
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect synthesis of chiral β-arylamines via additive-free asymmetric reductive amination enabled by tunable bulky phosphoramidite ligands†
Jing
Wang‡
a,
Wenji
Wang‡
a,
Haizhou
Huang
a,
Zhiqing
Ma
 b and
Mingxin
Chang
*ab
b and
Mingxin
Chang
*ab
aCollege of Chemistry & Pharmacy, Northwest A&F University, 22 Xinong Road, Yangling, Shaanxi 712100, PR China. E-mail: mxchang@nwsuaf.edu.cn
bCollege of Plant Protection, Shaanxi Research Center of Biopesticide Engineering & Technology, Northwest A&F University, 22 Xinong Road, Yangling, Shaanxi 712100, PR China
First published on 4th September 2024
Abstract
This report describes an additive-free iridium-catalyzed direct asymmetric reductive amination that enables the efficient synthesis of chiral β-arylamines, which are important pharmacophores present in a wide variety of pharmaceutical drugs. The reaction makes use of bulky and tunable phosphoramidite ligands for high levels of enantiomeric control, even for alkylamino coupling partners which lack secondary coordinating sites. The synthetic value of this succinct procedure is demonstrated by single-step synthesis of multiple drugs, analogs and key intermediates. Mechanistic investigations reveal an enamine-reduction pathway, in which H-bonding, steric repulsion, and CH-π and electrostatic interactions play important roles in defining the spatial environment for the “outer-sphere” hydride addition.
Introduction
Chiral β-arylamines are important pharmacophores present in a wide variety of pharmaceutical drugs and other biologically active compounds. For instance, 14 out of 200 top brand name drugs by US retail sales in 2021 contain chiral β-arylamino units.1 Some examples are lisdexamfetamine, tamsulosin, arformoterol, selegiline, silodosin, and sitagliptin (Scheme 1A). Consequently, it is of great significance to develop both atom- and step-economic methods for concise and efficient synthesis of such compounds.2 Two attractive transformations have emerged as highly potential approaches: the asymmetric hydrogenation (AH)3 of β-arylenamides4 and β-arylenamides5 (Scheme 1B-i), and the direct asymmetric reductive amination (ARA)6 of arylacetones7 (Scheme 1B-ii). In AH, extra steps are required to prepare and remove the amide group, and frequently the existence of the E/Z isomers will complicate the enantiocontrol.4a,b While ARA represents a superb choice in terms of substrate availability, cost-efficiency and the number of synthetic steps required for achieving the targeted product, particularly when H2 is utilized as the reducing agent, the massive diphenylmethyl group needs to be removed for further N-alkylation, which makes this option less attractive in the aspect of atom-economy.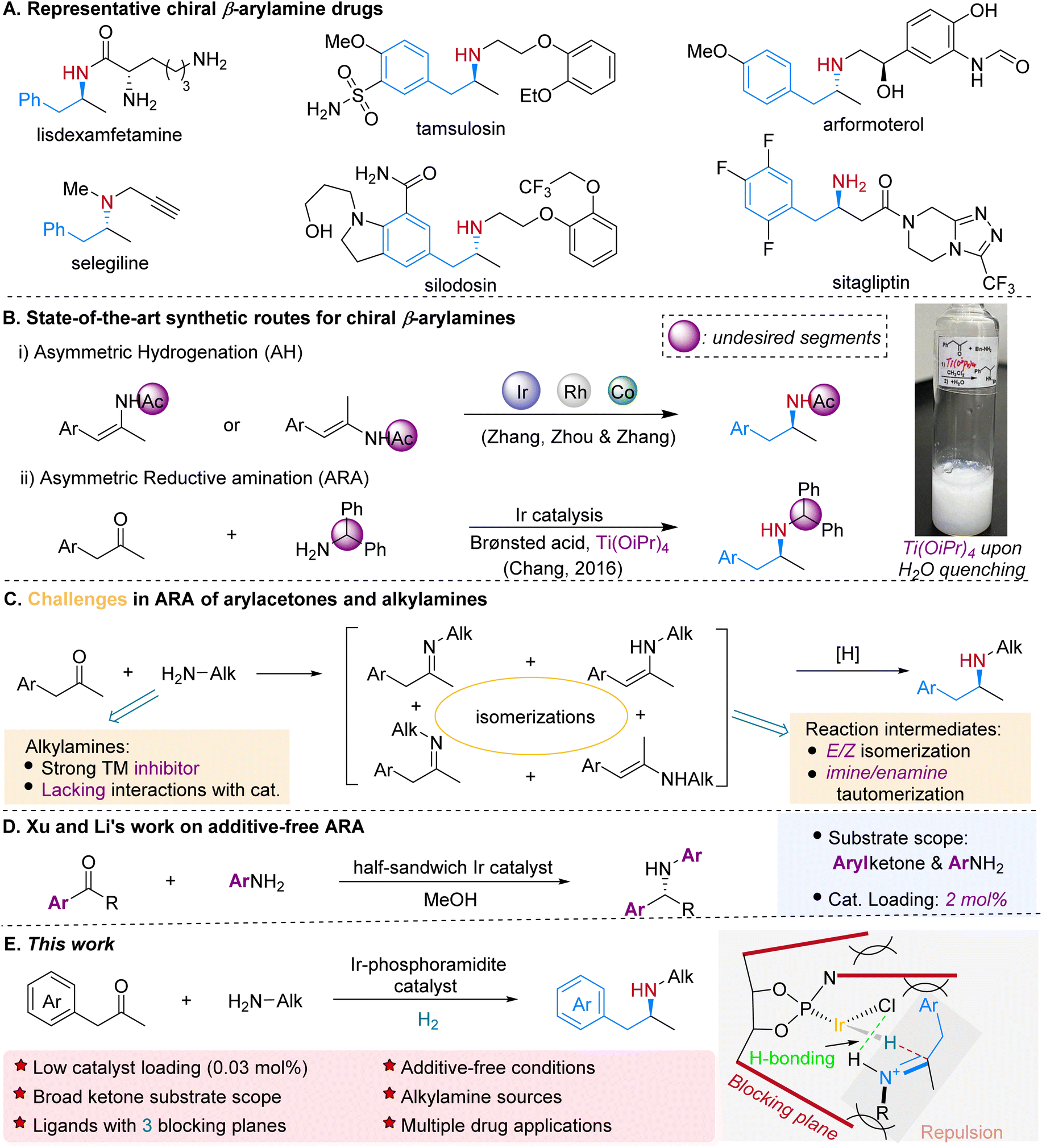 | ||
| Scheme 1 Relevance of chiral β-arylamines and efficient enantioselective synthetic strategies. | ||
In more than two decades, much progress has been achieved in the field of transition-metal-catalyzed ARA since the first example was reported by Blaser and co-workers in 1999,8 and is underlined by the applications in the production of sitagliptin7b and suvorexant.9 Nevertheless, factors including the inhibitory effects of nitrogen-containing species, especially alkyl amines (Scheme 1C), on transition-metals, the presence of multiple isomers (Scheme 1C), chemoselectivity and overalkylation significantly limit its prevailing application in synthesis of related chiral amines. As a result, less nucleophilic arylamines and ammonium cations are predominately utilized as the N-sources in ARA. And most of the times additives are required to alleviate the inhibitory effects and/or facilitate the formation of the imine intermediates. Titanium(IV) alkoxides, which boost the formation of the imine intermediates during the ARA process, are one class of mostly used Lewis acid additives, yet they tend to form gel-like fluid with non-settling fine particles upon quenching with water thus making the following workup difficult (Scheme 1B).7a,10 Recently Xu, Li and co-workers reported an excellent iridium-catalyzed additive-free transfer hydrogenative amination, while the substrates are limited to aromatic ketones and less inhibitory arylamino sources (Scheme 1D).11 Nevertheless, an additive-free and direct catalytic ARA system for the coupling of more general ketones and amines, particularly arylacetones and aliphatic N-sources, is highly desirable (Scheme 1C). Herein we demonstrate that the iridium complexes in situ generated from [Ir(cod)Cl]2 and bulky phosphoramidite ligands are efficient catalysts towards the direct asymmetric reductive amination of arylacetones and primary alkylamines (Scheme 1E). Under mild reaction conditions and without the addition of any additive, the prochiral ketones reductively couple with alkylamines effectively to form functionalized and structurally diverse chiral β-arylamines, including drugs, key pharmaceutical intermediates and their analogs.
Results and discussion
Iridium complexes are the most frequently employed catalysts in transition-metal catalyzed ARA due to their outstanding resistance to inhibitory effects, remarkable efficiency towards imine reduction3,12 and less chance of obtaining alcohol by-products from the ketone reduction.6 We set out our research on the reaction of 1-(4-methoxyphenyl)-propan-2-one (1a) and 2-phenylethylamine (2a) using [Ir(cod)Cl]2 as the metal precursor. In our previous research on the synthesis of β-arylamines via ARA utilizing diphenylmethylamine as the N-source,7a the catalyst generated from iridium and monodentate phosphoramidite chiral ligand13L1 displayed extraordinary reactivity and enantio-selectivity. Yet for the ARA reaction of a typical alkylamine 2a, it merely resulted in 58% yield and 23% ee (Table 1, entry 1). Without the addition of any additive, the reaction yield dramatically decreased to 16% (entry 2), and could be improved in the presence of molecular sieves (entry 3).|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solventb | Yield (%) | ee (%) |
a Reaction conditions: 1a 0.1 mmol, 2a (1.02 equiv.), solvent 1 mL. Yields and enantiomeric excess values were determined by chiral HPLC after the products were converted to the corresponding acetamides. TFE = 2,2,2-trifluoroethanol.
b PhMe/TFE = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
c CF3CO2H (30 mol%), Ti(OiPr)4 (30 mol%) and molecular sieves (4 Å, 25 mg) were added as in ref. 7a.
d Molecular sieves (4 Å, 25 mg) were added.
e [Ir]/L = 1:1.5.
f [Ir(cod)I]2 was used instead of [Ir(cod)Cl]2. :1.
c CF3CO2H (30 mol%), Ti(OiPr)4 (30 mol%) and molecular sieves (4 Å, 25 mg) were added as in ref. 7a.
d Molecular sieves (4 Å, 25 mg) were added.
e [Ir]/L = 1:1.5.
f [Ir(cod)I]2 was used instead of [Ir(cod)Cl]2.
|
||||
| 1c | L1 | CH2Cl2 | 58 | 23 |
| 2 | L1 | CH2Cl2 | 16 | <5 |
| 3d | L1 | CH2Cl2 | 25 | 32 |
| 4 | L1 | EtOAc | 58 | 5 |
| 5 | L1 | PhMe | <5 | — |
| 6 | L1 | MeOH | 68 | 34 |
| 7 | L1 | TFE | 90 | 50 |
| 8 | L2 | TFE | 89 | 23 |
| 9 | L3 | TFE | 78 | 34 |
| 10 | L4 | TFE | 58 | 62 |
| 11 | L5 | TFE | 85 | 79 |
| 12 | L6 | TFE | 91 | 75 |
| 13 | L5 | PhMe/TFE | 95 | 94 |
| 14c | L5 | PhMe/TFE | 98 | 92 |
| 15e | L5 | PhMe/TFE | 82 | 93 |
| 16f | L5 | PhMe/TFE | 81 | 94 |

In this reaction the solvent played an important role, as the protic solvents, methanol and particularly more polar 2,2,2-trifluoroethanol (TFE), delivered much higher yields and ee values than the non-protic ones (Table 1, entries 4–7). In the TFE environment, the formation of the imine intermediate, the proton transfer and the stabilization of the iminium cation could be all enhanced, thus leading to better reactivity.14 To employ the tunability of the phosphoramidite chiral ligands, H8-BINOL backbone based L2–L6 with different substituents at the 3,3′-position and/or amino parts were synthesized and examined. Although L3 with the 3,3′-Ph group alone did not provide better result, when the amino part of the phosphoramidite was changed from piperidine to tetrahydro-isoquinoline (L4), the enantiopurity of 3aa was improved by nearly 30% (entry 10). Increasing the bulkiness by introducing 1-naphthyl groups at the 3,3′-position helped to further elevate the ee value to 79% (L5, entry 11). A more spatial ligand L6 did not deliver better results (entry 11). The presence of a second solvent in the reaction system, toluene in this case, helped to achieve satisfactory results (entry 13). Molecular sieves were beneficial for the reaction yield, but there was a slight decrease in the ee value (entry 14). When the [Ir]/L ratio was lowered to 1:1.5, the reaction yield decreased to 82% (entry 15).
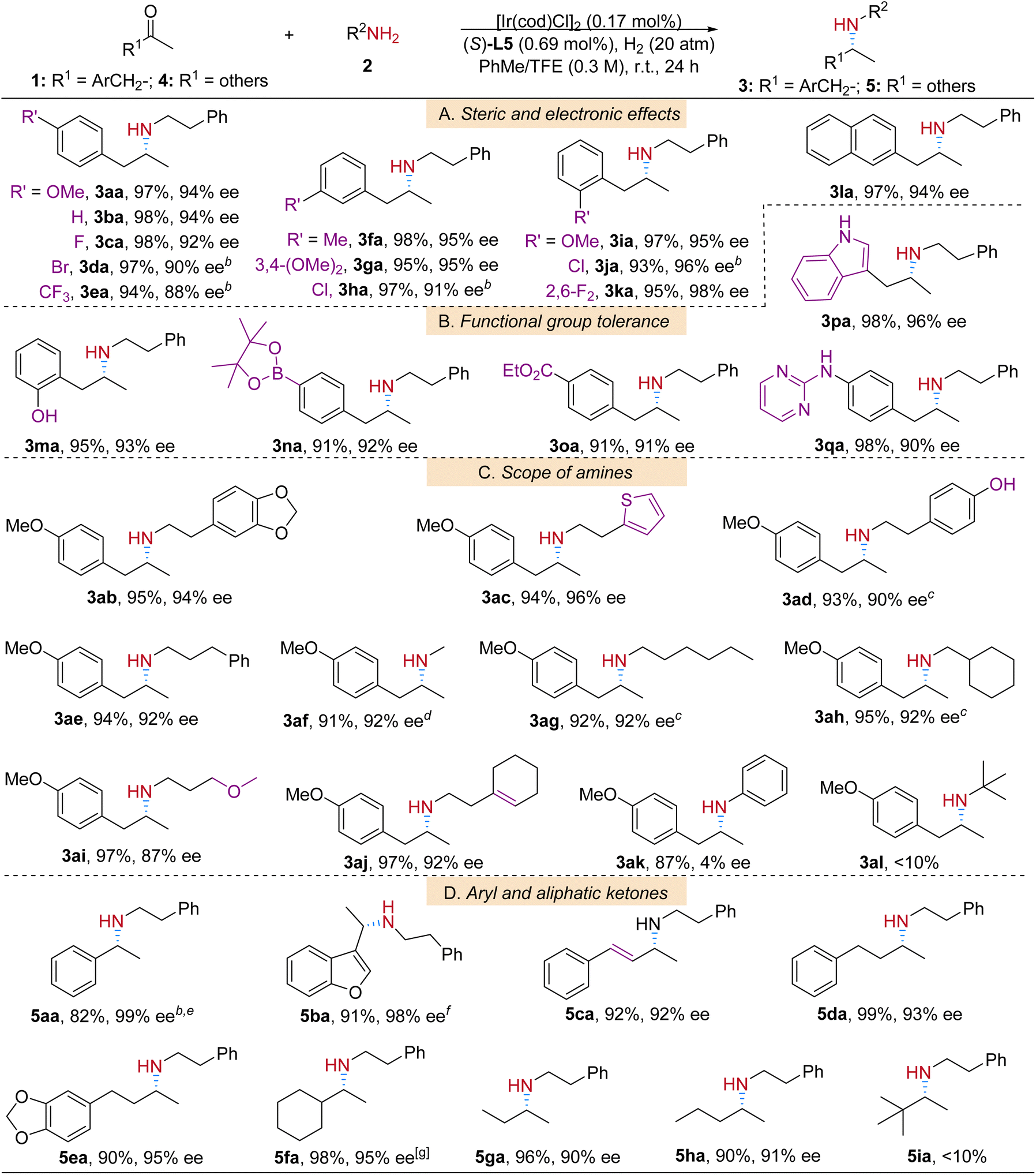
With the optimized reaction conditions in hand, the scope and limitations of the protocol were studied (Table 2). Substituted arylketones were first investigated. The electronic and steric properties of the aryl group did not significantly impact the reaction yields (Table 2A, products 3aa–3la); however, slightly higher enantioselectivity was observed for the electron-donating substituted β-arylamine products (3aa, 3fa, 3ga, and 3ia) than that with electron-withdrawing substituents (3ea and 3ha) when they are at para- or meta-positions; excellent yields and ees were achieved for all ortho-substituted products (3ia–3ka). To our delight, in the functional group tolerance examination the protic polar hydroxyl group, one of the challenging functional groups in drug synthesis,6d was well tolerated in our reaction system (Table 2B, product 3ma). Other common groups, borate (3na), ester (3oa) and indolyl (3pa), were also compatible with the catalytic conditions. It is worth mentioning that substrate 1q, which contains both heteroaromatic pyrimidinyl and protic amino groups, was smoothly transformed into product 3qa in an excellent yield and good enantioselectivity.
|
a Reaction conditions: 1/4 0.3 mmol, 2 (1.02 equiv.), PhMe/TFE = 3:1 (total 1 mL), room temperature, 24 h; isolated yields; enantiomeric excess values were determined by chiral HPLC after the products were converted to the corresponding acetamides.
b Molecular sieves (4 Å, 25 mg) were added.
c
L6 was used instead of L5.
d MeNH2·HCl was used.
e Reaction temperature was 60 °C.
f Reaction temperature was 40 °C.
g Reaction temperature was 80 °C.
|
|---|
|
Then a variety of alkylamine coupling partners with diverse structures (Table 2C, products 3ab–3al) were examined in the reactions with 1a. The amines containing both phenolic (–PhOH) and alkenyl groups successfully reductively coupled with 1a to yield 3ad and 3aj respectively. It is important to note that amines with pure alkyl substituents also resulted in corresponding products (3af–3ah) in good yields and ees. For n-hexylamine 2g and cyclohexylmethanamine 2h substrates, bulkier ligand L6 was applied for better enantioselectivity.15 In the reaction of aniline 2k as the amine source, product 3ak was obtained in a satisfactory yield, yet in near racemic form. And for sterically hindered t-butylamine 2l, the reaction barely proceeded.
After having successfully performed the ARA reactions of 1-arylpropan-2-ones and amines, we were interested in the applicability of this additive-free catalytic system in ketones with other structures. Fortunately, with the help of molecular sieves at higher reaction temperature, the iridium-L5 catalytic system worked very well towards aromatic and heteroaromatic ketones 4a and 4b (Table 2D, products 5aa and 5ba). Alkyl ketones are challenging substrates in transition-metal catalyzed asymmetric transformations because of lack of secondary interactions with the catalytic complexes. It is especially difficult to differentiate two similar groups in terms of steric and electronic properties.16 In this regard it is satisfying that related reaction intermediates for such ketones were well adapted in our catalytic environment and led to corresponding products 5da–5ha in excellent yields and high ee values. Even the methyl ethyl ketone substrate 5g is enantioselectively recognized by the Ir-L5 catalyst and leads to product 5ga in 90% ee. Sterically hindered 3,3-dimethylbutan-2-one 4i was not suitable under the standard reaction conditions.
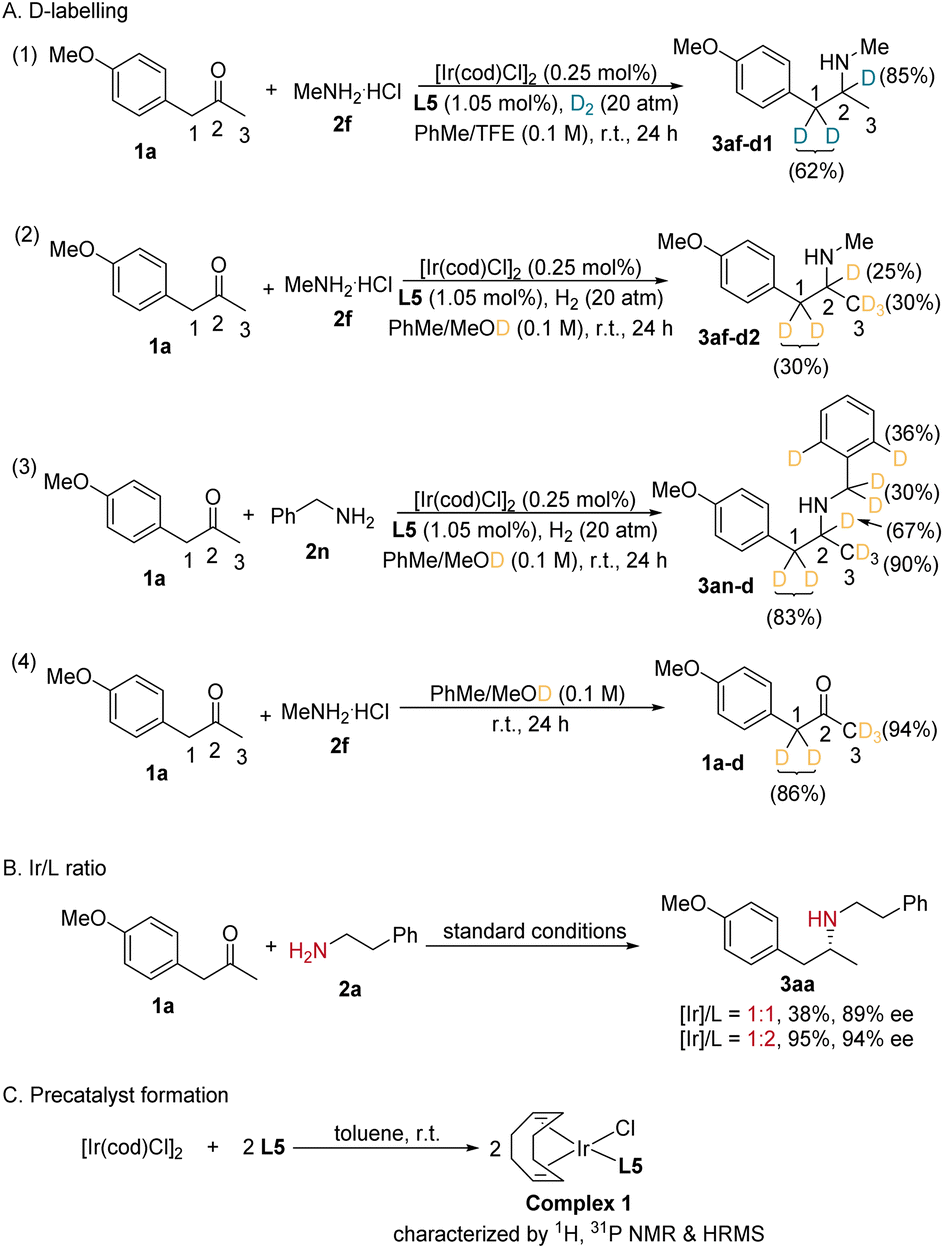
To demonstrate the practical utility and potential for the synthesis of related chiral amino pharmaceuticals using our protocol, a couple of gram-scale experiments were performed (Scheme 2). Catalyzed by 0.03 mol% of the Ir-L5 complex, 3 mmol of 1a was efficiently converted to 1.09 g of 3aa in 82% yield and 91% ee, while the addition of molecular sieves could elevate the reaction yield to 91%. In the second gram-scale reaction, 1.07 g of tamsulosin analog 3rm was synthesized in one single step from the reductive coupling of sulfamidoarylacetone precursor 1r and amine 2m (Scheme 2B). This method was also applied in the two-step synthesis of a MAO-B inhibitor for the treatment of Parkinson's disease, selegiline (Scheme 2C). Similarly, the analog of the key intermediate of silodosin was obtained via this procedure in a satisfactory yield and enantioselectivity (Scheme 2D). To gain insights into the reaction pathways, particularly on whether the reduction proceeds through an imine or enamine intermediate, we conducted an isotopic labelling experiment. For the reaction of 1a and 2f, from the 1H NMR integration, the deuterium abundance at the chiral center C2 of 3af was 85% and that on C1 was 62% with deuterium gas as the reductant (Scheme 3A-1). When utilizing MeOD/PhMe as the reaction solvent and normal H2, deuterium was found to partially incorporate into all the C1, C2 and C3 positions (Scheme 3A-2). As for the reaction of 1a and benzylamine 2n with MeOD as the D source, deuterium is not only present at the C1, C2 and C3 positions, but also at the benzyl and ortho-positions of the N-benzyl group of 3an (Scheme 3A-3). At the same time, the deuterium incorporation ratios for 3an at the C1, C2 and C3 positions are much higher than those for 3af. The above results indicate an enamine reduction pathway. The extra deuterium atom on C1 probably stems from the H/D exchange via a ketone/enol tautomerization (Scheme 3A-4), and the heterolytic cleavage of H2 during its addition to the iridium center, which also explains the source of the 15% hydrogen atom on C2 (Scheme 3A-1). Although the optimized [Ir]/L5 ratio was 1:2, with an [Ir]/L5 ratio of 1:1, product 3aa was obtained in only slightly lower ee, albeit in a much lower yield (Scheme 3B). In the formed complex from [Ir(cod)Cl]2 and L5, only 1 equiv. of L5 coordinates to Ir, even in the presence of a large excess of the phosphoramidite ligand (Scheme 3C).
 | ||
| Scheme 2 Gram-scale reactions and applications. Conditions (a) K2CO3 (3 equiv.), MeCN, 80 °C, 16 h. Condition variation (b) 40 °C, 40 atm H2, MS. | ||
 | ||
| Scheme 3 Mechanistic studies. | ||
 | ||
| Scheme 4 Proposed reaction pathways and energy profiles. | ||
DFT calculations for the reaction of 1b and methylamine (2f) using the Gaussian 16 program were performed to better understand the underlying reaction features and the origins of enantio-selectivity of this iridium catalysis. Based on the literature on iridium catalysis of the hydrogenation of imines14,17 and calculation results compared with “inner-sphere” H-addition (see the ESI†), possible reaction pathways were proposed (Scheme 4), which featured an “outer-sphere” H-addition as the key step (Scheme 4A, left). One of the major interactions between the substrate and the catalytic complex is the H-bonding, arising from the protonated enamine and the chloride on the iridium center. No obvious ee difference was observed when [Ir(cod)I]2 was used instead of [Ir(cod)Cl]2, probably because besides the chloride anion, the Ir–H(δ−) could also form a H-bond with the H+ from int-2.17c Although only one of the two 1-naphthyl groups of the phosphoramidite ligand L5 directly interacts with int-2, the other one plays its role cooperatively with the tetrahydroisoquinilinyl (THIQ) moiety. It affects the spatial position of the THIQ unit through a steric repulsion interaction, which subsequently confines the position of the phenyl group of the substrate in a similar manner. That is, both the two 1-naphthyl groups and the THIQ unit act collaboratively as blocking planes to define the steric environment of the substrate int-2 (Scheme 1E; Scheme 4A, left). The electrostatic interaction among the hydrogens, C(1)–Hδ+ of THIQ, Ir–Hδ−, and CBn–Hδ+ and CAr–Hδ+ of int-2, also helps to ensure the stability of the transition state (Scheme 5A, right). Surprisingly, the hydride cis to the phosphoramidite ligand also is able to add to int-2 and form the desired (R)-3bf product with even slightly lower Gibbs free energy for the transition state than that of the trans-addition. In this model, there is no H-bonding interaction. Instead, the (NCH2)–H of the iminium ion and the phenyl group of THIQ attract each other via a CH-π interaction (Scheme 5B, left). Similarly, there is electrostatic interaction between (Ir)–Hδ− and Clδ− and two hydrogens of the methyl group of int-2 (Scheme 5B, right). The rotamers resulting from the rotation of two 1-naphthyl groups around the C–C bonds linked to the H8-BINOL backbone also influence the stability of the transition states, with the (Sa,Sa)-isomer favored by 1.8 kcal mol−1 over the (Ra,Ra)-isomer (Scheme 5C).
 | ||
| Scheme 5 H-Addition transition states & rotamers. | ||
Conclusions
In summary, we have developed an efficient direct asymmetric reductive amination for the precise synthesis of chiral N-alkyl β-arylamines. Catalyzed by the in situ generated complexes from iridium and fine-tuning phosphoramidite ligands under additive-free and mild conditions, a variety of prochiral ketones with diverse structures smoothly reductively coupled with alkylamines to form α-aryl-, β-aryl-and alkyl chiral amines. DFT studies reveal that the two naphthyl groups at the 3,3′-positions and the tetrahydroisoquinolinyl substituent of the applied phosphoramidite ligand act as three blocking planes. Along with the H-bonding and multiple electrostatic interactions, the enamine intermediates are spatially well confined in the catalytic environment, thus leading to products with excellent enantioselectivity. This general protocol should find extensive utility for direct and precise chiral amine synthesis and shed light on better catalyst design.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. W. established the reaction conditions. W. W. performed the DFT calculations. J. W. and H. H. expanded the substrate scope. M. C. conceived and supervised the project and wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (21772155), Natural Science Basic Research Program of Shaanxi (Program No. 2024JC-JCQN-17), Innovation Capability Support Program of Shaanxi (Program No. 2024RS-CXTD-67) and Scientific Fund of Northwest A&F is gratefully acknowledged. We greatly acknowledge HPC of Northwest A&F University for the DFT calculations carried out in this work and Dr Xiuhuan Li of State Key Laboratory of Crop Stress Biology for Arid Areas at Northwest A&F University for her kind help in the NMR spectroscopy.Notes and references
- M. Qureshi and J. Njardarson, Compiled and produced by the Njardarson group, The University of Arizona, 2022 Search PubMed.
- (a) Chiral Amine Synthesis: Methods, De-velopments and Applications, ed. T. Nugent, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) W. Li, X. Zhang, Stereoselective Formation of Amines, Springer, Berlin, Heidelberg, 2014, vol. 343 CrossRef; (c) A. Trowbridge, S. Walton and M. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- (a) J. Xie, S. Zhu and Q. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (b) D. Ager, A. de Vries and J. de Vries, Chem. Soc. Rev., 2012, 41, 3340–3380 RSC; (c) C. Seo and R. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS; (d) J. Barrios-Rivera, Y. Xu, M. Wills and V. Vyas, Org. Chem. Front., 2020, 7, 3312–3342 RSC; (e) R. Ab-dine, G. Hedouin, F. Colobert and J. Wencel-Delord, ACS Catal., 2021, 11, 215–247 CrossRef CAS; (f) A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269–339 CrossRef PubMed.
- (a) J. Chen, W. Zhang, H. Geng, W. Li and X. Zhang, Angew. Chem., Int. Ed., 2009, 48, 800–802 CrossRef CAS PubMed; (b) G. Liu, X. Liu, Z. Cai, G. Jiao, G. Xu and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 4235–4238 CrossRef CAS PubMed; (c) T. Chen, Y. Zou, Y. Hu, Z. Zhang, H. Wei, L. Wei and W. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303488 CrossRef CAS PubMed.
- S. Li, K. Huang and X. Zhang, Chem. Commun., 2014, 50, 8878 RSC.
- (a) C. Wang and J. Xiao, Top. Curr. Chem., 2014, 343, 261–282 CrossRef CAS PubMed; (b) O. Afanasyev, E. Kuchuk, D. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed; (c) S. Roughley and A. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed; (d) D. Blake-more, L. Castro, I. Churcher, D. Rees, A. Thomas, D. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed; (e) T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed; (f) K. Murugesan, T. Senthamarai, V. Chandrashekhar, K. Natte, P. Kamer, M. Beller and R. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC; (g) Y. Tian, L. Hu, Y. Wang, X. Zhang and Q. Yin, Org. Chem. Front., 2021, 8, 2328–2342 RSC; (h) N. Reshi, V. Saptal, M. Beller and J. Bera, ACS Catal., 2021, 11, 13809–13837 CrossRef CAS.
- (a) H. Huang, X. Liu, L. Zhou, M. Chang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5309 CrossRef CAS PubMed; (b) D. Stein-huebel, Y. Sun, K. Matsumura, N. Sayo and T. Saito, J. Am. Chem. Soc., 2009, 131, 11316–11317 CrossRef CAS PubMed.
- H. Blaser, H. Buser, H. Jalett, B. Pugin and F. Spin-dler, Synlett, 1999, 867–868 CrossRef CAS.
- N. Strotman, C. Baxter, K. Brands, E. Cleator, S. Krska, R. Reamer, D. Wallace and T. Wright, J. Am. Chem. Soc., 2011, 133, 8362–8371 CrossRef CAS PubMed.
- (a) Y. Chi, Y. Zhou and X. Zhang, J. Org. Chem., 2003, 68, 4120–4122 CrossRef CAS PubMed; (b) H. Zhou, Y. Liu, S. Yang, L. Zhou and M. Chang, Angew. Chem., Int. Ed., 2017, 56, 2725–2729 CrossRef CAS PubMed; (c) Z. Wu, S. Du, G. Gao, W. Yang, X. Yang, H. Huang and M. Chang, Chem. Sci., 2019, 10, 4509–4514 RSC; (d) L. Hu, Y. Zhang, Q. Zhang, Q. Yin and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5321–5325 CrossRef CAS PubMed; (e) Z. Wu, W. Wang, H. Guo, G. Gao, H. Huang and M. Chang, Nat. Commun., 2022, 13, 3344 CrossRef CAS PubMed.
- Y. Gao, Z. Wang, X. Zhang, M. Zhao, S. Zhang, C. Wang, L. Xu and P. Li, Angew. Chem., Int. Ed., 2023, 62, e2023037 Search PubMed.
- F. Spindler and H.-U. Blaser in Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, vol. 3, p. 1193 Search PubMed.
- (a) A. Minnaard, B. Feringa, L. Lefort and J. de Vries, Acc. Chem. Res., 2007, 40, 1267–1277 CrossRef CAS PubMed; (b) W. Fu and W. Tang, ACS Catal., 2016, 6, 4814–4858 CrossRef CAS.
- J. Wang, W. Wang, X. Yang, J. Liu, H. Huang and M. Chang, Sci. China: Chem., 2023, 64, 518–525 CrossRef.
- B. Mitschke, M. Turberg and B. List, Chem, 2020, 6, 2515–2532 CAS.
- (a) F.-H. Zhang, F.-J. Zhang, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Nat. Catal., 2020, 3, 621–627 CrossRef CAS; (b) H. Zhou, Y. Zhou, H. Y. Bae, M. Leutzsch, Y. Li, C. K. De, G.-J. Cheng and B. List, Nature, 2022, 605, 84–89 CrossRef CAS PubMed; (c) M. Wang, S. Liu, H. Liu, Y. Wang, Y. Lan and Q. Liu, Nature, 2024, 631, 556–562 CrossRef CAS PubMed.
- (a) C. Cui, H. Chen, S. Li, T. Zhang, L. Qu and Y. Lan, Chem. Rev., 2020, 412, 213–251 Search PubMed; (b) G. Dobereiner, A. Nova, N. Schley, N. Hazari, S. Miller, O. Eisenstein and R. Crab-tree, J. Am. Chem. Soc., 2011, 133, 7547–7562 CrossRef CAS PubMed; (c) W. Zhao, W. Wang, H. Zhou, Q. Liu, Z. Ma, H. Huang and M. Chang, Angew. Chem., Int. Ed., 2023, 62, e202308836 CrossRef CAS PubMed; (d) C. Li, B. Villa-Marcos and J. Xiao, J. Am. Chem. Soc., 2009, 131, 6967–6969 CrossRef CAS PubMed; (e) W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan, L. Lian, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1668–1672 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures and spectra. See DOI: https://doi.org/10.1039/d4sc04416a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |