Trans vs. cis: a computational study of enasidenib resistance due to IDH2 mutations†
Received
17th April 2024
, Accepted 25th June 2024
First published on 27th June 2024
Abstract
Isocitrate dehydrogenase 2 (IDH2) is a homodimeric enzyme that plays an important role in energy production. A mutation R140Q in one monomer makes the enzyme tumourigenic. Enasidenib is an effective inhibitor of IDH2/R140Q. A secondary mutation Q316E leads to enasidenib resistance. This mutation was hitherto only found in trans, i.e. where one monomer has the R140Q mutation and the other carries the Q316E mutation. It is not clear if the mutation only leads to resistance when in trans or if it has been discovered in trans only by chance, since it was only reported in two patients. Using molecular dynamics (MD) simulations we show that the binding of enasidenib to IDH2 is indeed much weaker when the Q316E mutation takes place in trans not in cis, which provides a molecular explanation for the clinical finding. This is corroborated by non-covalent interaction (NCI) analysis and DFT calculations. Whereas the MD simulations show a loss of one hydrogen bond upon the resistance mutation, NCI and energy decomposition analysis (EDA) reveal that a multitude of interactions are weakened.
1 Introduction
Drug resistance is a major clinical challenge, especially in cancer, bacterial, viral, fungal and parasitic diseases. In modern drug design, drugs are developed to bind molecular targets (most often proteins) with high specificity and affinity. Mutations that develop in the drug target upon treatment are a major cause for resistance.1,2 Understanding the cause for drug resistance is essential for combating it and developing better drugs. Insights from physical chemistry can be highly useful in this respect. For example, in a study of resistance mutations to dasatinib that targets the protein ABL1, it was found that three such mutations have different mechanisms whereby resistance was driven by enthalpy, entropy or a combination of those.3 Other studies, theoretical and experimental, pointed to the importance of kinetics in enabling drug resistance.4–7
IDH2 is a homodimeric enzyme that, upon mutations in residues Arg140 or Arg172 leads to the formation of (R)-2-hydroxyglutarate (2HG), a metabolite which is not normally formed in the body. Accumulation of 2HG can drive several cancers. Specifically, a mutation R140Q in one of the two copies of the enzyme is associated with acute myeloid leukaemia (AML).8 Treatment with enasidenib, which specifically targets IDH2/R140Q over the wild-type enzyme, has been shown effective in such cases.9,10 Secondary mutations in IDH2, Q316E and I319M have been observed in patients treated by enasidenib and were suggested to cause drug resistance.10–12 Of note, AML is a rare disease and mutations in IDH2 are only observed in a subset of patients, thus reports on resistance mutations to enasidenib were few.
Interestingly, in the two cases where resistance mutations were reported, the resistance mutation was in trans to the activating R140Q mutation.12 However, no mechanism was suggested to explain this finding. Moreover, the small number of patients that were hitherto subjected to treatment with enasidenib and for which data on resistance is available is not enough to conclude whether or not mutations must be in trans. Due to the scarcity of clinical information, computational studies can be insightful to examine if the binding of the drug is indeed weakened in trans but not in cis. We therefore employed molecular dynamics (MD) simulations to study the binding of enasidenib to IDH2/R140Q and to IDH2/R140Q/Q316E in cis and in trans. We have further studied the binding by examining how the drug binds to its nearby environment using density functional theory (DFT), non-covalent interactions (NCI) analysis and energy decomposition analysis (EDA) to gain physical insights on the binding of the drug in the single and double mutants.
2 Computational methods
2.1 Structure preparation
Structure preparations were conducted in CHARMM-GUI solution builder.13–15 The crystal structure for R140Q-mutated IDH2 (PDB id 5I9616) was downloaded from Protein Data Bank (PDB).17 This structure had the R140Q mutation on both chains (A and B, each representing one monomer). Residue Gln140 was modified back to Arg on chain A. Residues 43 to 449 were included in the analysis as these were visible in both chains. The systems were prepared by removing everything except the protein, enasidenib, the cofactor NADPH and calcium ions. Residue 316 was either (1) not modified (2) modified in cis to Glu, i.e. in chain B or (3) modified in trans to Glu. A cubic water box was applied with distance 12 Å from the protein. To mimic the conditions in cells, the systems were neutralized and simulated in a KCl solution (0.15 mol dm−3). The pressure was kept at 1 bar during the simulations.
2.2 MD simulations setup and forcefield
The simulations employed CHARMM3618 as force field for the protein and CGENFF19 for the drug. Water molecules were simulated with the TIP3P20 model. GROMACS version 2022.321,22 was used throughout all of the protein simulations. Particle–mesh Ewald (PME) was used to treat long-range electrostatic interactions.23 The temperature was kept constan at T = 310 K by the velocity rescaling thermostat24 (τt = 0.1 ps). In order to constrain hydrogen bonds the LINCS constraint algorithm was used.25 The SETTLE algorithm26 was employed to constrain the water molecules. The cut-off distance for Coulomb and van der Waals interactions was set to 1.2 nm. The vdW potential was smoothly switched off starting at a distance of 1.0 nm between atoms.
The energy minimization procedure was run until maximum force was below 1000 kJ mol−1 nm−1 on every atom. Following this, a 20 ps simulation with positional restraints on solute atoms was performed as the first step towards system equilibration. The restrains were removed and a second equilibration simulation was carried out for 5 ns. Finally a 100 ns production simulation was run. Each system (wt and two mutants) was simulated in quadruplicate.
The pressure was kept constant by use of C-rescale algorithm27 during equilibration and the Parrinello–Rahman algorithm28 during production runs. The production simulations were carried out at the PDC high-performance computer system at KTH, Sweden.
2.3 Analysis of the MD simulations
The analysis was performed in GROMACS. Visual molecular dynamics (VMD)29 was used for visualization. Median and maximal root mean square deviations (RMSD) values are given in the ESI.† Analysis of the structural similarity between the monomers was performed as follows. Firstly, ten equally-spaced structures were extracted from each simulation for each monomer, yielding overall 40 structures. These were thereafter aligned in pairs on their Cα atoms using the gmx confrms tool that only accepts single structures as input (no trajectories).
2.4 Estimation of the protein–drug binding free energies with linear interaction energy
Estimation of the protein–drug binding energies were performed here to supplement the finding from MD simulations. To this aim, we used the linear interaction energy (LIE) method, which has been developed to estimate free energies of binding from interaction energies calculated from MD simulations.30 LIE was preferred over more sophisticated enhanced sampling methods such as the use of pulling simulations31 or non-equilibrium simulations32–34 due to its simplicity, stability and lower computational cost.
The free energy is estimated as:35
| | | ΔGb ≈ α(〈El–pvdW〉 − 〈El–svdW〉) + β(〈El–pCoul〉 − 〈El–sCoul〉) + γ | (1) |
α, β and γ are parameters, whereas El–p and El–s are the ligand–protein and ligand–solvent interaction energies (vdW or Coulomb). In practice, interaction energies are averaged from simulations of drug bound to a protein and in solvent giving the 〈E〉 values in Eq. 1. A simulation of the drug in solvent was ran separately, using the same number of water molecules as in the simulations of the complex, to calculate the El–s values. From Linear Response Theory, β = 0.5.30α = 0.16 had been used when LIE was developed and was used here as well. We tuned γ to reproduce the Gibbs energy of binding enasidenib to the R140Q mutant. The energy was estimated as the median value from the measurements of IC50 reported in the BindingDB database,36 yielding −8.9 kcal mol−1 (corresponding to an IC50 of 262 nM). Using the extracted value of γ, the binding energies were calculated for the R140Q/Q316E mutants. The relative error was calculated by estimating the standard deviations for the El–pvdW and El–pCoul energies and dividing these by the corresponding averages to get the relative error. The larger of these values was multiplied by the calculated free energy, as in Eqn 2 (σ in this equation refers to the standard deviation).
| |  | (2) |
2.5 DFT calculations of the binding free energy of the drug and adjacent residues
The method for the binding energy calculations was described in ref. 3 and 37. A highly simplified model was used where the protein, drug and complex assumed the exact same conformation and only residues hydrogen bonding to the drug were considered. This neglects many important interactions while also reduces artifacts from strain to the protein. To build the DFT models, five snapshots from each simulation were extracted at equidistant time intervals (20, 40, 60, 80 and 100 ns) for a total of 20 energy calculations for each mutation. The residues were truncated and hydrogens were added at specific positions (Table 1 and Fig. 1). Avogadro version 1.2.038 was used for this purpose.
Table 1 Positions of added hydrogens for the DFT calculations
|
|
R140Q/Q316Ecis |
| Gln316 |
Truncated at ATOM 4408 CG, valence filled |
| Glu316 |
Truncated at ATOM 4408 CG, valence filled |
|
|
|
|
|
R140Q/Q316Etrans |
|
|
|
| Glu316 |
Truncated at ATOM 10852 CG, valence filled |
| Gln316 |
Truncated at ATOM 10850 CG, valence filled |
 |
| | Fig. 1 Enasidenib in complex with R140Q mutated IDH2. PDB id 5I96. (a) A schematic view of the binding site for R140Q/Q316Ecis. (b) Enasidenib's positions at the interface between the two chains of the protein. (c) Enasidenib at the binding site. | |
Once the structures were prepared, geometry optimization was carried out in ORCA version 5.0.439,40 with the M06 functional41 and the def2-SV(P) basis set42 with the solvent (water) represented as PCM.43 During optimization, heavy atoms were fixed to their initial positions. After the optimization was finished, binding free energy calculations were conducted. These calculations were performed with ωB97X-D/def2-TZVP/SMD.42,44,45 Basis set superposition error (BSSE) was included. The binding energy was then obtained by the relations:
| | | ΔGbind = Gcomplex − (Genasidenib + GR140Q/Q316EIDH2) | (3) |
and
| | | ΔGbind = Gcomplex − (Genasidenib + GR140QIDH2) | (4) |
where
Gcomplex,
Genasidenib,
GR140QIDH2 and
GR140Q/Q316EIDH2 are the electronic ground state free energies in SMD for each quantity. Δ
G was taken as the arithmetic mean value of the 20 energy values for each mutation. ΔΔ
G was then calculated by
| |  | (5) |
| | 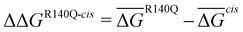 | (6) |
| |  | (7) |
2.6 Energy decomposition analysis
Energy decomposition analysis (EDA) was performed in XEDA46 using GKSEDA47–49 at the ωB97X-D/def2-TZVP level of theory in gas phase. The calculation matches the PCMEDA method48 which uses a set of energy calculations to divide the interaction energies between two or more monomers in a complex into contributions from electrostatics, exchange, repulsion, polarisation, desolvation, correlation and dispersion. The desolvation components were calculated separately in ORCA39,40 since PCMEDA assumes the same cavity size for all atoms, which might be inappropriate for protein–drug interactions.3 Small differences between values calculated with XEDA and ORCA occur due to the different grids employed by the two packages and (to a small extent) since ORCA employs the RIJCOSX approximation.
2.7 Extracting representative structures from the simulations
Representative structures for further analysis were generated by clustering all snapshots in the trajectory using gmx cluster. The Gromos algorithm50 was used for clustering with a cutoff of 0.15 nm. The central structure of the most occupied cluster was selected as representative.
2.8 Non-covalent interactions
The NCI analysis was performed in NCI plot version 4.51 The structures were taken from clustering analysis (vide supra) and include only the same representations as used in the DFT calculations. The NCI analysis for each of these structures was then plotted in the same figure using MATLAB version R2022b.
3 Results
3.1 Molecular dynamics simulations clearly indicate reduced protein–drug binding in trans but not in cis
The extensive sampling afforded by MD simulations enabled us to follow on of the number of hydrogen bonds and contacts between the drug and the protein (Table 2). These values revealed a very clear effect of the mutation in trans, which led to a loss of two hydrogen bonds and ∼30 protein–drug contacts. As an additive force-field was used in the MD simulations, it could be expected that the loss of hydrogen bonds would contribute to the electrostatic contribution to the binding energy becoming less favourable, whereas the loss of contacts can be expected to make the Lennard-Jones (LJ) contribution less favourable. Interestingly, analysis of the interaction energies shows only a change in electrostatics (Table 3). Furthermore, despite an overall lower number of hydrogen bonds when Gln316 was mutated in cis, the interaction energies of the cis mutant were very similar to those observed in the wild type. This suggests that the cis mutation does not lead to resistance.
Table 2 Average number of hydrogen bonds between enasidenib and IDH2 for the different mutations from MD simulations
| Measurement |
R140Q |
R140Q/Q316Ecis |
R140Q/Q316Etrans |
|
The deviations from average are smaller than 0.1 for the hydrogen bonds and 1.0 for the contacts and are hence not shown.
|
| IDH2-drug H-bondsa |
3.9 |
3.0 |
1.7 |
| IDH2-drug contactsa |
493 |
497 |
464 |
Table 3 Protein–drug interaction energies calculated from MD simulations of IDH2 with enasidenib (values in kcal mol−1 with standard deviations for the interaction energies and error estimations for ΔΔGb in parentheses). ΔΔGb is the Gibbs energy difference for drug binding calculated with LIE
| Measurement |
R140Q |
R140Q/Q316Ecis |
R140Q/Q316Etrans |
| Coul-SR |
−24.1 (3.8) |
−25.3 (6.9) |
−14.3 (2.9) |
| LJ-SR |
−45.7 (2.9) |
−46.6 (3.1) |
−45.3 (3.3) |
| ΔΔGb |
0 |
−0.1 (2.0) |
+2.5 (1.4) |
Examining the hydrogen bonds involving residue 316 shows some difference between the mutants in cis and trans (Table 4). In the IDH2 R140Q variant, Residue Gln316 is hydrogen bonded to residues Asp312, Gly313 and Leu320 in the first monomer (with arginine in position 140) and to Asp312 and Leu320 in the second. Notably in the cis mutant, the carboxylate of Glu316 accept a hydrogen bond from Trp306 whereas in the trans mutant, it forms a salt bridge with Arg149. This is one structural reason for the reduction in hydrogen-bonding capacity between the trans mutant and the drug.
Table 4 Average number of hydrogen bonds involving residue 316 and selected protein residues in the simulations. Only residues with average number of hydrogen bonds above 0.1 are shown
| Variant |
Residue 140 in monomer |
Residue 316 in monomer |
H-Bonding residue |
No. of H-bondsa |
|
The deviations from average are smaller than 0.1 for the hydrogen bonds and are hence not shown.
|
| R140Q |
Arg140 |
Gln316 |
Asp312 |
1.4 |
| Gly313 |
0.6 |
| Leu320 |
0.9 |
| Gln140 |
Gln316 |
Asp312 |
1.3 |
| Leu320 |
0.9 |
|
|
|
Cis
|
Arg140 |
Gln316 |
Asp312 |
0.8 |
| Leu320 |
0.9 |
| Gln140 |
Glu316 |
Asp312 |
0.9 |
| Leu320 |
0.9 |
| Trp306 |
1.0 |
|
|
|
Trans
|
Arg140 |
Glu316 |
Gly313 |
0.3 |
| Leu320 |
0.4 |
| Arg149 |
1.1 |
| Gln140 |
Gln316 |
Asp312 |
1.1 |
| Leu320 |
1.0 |
Linear interaction energy (LIE)35 was used to approximate the free energy change upon mutation based on MD simulations. In agreement with the reported difference in interaction energies, the results (Table 3) show essentially no difference between the R140Q/Q316Ecis mutant and the R140Q IDH2 variant. The R140Q/Q316Etrans mutant show decreased binding of about 2.5 kcal mol−1.
3.2 Structural changes observed in the molecular dynamics simulations explain the reduced binding upon trans mutation
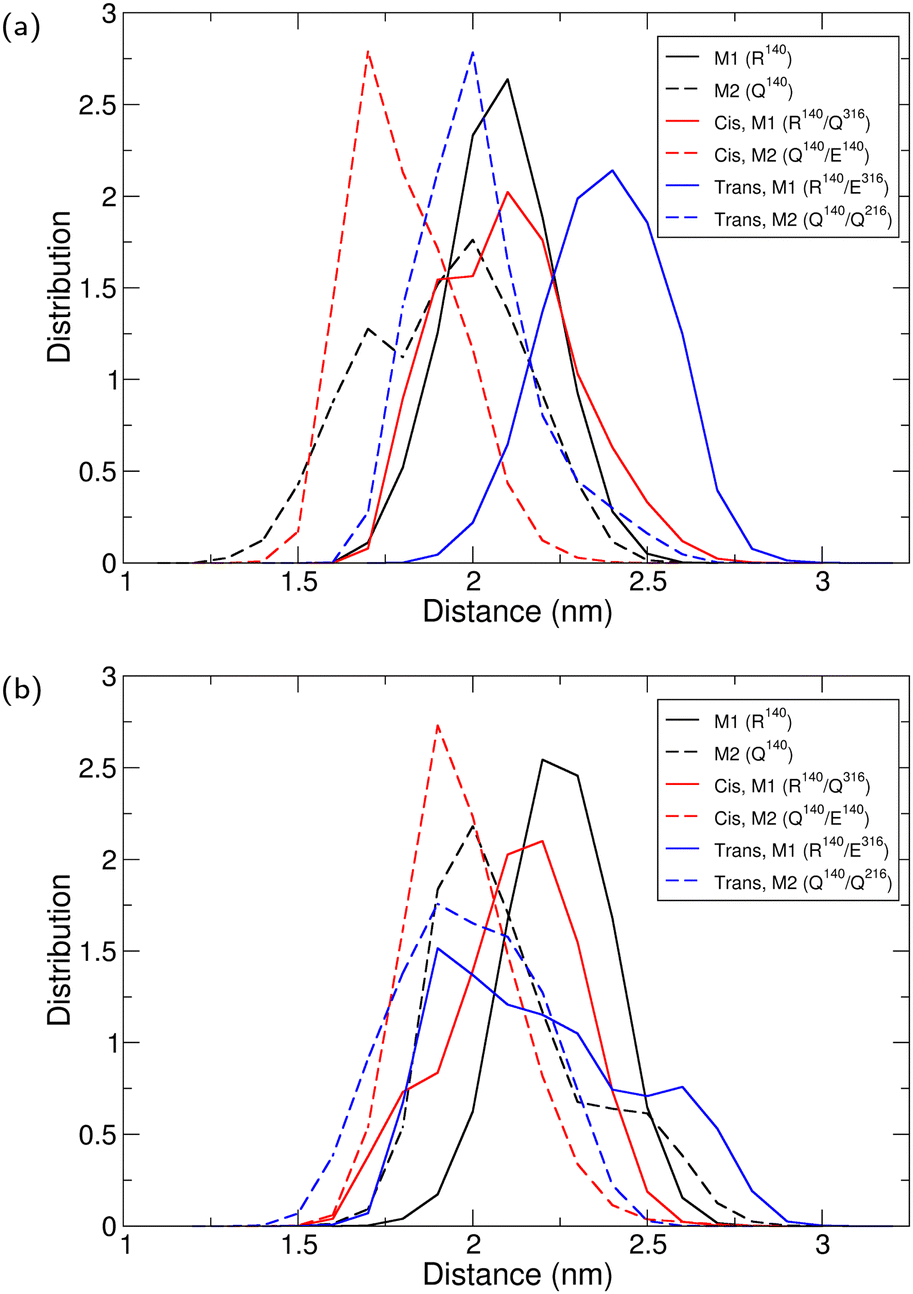
To examine the structural changes between the different IDH2 variants as observed in the simulations, we start by examination of the opening of the active site. In crystal structures of IDH, the opening of the active site is measured by the distances between the residues that correspond to Ile75 in one monomer and Leu250 in the other in human IDH1.52 In human IDH2, these residues are Ile116 and Leu298. In the crystal structure, these distances are 2.20 and 1.99 nm. The distribution of the distances in the simulations of IDH2/Arg140|IDH2/Gln140 (apo structure) show two somewhat asymetric openings with maxima at 2.10 and 1.99 nm (Fig. 2A, black lines). The active site becomes closer at the narrow side when Gln316 is mutanted in cis (Fig. 2A, red lines, maxima at 2.10 and 1.70 nm). The trans-mutant shows a wider opening at the wide side (Fig. 2A, blue, with maxima at 2.40 and 2.00 nm). Only small changes are observed when the drug is bound to the R140Q variant or cis monomer (Fig. 2B, black and red lines). When residue 316 is mutanted in trans, both sides tend to be closed (with maxima of their distance distributions at 1.90 nm), but one distribution is much wider (Fig. 2B, blue; the solid line shows a wider distribution). Overall, the trans mutant shows considerable deviations from the original variant, especially when the drug is bound to it.
 |
| | Fig. 2 Distance distribution for the opening of the active site in IDH, as calculated from the distance between the Cα atoms of residues Ile116 and Leu298. M1 is monomer 1 and M2 is monomer 2, referring to the location of Ile116. (a) Apo IDH2. (b) Drug-bound IDH2. | |
The structure of IDH protein also includes a back-cleft that can be in a closed (active) or open (inactive) state.52 The opening of the back cleft was defined by residues Met199 and His342 on each monomer in IDH1. The corresponding residues in IDH2 are Tyr238 and His381. Unlike the active site opening, the back cleft opening was rather similar in all simulations (Fig. S1, ESI†).
Given that the opening of the active site between the monomers has been modified in the drug resistant trans mutant but the opening of the back cleft within the monomers did not, we have also carried out an analysis wherein the monomers were aligned with each other. This analysis also shows that the deviation between the monomers is about 0.25 nm in the free and 0.22 nm in the drug-bound state, showing a larger similarity in the latter case except for the trans mutant where it is ∼0.25 nm regardless of the drug-binding (Table 5). The deviations between the monomers can be smaller than the overall root mean square deviation (RMSD) for the whole complex (Table S1, ESI†) because the values for the whole complex require the alignment of the two dimers together.
Table 5 Root mean square deviations between monomer 1 and monomer 2 (averages over 40 calculations). Values are averages in nm with standard deviations in parentheses corresponding to the last significant digit, i.e. 0.24(5) means that the average is 0.24 nm and the standard deviation 0.05 nm
| Protein |
RMSD |
| R140Q apo |
0.24(5) |
|
Cis apo |
0.25(5) |
|
Trans apo |
0.25(6) |
| R140Q holo |
0.22(4) |
|
Cis holo |
0.22(5) |
|
Trans holo |
0.25(6) |
3.3 NCI analysis reveals differences in the trans mutant
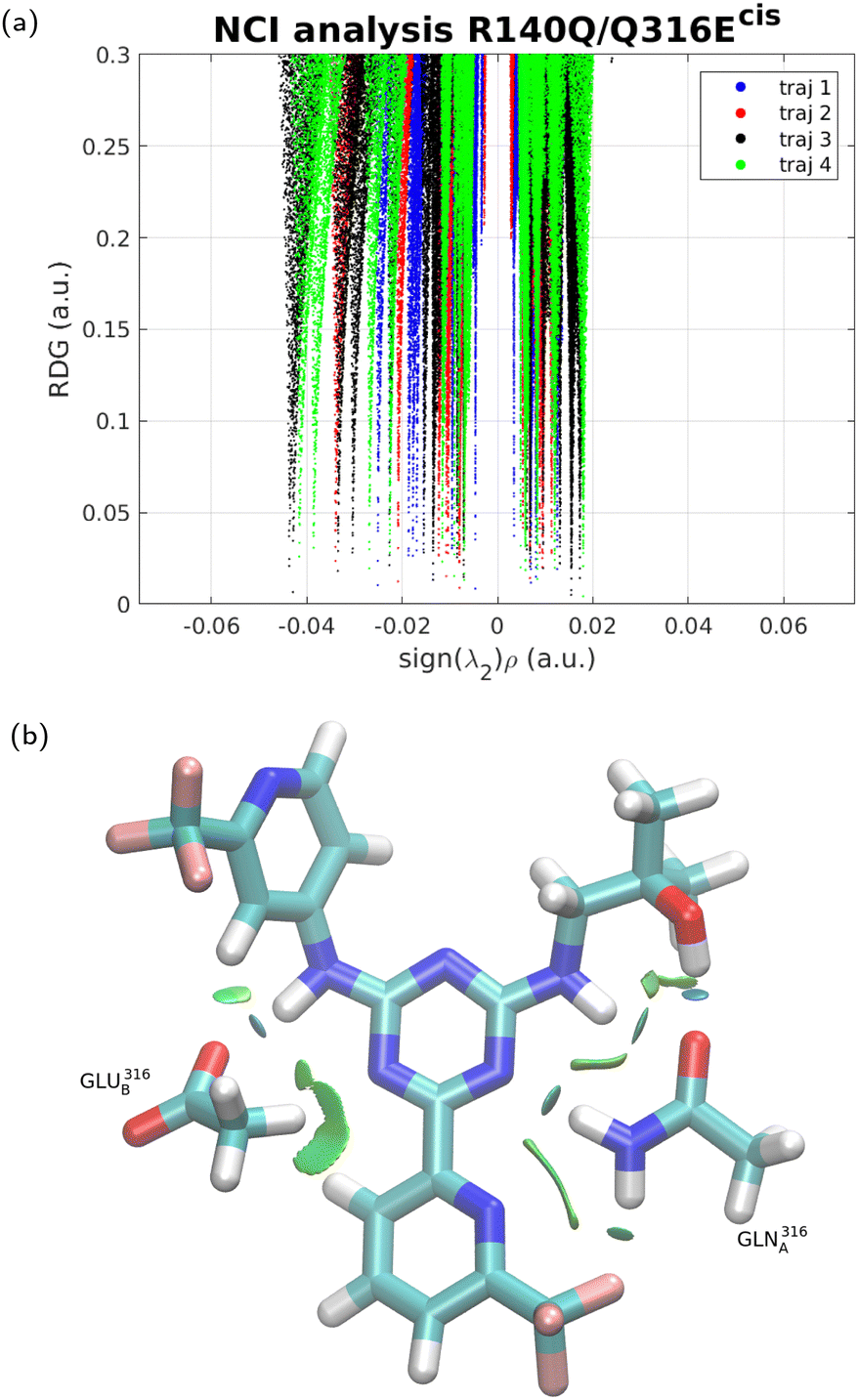
To further examine the difference between the three forms of the IDH protein in terms of binding to the inhibitor, NCI interaction analysis51 was performed for each of the four trajectories in each case, and the results are shown in Fig. 3–5. In NCI analysis, repulsive interactions are shown by positive values along the X-axis and attractive ones by negative values. The results show little if any difference between IDH2/R140Q and IDH2/R140Q/Q316Ecis. The IDH2/R140Q/Q316Etrans variant displays slightly smaller repulsion interactions with respect to the other two variants, and clearly less emphasized attraction (no interaction with sign(λ2) < −0.04), corroborating the findings from the MD simulations.
 |
| | Fig. 3 NCI analysis for the interaction between enasidenib IDH2 mutant R140Q. The analysis was performed separately on four independent MD trajectories. An example of the structure from one run is shown in frame (b), with areas of interaction in green. (a) Reduced density gradient. (b) Structural plot. | |
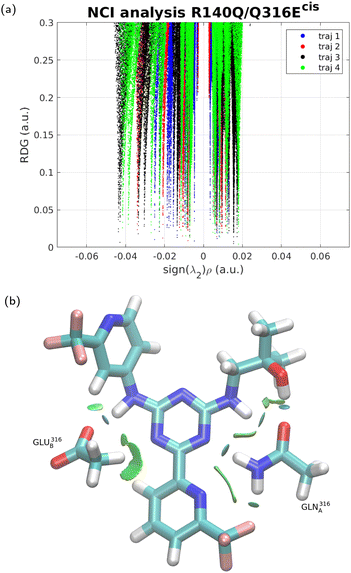 |
| | Fig. 4 NCI analysis for the interaction between enasidenib IDH2 mutant R140Q/Q316Ecis. The analysis was performed separately on four independent MD trajectories. An example of the structure from one run is shown in frame (b), with areas of interaction in green. (a) Reduced density gradient. (b) Structural plot. | |
 |
| | Fig. 5 NCI analysis for the interaction between enasidenib IDH2 mutant R140Q/Q316Etrans. The analysis was performed separately on four independent MD trajectories. An example of the structure from one run is shown in frame (b), with areas of interaction in green. (a) Reduced density gradient. (b) Structural plot. | |
3.4 DFT calculations of interaction energies and energy decomposition analysis of the drug binding
The interaction energies calculated from the MD simulations do not include quantum mechanical (QM) effects. To this end, we approximated the binding free energies using small model of the binding site with full DFT, following our earlier studies that showed the reliability of such approach.3,53 The results (Table 3) were in agreement with the interaction energies calculated in the MD simulations and clearly show that the binding free energy becomes much less favourable upon Q316E mutation in trans but not in cis (Table 6).
Table 6 Binding free energies (kcal mol−1) estimated for a binding site model of IDH2 with enasidenib. The values were calculated with DFT for the R140Q, R140Q/Q316Ecis (cis) and R140Q/Q316Etrans (trans) mutants. CI = confidence interval. The QM calculations were performed in ORCA39,40 using the ωB97X-D functional,45 the def2-TZVP basis set42 and the solvent model SMD.44 The values were calculated from 20 snapshot structures taken from the MD simulations
|
|
ΔGR140Q (n = 20) |
ΔGcis (n = 20) |
ΔGtrans (n = 20) |
| Mean |
−17.6 (3.6) |
−16.1 (3.6) |
−9.7 (2.0) |
| CI (95%) |
[−19.3, −15.9] |
[−17.8, −14.4] |
[−10.6, −8.8] |
|
|
ΔΔGtrans–cis |
ΔΔGcis–R140Q |
ΔΔGtrans–R140Q |
|
|
+6.3 |
+1.6 |
+7.9 |
| CI (95%) |
[4.5, 8.3] |
[−0.8, 3.8] |
[6.0, 9.8] |
Analysis of the MD simulations suggests that the reduced affinity of the drug to the Q316Etrans mutant is due to loss of electrostatic interactions (Table 3). To get further insights into the different contributions and consider interactions that are not represented in the forcefield, we decomposed the energies calculated by DFT using the PCMEDA approach47–49 (Fig. 6). The contribution of favourable electrostatics is indeed much reduced for the complex with the Q316Etrans mutant even in EDA. However, the EDA calculation reveal a more complex picture, where in effect all interactions are reduced, negative and positive, which is in agreement with the NCI calculations. The binding of the drug is dominated by electrostatics and polarization regardless of the mutation.
 |
| | Fig. 6 Energy decomposition analysis for the interaction between enasidenib and the IDH2 mutants R140Q, R140Q/Q316Ecis and R140Q/Q316Etrans. PCMEDA was performed in each case for a structure where the interactions energies were the largest in absolute value. EL – electrostatics, EX + REP − exchange + repulsion, POL – polarization, DISP – dispersion, CORR – DFT correlation, DES – desolvation, TOT – total. EDA calculations in gas phase were performed in XEDA46 and desolvation was calculated using ORCA.39,40 | |
4 Discussion
In this study we show, using MD simulations, that the previous finding of a resistance mutation Q316E in trans in two patients upon treatment with enasidenib is likely due to the protein–drug interaction rather than the small number of patients. MD simulations, NCI analysis and DFT calculations all show that the mutation in trans, i.e. in a different copy of the dimer than the activating mutation R140Q, leads to less favourable protein–drug interactions. The MD simulations show clearly that a hydrogen bond is lost between the drug and the protein, and that the electrostatic interaction is weakened. EDA analysis shows that this is the results of a weakening of multiple interactions (electrostatics, polarization, correlation and dispersion).
Analysis of the MD simulations showed modifications to the opening of the active site in the mutated IDH upon the Q316E mutation in trans. It remains to be seen if these changes might also affect the catalytic efficiency of the enzyme. In the apo structure, the two monomers are somewhat asymetric (RMSD of ∼0.25 nm between them) without clear changes when IDH2 was mutated. This indicates that modifications are mainly in the bound structure, which might make it more difficult to design drugs that would overcome resistance due to the Q316E mutation. On the other end, a comparison of the mutants reveal that maintaining the hydrogen bonds between the protein and the drug is a crucial aspect. Finally, this study suggests that any efforts to curb drug resistance in a dimeric structure should consider on which monomer the mutations take place.
Author contributions
Erik Arvidsson: investigation. Erik Lindahl: investigation, methodology, visualization, writing – original draft, writing – review & editing. Ran Friedman: conceptualization, data curation, funding acquisition, investigation, methodology, project administration, resources, supervision, writing – review & editing.
Data availability
All procedures are detailed in the article. Input files for energy calculations, EDA and NCI are freely available at: https://dx.doi.org/10.6084/m9.figshare.25205792.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Most calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at PDC, partially funded by the Swedish Research Council through grant agreement no. 2018-05973. EDA calculations were performed on the Xiamen Atomistic Computing Suite (XACS).
Notes and references
- R. Friedman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 12, e1563 Search PubMed
 .
.
- N. Kurt Yilmaz and C. A. Schiffer, Chem. Rev., 2021, 121, 3235–3237 CrossRef CAS PubMed .
- M. J. Dávila-Rodrguez, T. S. Freire, E. Lindahl, I. Caracelli, J. Zukerman-Schpector and R. Friedman, Chem. Commun., 2020, 56, 6727–6730 RSC .
- G. Jindal, D. Mondal and A. Warshel, J. Phys. Chem. B, 2017, 121, 6831–6840 CrossRef CAS PubMed .
- P. S. Georgoulia, G. Todde, S. Bjelic and R. Friedman, Biochim. Biophys. Acta, Gen. Sub., 2019, 1863, 732–741 CrossRef CAS PubMed .
- P. S. Georgoulia, S. Bjelic and R. Friedman, FEBS J., 2020, 287, 3200–3220 CrossRef CAS PubMed .
- I. Choudhuri, A. Biswas, A. Haldane and R. M. Levy, J. Phys. Chem. B, 2022, 126, 10622–10636 CrossRef CAS PubMed .
- R. Chowdhury, K. K. Yeoh, Y.-M. Tian, L. Hillringhaus, E. A. Bagg, N. R. Rose, I. K. H. Leung, X. S. Li, E. C. Y. Woon, M. Yang, M. A. McDonough, O. N. King, I. J. Clifton, R. J. Klose, T. D. W. Claridge, P. J. Ratcliffe, C. J. Schofield and A. Kawamura, EMBO Rep., 2011, 12, 463–469 CrossRef CAS PubMed .
- E. S. Kim, Drugs, 2017, 77, 1705–1711 CrossRef CAS PubMed .
- E. M. Stein, C. D. DiNardo, A. T. Fathi, D. A. Pollyea, R. M. Stone, J. K. Altman, G. J. Roboz, M. R. Patel, R. Collins, I. W. Flinn, M. A. Sekeres, A. S. Stein, H. M. Kantarjian, R. L. Levine, P. Vyas, K. J. MacBeth, A. Tosolini, J. VanOostendorp, Q. Xu, I. Gupta, T. Lila, A. Risueno, K. E. Yen, B. Wu, E. C. Attar, M. S. Tallman and S. de Botton, Blood, 2019, 133, 676–687 CrossRef CAS PubMed .
- E. M. Stein, C. D. DiNardo, D. A. Pollyea, A. T. Fathi, G. J. Roboz, J. K. Altman, R. M. Stone, D. J. DeAngelo, R. L. Levine, I. W. Flinn, H. M. Kantarjian, R. Collins, M. R. Patel, A. E. Frankel, A. Stein, M. A. Sekeres, R. T. Swords, B. C. Medeiros, C. Willekens, P. Vyas, A. Tosolini, Q. Xu, R. D. Knight, K. E. Yen, S. Agresta, S. de Botton and M. S. Tallman, Blood, 2017, 130, 722–731 CrossRef CAS PubMed .
- A. M. Intlekofer, A. H. Shih, B. Wang, A. Nazir, A. S. Rustenburg, S. K. Albanese, M. Patel, C. Famulare, F. M. Correa, N. Takemoto, V. Durani, H. Liu, J. Taylor, N. Farnoud, E. Papaemmanuil, J. R. Cross, M. S. Tallman, M. E. Arcila, M. Roshal, G. A. Petsko, B. Wu, S. Choe, Z. D. Konteatis, S. A. Biller, J. D. Chodera, C. B. Thompson, R. L. Levine and E. M. Stein, Nature, 2018, 559, 125–129 CrossRef CAS PubMed .
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed .
- B. R. Brooks, C. L. Brooks, 3rd, A. D. Mackerell, Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed .
- J. Lee, X. Cheng, J. M. Swails, M. S. Yeom, P. K. Eastman, J. A. Lemkul, S. Wei, J. Buckner, J. C. Jeong, Y. Qi, S. Jo, V. S. Pande, D. A. Case, C. L. I. Brooks, A. D. J. MacKerell, J. B. Klauda and W. Im, J. Chem. Theory Comput., 2016, 12, 405–413 CrossRef CAS PubMed .
- K. Yen, J. Travins, F. Wang, M. D. David, E. Artin, K. Straley, A. Padyana, S. Gross, B. DeLaBarre, E. Tobin, Y. Chen, R. Nagaraja, S. Choe, L. Jin, Z. Konteatis, G. Cianchetta, J. O. Saunders, F. G. Salituro, C. Quivoron, P. Opolon, O. Bawa, V. Saada, A. Paci, S. Broutin, O. A. Bernard, S. de Botton, B. S. Marteyn, M. Pilichowska, Y. Xu, C. Fang, F. Jiang, W. Wei, S. Jin, L. Silverman, W. Liu, H. Yang, L. Dang, M. Dorsch, V. Penard-Lacronique, S. A. Biller and S.-S. M. Su, Cancer Discovery, 2017, 7, 478–493 CrossRef CAS PubMed .
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed .
- R. B. Best, X. Zhu, J. Shim, P. E. Lopes, J. Mittal, M. Feig and A. D. Mackerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed .
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed .
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS .
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed .
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef .
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS .
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed .
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS .
- S. Miyamoto and P. A. Kollman, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS .
- M. Bernetti and G. Bussi, J. Chem. Phys., 2020, 153, 114107 CrossRef CAS PubMed .
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS .
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–28 Search PubMed .
- J. Åqvist, C. Medina and J.-E. Samuelsson, Protein Eng., Des. Sel., 1994, 7, 385–391 CrossRef PubMed .
- J. A. Lemkul and D. R. Bevan, J. Phys. Chem. B, 2010, 114, 1652–1660 CrossRef CAS PubMed .
- S. Kim, H. Oshima, H. Zhang, N. R. Kern, S. Re, J. Lee, B. Roux, Y. Sugita, W. Jiang and W. Im, J. Chem. Theory Comput., 2020, 16, 7207–7218 CrossRef CAS PubMed .
- V. Gapsys, A. Yildirim, M. Aldeghi, Y. Khalak, D. van der Spoel and B. L. de Groot, Commun. Chem., 2021, 4, 61 CrossRef CAS PubMed .
- T. S. Freire, I. Caracelli, J. Zukerman-Schpector and R. Friedman, Phys. Chem. Chem. Phys., 2023, 25, 6175–6183 RSC .
-
H. Gutiérrez-de Terán and J. Åqvist, Linear Interaction Energy: Method and Applications in Drug Design, Springer, New York, 2011, vol. 819, pp. 305–323 Search PubMed .
- T. Liu, Y. Lin, X. Wen, R. N. Jorissen and M. K. Gilson, Nucleic Acids Res., 2007, 35, D198–D201 CrossRef CAS PubMed .
- B. Oruganti, E. Lindahl, J. Yang, W. Amiri, R. Rahimullah and R. Friedman, J. Biol. Chem., 2022, 298, 102238 CrossRef CAS PubMed .
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS .
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed .
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed .
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed .
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC .
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef .
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed .
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC .
- Z. Tang, Y. Song, S. Zhang, W. Wang, Y. Xu, D. Wu, W. Wu and P. Su, J. Comput. Chem., 2021, 42, 2341–2351 CrossRef CAS PubMed .
- P. Su, H. Liu and W. Wu, J. Chem. Phys., 2012, 137, 034111 CrossRef PubMed .
- P. Su, Z. Jiang, Z. Chen and W. Wu, J. Phys. Chem. A, 2014, 118, 2531–2542 CrossRef CAS PubMed .
- P. Su, Z. Tang and W. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1460 CAS .
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS .
- R. Boto, F. Peccati, R. Laplaza, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, J. Chem. Theory Comput., 2019, 16, 4150–4158 CrossRef PubMed .
- X. Xu, J. Zhao, Z. Xu, B. Peng, Q. Huang, E. Arnold and J. Ding, J. Biol. Chem., 2004, 279, 33946–33957 CrossRef CAS PubMed .
- R. Friedman, J. Phys. Chem. B, 2021, 125, 2251–2257 CrossRef CAS PubMed .
|
| This journal is © the Owner Societies 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a