical properties of long-chain polyester copolymers could be modulated by the relative quantity of linear and branched motifs in their compositions. The formation of amorphous phases, mainly driven by the addition of branched motifs, afforded high stretchability, distinguishing them from their related homopolymers. The combination of relatively high crystallinity and enhanced stretchability resulted in a significant increase in toughness for the copolymers. Particularly noteworthy was the composition of P(AA0.3-co-HHA0.7), which exhibited high toughness and Young's modulus and, due to chemical crosslinking, was also characterized by a high energy dissipating capability. These data clearly demonstrate that copolymerization of bio-inspired long-chain polyesters offers a viable strategy to produce inherently degradable polymeric materials possessing comparable performance to current commodity materials.Experimental section
Materials
9,10,16-Trihydroxyhexadecanoic acid (AA, 95%, Alfa Aesar) and 16-hydroxyhexadecanoic acid (HHA, >98.0%, TCI) were used without further purification. All solvents were purchased from Sigma-Aldrich. Low density polyethylene (LDPE) sheet (1 mm thickness) was purchased from McMaster-Carr.Characterization
 where R and T are the ideal gas constant and temperature in °K, respectively; d is the density of long-chain polyester ∼0.95 g cm−3;
where R and T are the ideal gas constant and temperature in °K, respectively; d is the density of long-chain polyester ∼0.95 g cm−3;  is the elastic modulus at 100 °C. Incremental loading and unloading cycle tests were performed using a DMA 850 in tensile mode at 25 °C. The same rectangular-shaped samples were tested, and the gauge length was fixed at 6 mm. Six cycles with increasing tensile strain (8.3, 16.6, 33.3, 50, 66.7 and 83.3%) were performed. Each cycle had two stages: stretch to the targeted strain amplitude and unload to zero displacement at 1 mm min−1 stretching rate.
sinθ)/λ after subtraction of background scattering. The stretched samples were measured ex situ, where samples were stretched on Instron until they were maximally stretched and positioned on the sample rack with the stretching direction perpendicular to the X-ray beam direction.
is the elastic modulus at 100 °C. Incremental loading and unloading cycle tests were performed using a DMA 850 in tensile mode at 25 °C. The same rectangular-shaped samples were tested, and the gauge length was fixed at 6 mm. Six cycles with increasing tensile strain (8.3, 16.6, 33.3, 50, 66.7 and 83.3%) were performed. Each cycle had two stages: stretch to the targeted strain amplitude and unload to zero displacement at 1 mm min−1 stretching rate.
sinθ)/λ after subtraction of background scattering. The stretched samples were measured ex situ, where samples were stretched on Instron until they were maximally stretched and positioned on the sample rack with the stretching direction perpendicular to the X-ray beam direction.
Preparation of long-chain polyesters
Long-chain polyesters were prepared as follows: AA and HHA were melted at 100 °C and poured into pre-heated (∼110 °C) Petri dishes coated with PTFE releasing agent. Next, the samples were degassed in a vacuum oven at 110 °C for 2 h and further heated at 150 °C overnight under vacuum to achieve polycondensation reactions. For the copolymers, monomer mixtures were stirred for 30 min at 100 °C to ensure good homogenization prior to polymerization.Hydrolysis of long-chain polyester copolymers
Copolymers P(AA-co-HHA) (∼0.2 g) were suspended in 5 mL of 1:1 v/v% H2O/EtOH solutions containing 6 M NaOH in 20 mL vials equipped with magnetic stirring bars. The reaction mixtures were heated at 80 °C for 16 h. After cooling, an aliquot of each reaction mixture was removed and diluted in MeOD for analysis by 1H NMR spectroscopy.
Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was sponsored by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory managed by UT-Battelle LLC for the U.S. DOE. J. C. F. was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. The authors acknowledge Wim Bras for assistance in interpreting X-ray scattering data.References
- U. S. E. P. Agency, Plastics: Material-Specific Data, https://www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/plastics-material-specific-data, accessed, April 2, 2024 Search PubMed.
- N. Patil, C. Jérôme and C. Detrembleur, Recent advances in the synthesis of catechol-derived (bio)polymers for applications in energy storage and environment, Prog. Polym. Sci., 2018, 82, 34–91 CrossRef CAS.
- A. Morales, J. Labidi, P. Gullón and G. Astray, Synthesis of advanced biobased green materials from renewable biopolymers, Curr. Opin. Green Sustainable Chem., 2021, 29, 100436 CrossRef CAS.
- A. George, M. R. Sanjay, R. Srisuk, J. Parameswaranpillai and S. Siengchin, A comprehensive review on chemical properties and applications of biopolymers and their composites, Int. J. Biol. Macromol., 2020, 154, 329–338 CrossRef CAS PubMed.
- T. L. de Albuquerque, J. E. Marques Júnior, L. P. de Queiroz, A. D. S. Ricardo and M. V. P. Rocha, Polylactic acid production from biotechnological routes: a review, Int. J. Biol. Macromol., 2021, 186, 933–951 CrossRef CAS PubMed.
- K. J. Jem and B. Tan, The development and challenges of poly (lactic acid) and poly (glycolic acid), Adv. Ind. Eng. Polym. Res., 2020, 3(2), 60–70 Search PubMed.
- O. Platnieks, S. Gaidukovs, V. K. Thakur, A. Barkane and S. Beluns, Bio-based poly (butylene succinate): recent progress, challenges and future opportunities, Eur. Polym. J., 2021, 161, 110855 CrossRef CAS.
- C. Utsunomia, Q. Ren and M. Zinn, Poly(4-Hydroxybutyrate): Current State and Perspectives, Front. Bioeng. Biotechnol., 2020, 8 DOI:10.3389/fbioe.2020.00257.
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a circular economy, Nat. Rev. Mater., 2022, 7(2), 117–137 CrossRef PubMed.
- F. Luzi, L. Torre, J. M. Kenny and D. Puglia, Bio- and Fossil-Based Polymeric Blends and Nanocomposites for Packaging: Structure–Property Relationship, Materials, 2019, 12(3), 471 CrossRef CAS PubMed.
- F. Stempfle, P. Ortmann and S. Mecking, Long-Chain Aliphatic Polymers to Bridge the Gap between Semicrystalline Polyolefins and Traditional Polycondensates, Chem. Rev., 2016, 116(7), 4597–4641 CrossRef CAS PubMed.
- J. Cai, C. Liu, M. Cai, J. Zhu, F. Zuo, B. S. Hsiao and R. A. Gross, Effects of molecular weight on poly(ω-pentadecalactone) mechanical and thermal properties, Polymer, 2010, 51(5), 1088–1099 CrossRef CAS.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Closed-loop recycling of polyethylene-like materials, Nature, 2021, 590(7846), 423–427 CrossRef PubMed.
- M. Batsale, D. Bahammou, L. Fouillen, S. Mongrand, J. Joubès and F. Domergue, Biosynthesis and Functions of Very-Long-Chain Fatty Acids in the Responses of Plants to Abiotic and Biotic Stresses, Cells, 2021, 10(6), 1284 CrossRef CAS PubMed.
- Y. Li-Beisson, G. Verdier, L. Xu and F. Beisson, Cutin and Suberin Polyesters, in eLS, 2016, pp. 1–12 Search PubMed.
- J. A. Heredia-Guerrero, A. Heredia, R. García-Segura and J. J. Benítez, Synthesis and characterization of a plant cutin mimetic polymer, Polymer, 2009, 50(24), 5633–5637 CrossRef CAS.
- G. Tedeschi, S. Guzman-Puyol, L. Ceseracciu, J. J. Benitez, P. Cataldi, M. Bissett, A. Heredia, A. Athanassiou and J. A. Heredia-Guerrero, Sustainable, high-barrier polyaleuritate/nanocellulose biocomposites, ACS Sustainable Chem. Eng., 2020, 8(29), 10682–10690 CrossRef CAS.
- J. A. Heredia-Guerrero, A. Heredia, E. Domínguez, R. Cingolani, I. S. Bayer, A. Athanassiou and J. J. Benítez, Cutin from agro-waste as a raw material for the production of bioplastics, J. Exp. Bot., 2017, 68(19), 5401–5410 CrossRef CAS PubMed.
- S. Singha, V. Gowda and M. S. Hedenqvist, Plant Cuticle-Inspired Polyesters as Promising Green and Sustainable Polymer Materials, ACS Appl. Mater. Interfaces, 2021, 3(8), 4088–4100 CAS.
- V. Polisetti, S. Subramaniyan, S. Singha, M. Hakkarainen, A. J. Svagan and M. S. Hedenqvist, Plant Cutin-Inspired Co- and Terpolyesters as Potential Packaging Materials, ACS Sustainable Chem. Eng., 2024, 12(21), 8001–8009 CrossRef CAS.
- P. R. Anusuyadevi, S. Singha, D. Banerjee, M. P. Jonsson, M. S. Hedenqvist and A. J. Svagan, Synthetic Plant Cuticle Coating as a Biomimetic Moisture Barrier Membrane for Structurally Colored Cellulose Films, Adv. Mater. Interfaces, 2023, 10(7), 2202112 CrossRef CAS.
- J. J. Benitez, S. Guzman-Puyol, M. A. Cruz-Carrillo, L. Ceseracciu, A. Gonzalez Moreno, A. Heredia and J. A. Heredia-Guerrero, Insoluble and Thermostable Polyhydroxyesters From a Renewable Natural Occurring Polyhydroxylated Fatty Acid, Front. Chem., 2019, 7, 643 CrossRef CAS PubMed.
- S. K. Kumar, B. C. Benicewicz, R. A. Vaia and K. I. Winey, 50th anniversary perspective: are polymer nanocomposites practical for applications?, Macromolecules, 2017, 50(3), 714–731 CrossRef CAS.
- J. J. Benítez, J. A. Heredia-Guerrero, M. A. Cruz-Carrillo, M. J. Barthel, H. E. Knicker and A. Heredia, Insolubilization and thermal stabilization of a long-chain polyester by noncatalyzed melt-polycondensation synthesis in air, J. Appl. Polym. Sci., 2017, 134(1), 44350 CrossRef.
- M. Jia, J. Liu, Y. Gnanou and X. Feng, Triblock Copolymers from CO2, Propylene Oxide, and p-Tosyl Isocyanate of Higher Toughness than Polyethylenes, Macromolecules, 2023, 56(10), 3631–3640 CrossRef CAS.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Bio-based polyesters: recent progress and future prospects, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- P. J. Flory, Molecular Size Distribution in Three Dimensional Polymers. II. Trifunctional Branching Units, J. Am. Chem. Soc., 1941, 63(11), 3091–3096 CrossRef CAS.
- W. H. Stockmayer, Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking, J. Chem. Phys., 1944, 12(4), 125–131 CrossRef CAS.
- J. J. Benítez, P. M. Castillo, J. C. Del Río, M. León-Camacho, E. Domínguez, A. Heredia, S. Guzmán-Puyol, A. Athanassiou and J. A. Heredia-Guerrero, Valorization of Tomato Processing by-Products: Fatty Acid Extraction and Production of Bio-Based Materials, Materials, 2018, 11(11), 2211 CrossRef PubMed.
- A. Heredia, J. A. Heredia-Guerrero, E. Domínguez and J. J. Benítez, Cutin synthesis: a slippery paradigm, Biointerphases, 2009, 4(1), 1–3 CrossRef PubMed.
- J. A. Heredia-Guerrero, J. J. Benítez and A. Heredia, Self-assembled polyhydroxy fatty acids vesicles: a mechanism for plant cutin synthesis, Bioessays, 2008, 30(3), 273–277 CrossRef CAS PubMed.
- L. P. Kuhn, The Hydrogen Bond. I. Intra-and Intermolecular Hydrogen Bonds in Alcohols, J. Am. Chem. Soc., 1952, 74(10), 2492–2499 CrossRef CAS.
- L. Bellamy, The Infra-red Spectra of Complex Molecules, Springer Science & Business Media, 2013 Search PubMed.
- M. Marc, C. Lopez, N. Viller, X. Falourd, M. Fanuel, D. Marion, E. Leroy, B. Bakan and D. Lourdin, From Tomato Pomaces Biorefinery to Biobased Shape-Memory Semicrystalline Polyester Networks, ACS Sustainable Chem. Eng., 2024, 12(6), 2191–2202 CrossRef CAS.
- K. Tashiro, S. Sasaki and M. Kobayashi, Structural investigation of orthorhombic-to-hexagonal phase transition in polyethylene crystal: the experimental confirmation of the conformationally disordered structure by X-ray diffraction and infrared/Raman spectroscopic measurements, Macromolecules, 1996, 29(23), 7460–7469 CrossRef CAS.
- K. Min, J. D. Cuiffi and R. T. Mathers, Ranking environmental degradation trends of plastic marine debris based on physical properties and molecular structure, Nat. Commun., 2020, 11(1), 727 CrossRef CAS PubMed.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Plastics of the Future? The Impact of Biodegradable Polymers on the Environment and on Society, Angew. Chem., Int. Ed., 2019, 58(1), 50–62 CrossRef CAS PubMed.
- R. Hiss, S. Hobeika, C. Lynn and G. Strobl, Network stretching, slip processes, and fragmentation of crystallites during uniaxial drawing of polyethylene and related copolymers. A comparative study, Macromolecules, 1999, 32(13), 4390–4403 CrossRef CAS.
Footnotes |
| † This manuscript has been authored by UT-Battelle LLC under contract DE-AC05-00OR22725 with the U.S. Department of Energy (DOE). The U.S. government retains, and the publisher, by accepting the article for publication, acknowledges that the U.S. government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for U.S. government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan). |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00454j |
| This journal is © The Royal Society of Chemistry 2024 |