
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible C–H bond silylation with a neutral silicon Lewis acid†
Thaddäus
Thorwart
and
Lutz
Greb
 *
*
Ruprecht-Karls-Universität Heidelberg, Anorganisch-Chemisches Institut, Im Neuenheimer Feld 270, Heidelberg 69120, Germany. E-mail: greb@uni-heidelberg.de
First published on 14th September 2023
Abstract
The silicon–carbon bond is a valuable linchpin for synthetic transformations. However, installing Si–C functionalities requires metalated C-nucleophiles, activated silicon reagents (silylium ions, silyl radicals, and silyl anions), or transition metal catalysis, and it occurs irreversibly. In contrast, spontaneous C–H silylations with neutral silanes leading to anionic silicates, and their reversible deconstruction, are elusive. Herein, the CH-bond silylation of heterocycles or a terminal alkyne is achieved by reaction with bis(perfluoro(N-phenyl-ortho-amidophenolato))silane and 1,2,2,6,6-pentamethylpiperidine. Computational and experimental insights reveal a frustrated Lewis pair (FLP) mechanism. Adding a silaphilic donor to the ammonium silicate products induces the reformation of the C–H bond, thus complementing previously known irreversible C–H bond silylation protocols. Interestingly, the FLP “activated” N-methylpyrrole exhibits “deactivated” features against electrophiles, while a catalytic functionalization is found to be effective only in the absence of a base.
Introduction
Organo-silicon species are of high interest in materials science1–5 and biological research6–10 and represent strategic connections for carbon–carbon or carbon–heteroatom bond formations.11–13 The introduction of silyl groups in organic substrates is traditionally performed via stoichiometric metalation followed by the sequestering of the C-nucleophiles with R3SiX (Fig. 1a). During the past decades, significant progress has been made in catalytic C–H bond silylations.14–16 The available approaches can be classified into transition metal-catalyzed silylation (Fig. 1c), electrophilic aromatic substitution via silylium ions (Fig. 1b), nucleophilic substitution via silyl anions (Fig. 1d), silyl radical reactions (Fig. 1e), or approaches based on designed reagents as alternative silicon sources.15–17 Along every individual path, the corresponding silicon reagent X–SiR3 needs to be activated by either Si–X bond heterolysis, homolysis, or hypercoordination before reacting with the C–H bond. Examples of C–H silylation leading to anionic silicates without the need for prior activation of the silicon reagent are unknown. In contrast, such chemistry is well established for Lewis acidic boranes,18–25 and was recently extended to phosphonium ions.26 The primary cause for the lack of reactivity of neutral silanes is their poor electrophilicity, which turns the attack of prospective nucleophilic substrates kinetically unfeasible and the anionic silicate products thermodynamically unfavorable.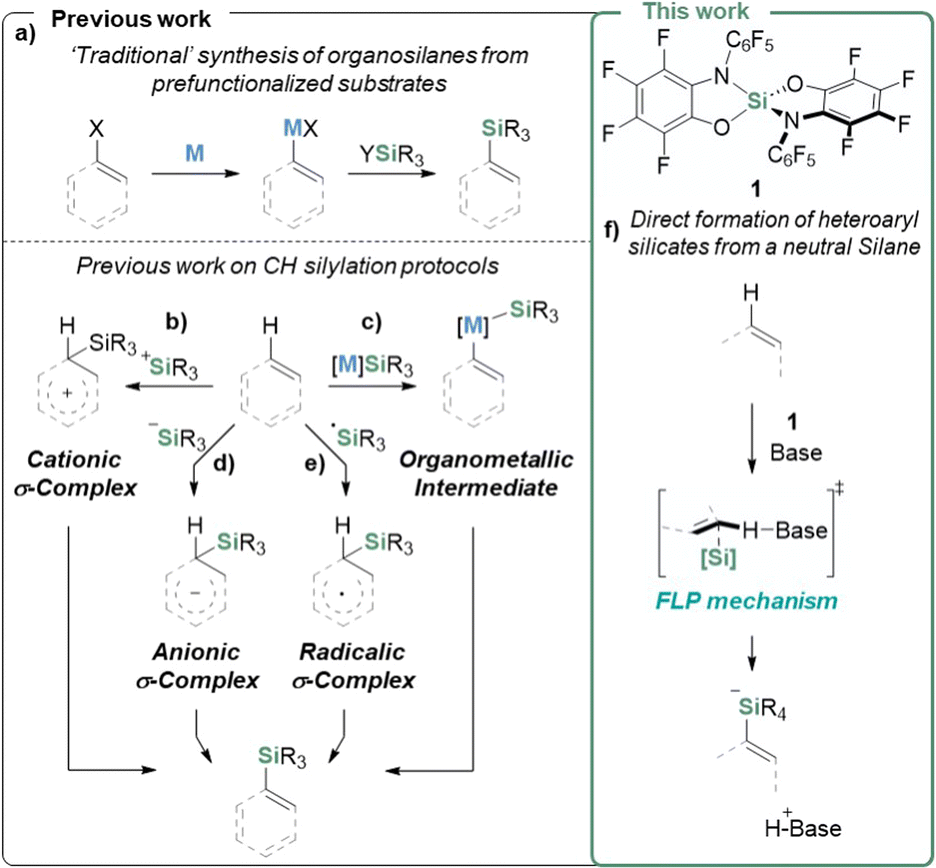 | ||
| Fig. 1 (a) Previously reported conventional silane syntheses, (b)–(e) direct C–H silylation strategies,15 and (f) FLP type silylation reported in this work. | ||
We recently launched a program on the chemistry of highly Lewis acidic silanes based on the catecholate ligand and advanced its use as a catalyst, e.g., in C–O bond metathesis.27
A dominant limitation of this substance class was its poor solubility stemming from the polymeric structure formed via reversible Si–O dynamic covalent chemistry.28 Thus, a second generation of highly Lewis acidic, non-oligomerizing bis(perfluoro(N-phenyl-ortho-amidophenolato))silane 1 was developed (Fig. 1f), allowing unprecedented H2 cleavage with a neutral silane.29 In the present study, we employ 1 for the first C–H bond silylation reaction with a neutral silane, delivering organosilicates by the co-action of a Brønsted base. Remarkably, this process is reversible and can redeliver the C–H bond upon the addition of silaphilic agents.
Results and discussion
Reacting N-methylindole with one equivalent of 1,2,2,6,6-pentamethylpiperidine (pmp) and 1 in CD2Cl2, resulted in a fast conversion at rt. NMR spectroscopy suggested clean transformation toward the N-methylindol-3-yl silicate [2a]− and the [pmpH]+ cation (Fig. 2a). The molecular structure of the proposed ion pair was confirmed from the SCXRD analysis of the crystals grown by gas diffusion of n-pentane into the reaction mixture (Fig. 2b).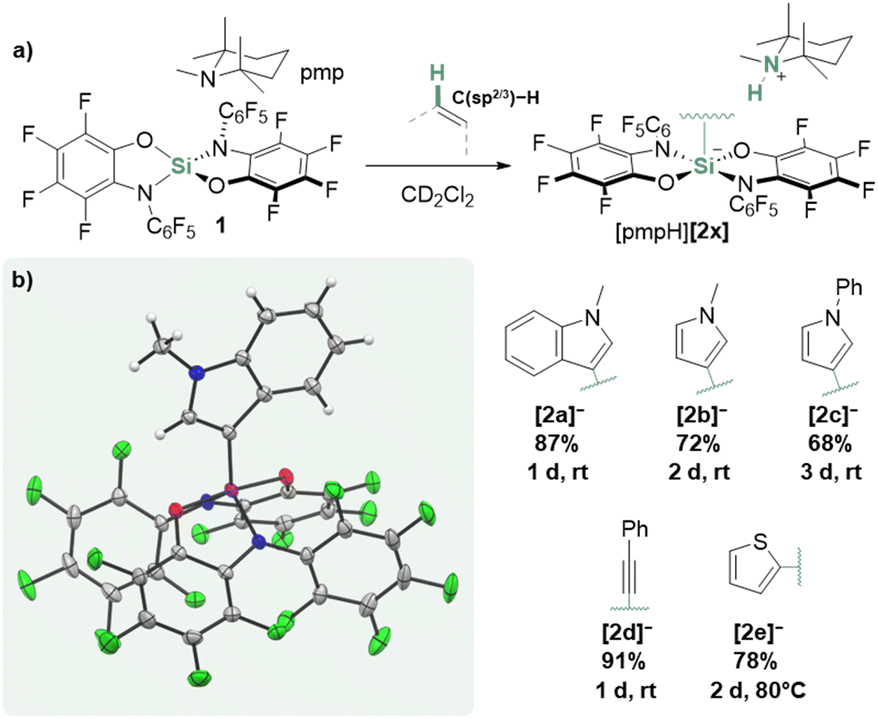 | ||
| Fig. 2 (a) General synthesis of silicates [pmpH][2x] from 1 and (b) SCXRD-derived structure of [2a]− ([pmpH]+ cation omitted for clarity, ellipsoids shown at 50% probability). | ||
Encouraged by this reactivity, related N-methyl- and N-phenylpyrrole were tested and found to analogously react to the corresponding silicates under mild conditions. A similar reaction was observed for phenylacetylene, leading to silicate [2d]−. For thiophene, an activation product could not be detected at rt; heating 1/pmp in neat thiophene to 80 °C resulted in the formation of the C–H silylated thiophene [2e]−. All products could be isolated up to 91% yields. While the C–H silylation of N-heterocycles30–33 and acetylenes34–43 that rely on the prior activation of the silicon reagents are established, we assumed a different mechanism in the present case.
Thus, the reaction was examined computationally (DLPNO-CCSD(T)/def2-TZVPP + SMD(CH2Cl2)//PBE0-D3(BJ)/def2-TZVPP, for details, see ESI†). The thermodynamic data demonstrated the stability of all products [pmpH][2x] (ΔG = −52.8 to −80.1 kJ mol−1). N-Methylpyrrole was investigated in detail concerning possible mechanisms. The initial deprotonation of the C–H bond by pmp was assumed implausible from the consideration of the respective pKa values (see Table S1, ESI†). Thus, a Friedel–Crafts-type process via the Wheland intermediate, as found with silylium ions,44 element-ligand-cooperation (ELC), as per the results of bis(catecholato) phosphonium ions,26 or an FLP process, in accordance with an intramolecular aminoborane,25 were considered viable. First, the interaction between the substrate and Lewis acid was investigated. The distance between the silicon center and N-methylpyrrole in the endergonic precomplex INT-2b (ΔG = 36 kJ mol−1) is relatively large, and the pyrrole carbon do not pyramidalize – overall not indicating the formation of a Wheland intermediate, but rather a van-der-Waals complex (Fig. 3, INT-2b). Four different ELC-type transition states are possible by combining the two basic sites in the ligand of 1 (O and N) with the 2- or 3-position at the pyrrole. Barriers of >150 kJ mol−1 disagree with the mild reaction conditions (Fig. 3, orange path). Instead, the FLP-type cleavage in the 3-position revealed a low barrier of ΔG‡ = 72 kJ mol−1, well in line with the observed reactivity at rt (Fig. 3, green path). The high selectivity is satisfactorily explained by a significantly higher barrier for the 2-position (ΔG‡ = 94 kJ mol−1, Fig. 3, blue path).
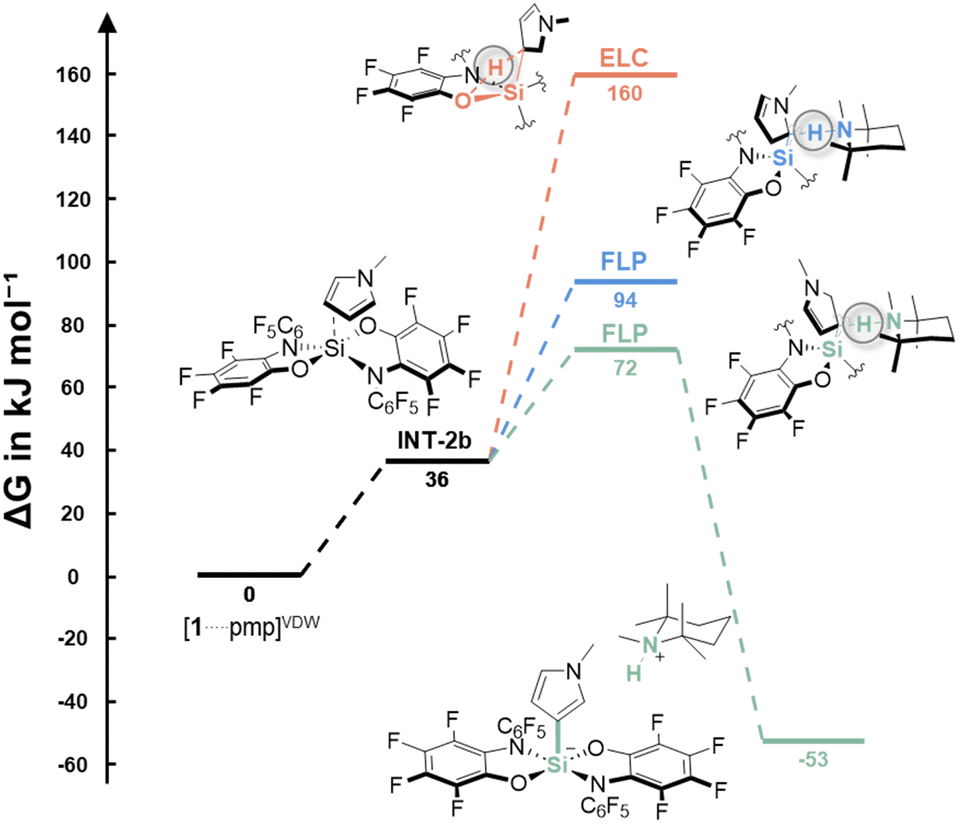 | ||
| Fig. 3 Free-energy profile for the C–H silylation of N-methylpyrrole with 1 and pmp (bottom, DLPNO-CCSD(T)/def2-TZVPP + SMD(CH2Cl2)//PBE0-D3(BJ)/def2-TZVPP). For details and the energies of other ELC transition states, see ESI.† | ||
Based on these insights, we concluded that the FLP mechanism is operative. For N-methylindole, an even lower barrier for bond cleavage in 3-position was revealed (ΔG‡ = 60 kJ mol−1), whereas C–H silylation at C2 was found to be less favored (ΔG‡ = 120 kJ mol−1). The C2 selectivity in thiophene is in line with a barrier of ΔG‡ = 98 kJ mol−1, and a significant barrier of ΔG‡ = 122 kJ mol−1 at C3. Given the simultaneous C–H deprotonation in the transition state, it is also interesting to compare the pKa values of the C2–H bonds in this context: thiophene = 33.0, N-methylindole = 38.1, and N-methylpyrrole = 39.5.45 Since the pKa values do not correlate with the computed barriers, it can be concluded that it is not the C–H bond acidity, but the donor-interaction with the silane that determines the barrier. Accordingly, for the even weaker donor benzene, a calculated barrier of ΔG‡ = 154 kJ mol−1 explains the absence of an activation product even at elevated temperatures.
To corroborate the C–H bond cleavage as the rate-determining step, we measured the kinetic isotope effect (KIE). Equimolar amounts of the respective 3-proteo- and 3-deutero- N-methylindole were dissolved in CD2Cl2 and reacted with 0.5 eq. of pmp and 1. The quantification of the H/D ratios before and after the reaction revealed a significantly faster reaction for the proteo derivative (KIE = 3.5, for details, see ESI†). This value is in line with an FLP-type C–H bond cleavage, as observed previously by Fontaine et al.25 Another experimental indication for the FLP-type mechanism was obtained by using 2,6-di-tert-butyl-pyridine as the base, in which case, no reaction occurred. The reactivity of FLPs is decisively influenced by weak association through secondary interactions,46,47 for which the choice of the base can be crucial.18 In a Friedel–Crafts or ELC mechanism, the base would not be involved in the rate-determining step and less sensitivity on their nature would be expected.
Despite considerable recent progress with group 14 FLPs,48–58 the cleavage of C–H bonds states a novum. Thus, we were interested in how other silanes would perform in such reactivity. We computed the FLP mechanism for SiCl4 as ‘archetypal’ silicon Lewis acid with N-methylpyrrole as a substrate. A barrier of ΔG‡ = 181 kJ mol−1 renders the reaction very unlikely. Accordingly, a control experiment with SiCl4/pmp did not result in any observable reaction after 7 d at rt or 2 d at 60 °C. To account for a stronger silicon Lewis acid, we computed the process with Si(catCl)2,72 which revealed a sufficiently low calculated barrier (ΔG‡ = 63 kJ mol−1). Indeed, similar reactivity could be observed for the FLP [Si(catCl)2]n/pmp, but with substantially prolonged reaction times (56% conversion after 5 d), compared to 1/pmp (78% after 16 h). This discrepancy arises from the polymeric nature of [Si(catCl)2]n, which causes extremely poor solubility in non-donor solvents.59 While the monomeric, Lewis superacidic Si(catCl)2 is easily formed with donor-substrates (e.g., ethers, amines), the cleavage of C–H bonds is favorable only with 1, which is monomeric and soluble right away.
Next, we probed the reactivity of the ammonium silicate products. Interestingly, when [pmpH][2a] was handled in strong donor solvents such as DMSO, we observed partial reformation of N-methylindole. Aiming for a complete reconstruction of the C–H bond, [pmpH][2a] was dissolved in CD2Cl2, and one equivalent of 1,3-dimethylimidazolidin-2-one (DMI) was added. Heating the mixture to 60 °C resulted in the formation (>90%) of N-methylindole and pmp, along with the DMI adduct of 1 (Fig. 4a). Experiments to transfer a benzyl group, instead of a proton, to the silylated C3 in [2a]− using trimethylbenzyl-ammonium chloride were unsuccessful, but again led to N-methylindole.
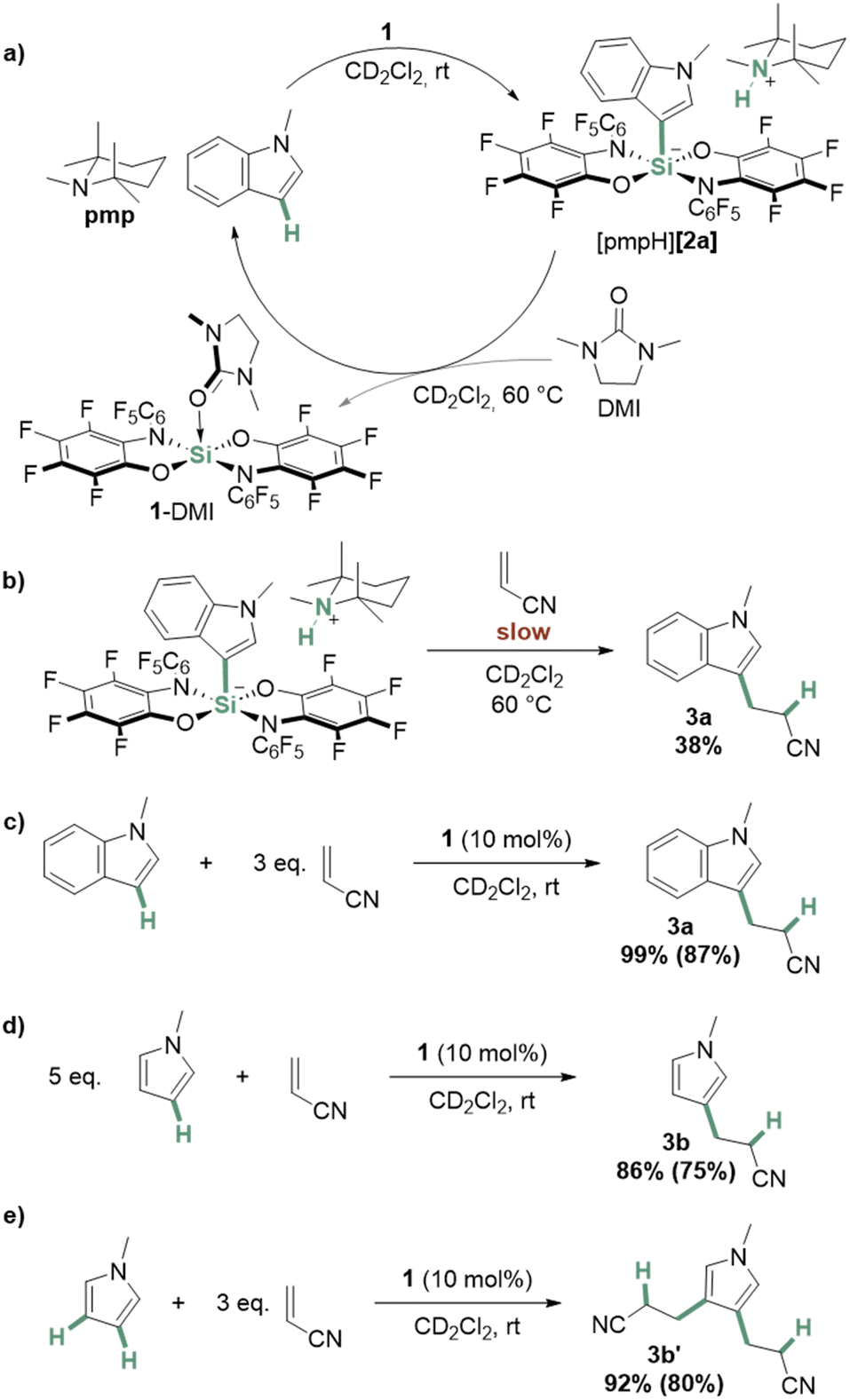 | ||
| Fig. 4 Schematic depiction of (a) the synthesis of [pmpH][2a] from N-methylindole and the reverse reaction triggered by DMI, (b) the reaction of acrylonitrile and [pmpH][2a], the addition of (c) N-methylindole and (d)/(e) N-methylpyrrole to acrylonitrile catalyzed by 1. Yields were determined against cyclooctane as an internal standard, yields in parentheses are isolated yields (details see ESI†). | ||
To probe the potential of the C–H bond silylation for subsequent electrophilic functionalization, the reaction of [pmpH][2a] with acrylonitrile was tested (Fig. 4b).60,61 The formation of 3a could be observed, but the process occurred only slowly and incompletely at 60 °C (38%, details see ESI†). Based on the computed energies (Fig. 3), we attributed the poor reactivity of [pmpH][2a] to its high thermodynamic stability. Thus, we considered the absence of a base to avoid this energetic sink. Indeed, the reaction between N-methylindole and acrylonitrile proceeded at rt to quantitative conversion using 10 mol% of 1 (Fig. 4c).
To our delight, we found 1 also to catalyze the one- and two-fold addition of N-methylpyrrole to acrylonitrile to yield 3b and 3b′ at room temperature (Fig. 4d and e, respectively). Of course, in the absence of a base, the Lewis acid activation of acrylonitrile by 1 is more likely than the involvement of a C–H silylated intermediate. Thus, the formation of [pmpH][2a] would represent a “bond activation” that corresponds to an overall substrate deactivation – a situation always to be suspected if one aims for catalysis with highly reactive reagents. Instead, the observed reactivity ultimately exploits the ability of this class of Lewis acids to bind even weakly donating nitriles62–66 – and unlocks a substrate class that is challenging to activate with less reactive catalysts.
Indeed, while the Lewis acid-mediated addition of N-heteroarenes to Michael systems has been investigated for nitroolefins or acrolein derivatives,67–70 only a few examples have been reported for catalytic additions to acrylonitrile. For the synthesis of 3a from N-methylindole, we only found a gold(III) catalyzed protocol resulting in a 52% yield.71 For less nucleophilic pyrroles, such reactivity is even more sparsely reported. Using osmium catalysis, 3b could be synthesized in 40% yield.72 Related protocols for the synthesis of 3b′ were, to the best of our knowledge, not reported.
Conclusions
A C–H bond cleavage using a neutral Lewis superacidic silane 1 and the amine pmp is introduced as a valuable extension to existing C–H silylation protocols. The process occurs via an FLP-type mechanism, which represents a novum for silicon Lewis acid–base pairs. Lewis acid 1 offers superior properties over other neutral silicon Lewis acids due to solubility-enhanced kinetic advantages, improving the young class of Lewis superacidic silanes. The bond cleavage at N-methylindole can be reversed using a donor, such as DMI. Because carbon-substituted bis(catecholato)silicates have proven to be versatile for generating C-radicals,73–78 or as transmetalation reagents in coupling reactions,79–81 strategic combinations with the herein-discovered C–H silylation can be considered. However, this study also emphasizes that an apparent “bond activation” does not necessarily correspond to “substrate activation”, as exemplified in the electrophilic alkylation of N-methylindole.Data availability
Cartesian coordinates of the computed structures and characterization data of the described compounds are available in the ESI.† Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (CCDC: 2279861).Author contributions
L. G. and T. T. devised the project and designed the experiments. T. T. carried out the calculations, performed the experimental work, and analyzed the data. L. G. and T. T. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster). Dr D. Hartmann is thanked for her assistance with structure refinement and finalization. Open Access funding enabled and organized by Projekt DEAL.References
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D. H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718–723 CrossRef CAS PubMed.
- C. Velasco-Santos, A. L. Martinez-Hernandez, M. Lozada-Cassou, A. Alvarez-Castillo and V. M. Castano, Nanotechnology, 2002, 13, 495–498 CrossRef CAS.
- F. Wang, Z. Xie, H. Zhang, C. Y. Liu and Y. G. Zhang, Adv. Funct. Mater., 2011, 21, 1027–1031 CrossRef CAS.
- Z. L. Zhang, Y. H. Wang, W. F. Ren, Q. Q. Tan, Y. F. Chen, H. Li, Z. Y. Zhong and F. B. Su, Angew. Chem., Int. Ed., 2014, 53, 5165–5169 CrossRef CAS PubMed.
- R. J. Fessenden and J. S. Fessenden, Adv. Drug Res., 1967, 4, 95–132 CAS.
- R. J. Fessenden and J. S. Fessenden, in Adv. Organomet. Chem., Elsevier, 1980, vol. 18, pp. 275–299 Search PubMed.
- S. Gately and R. West, Drug Dev. Res., 2007, 68, 156–163 CrossRef CAS.
- M. W. Mutahi, T. Nittoli, L. X. Guo and S. M. Sieburth, J. Am. Chem. Soc., 2002, 124, 7363–7375 CrossRef CAS PubMed.
- R. Tacke and U. Wannagat, Bioactive Organo-Silicon Compounds, Springer, Berlin, Heidelberg, 1979 Search PubMed.
- S. Rendler and M. Oestreich, Synthesis, 2005, 11, 1727–1747 Search PubMed.
- E. W. Colvin, Chem. Soc. Rev., 1978, 7, 15–64 RSC.
- W. P. Weber, Silicon Reagents for Organic Synthesis, Springer, Berlin, Heidelberg, 2012 Search PubMed.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- S. C. Richter and M. Oestreich, Trends Chem., 2020, 2, 13–27 CrossRef CAS.
- H. Khatua, S. Das, S. Patra and B. Chattopadhyay, Synthesis, 2023 DOI:10.1055/a-2110-4581.
- B. Neil, F. Lucien, L. Fensterbank and C. Chauvier, ACS Catal., 2021, 11, 13085–13090 CrossRef CAS.
- V. Iashin, K. Chernichenko, I. Pápai and T. Repo, Angew. Chem., Int. Ed., 2016, 55, 14146–14150 CrossRef CAS PubMed.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396–8397 CrossRef CAS PubMed.
- C. M. Mömming, S. Frömel, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 131, 12280–12289 CrossRef PubMed.
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- C. Jiang, O. Blacque and H. Berke, Organometallics, 2010, 29, 125–133 CrossRef CAS.
- K. Chernichenko, M. Lindqvist, B. Kótai, M. Nieger, K. Sorochkina, I. Pápai and T. Repo, J. Am. Chem. Soc., 2016, 138, 4860–4868 CrossRef CAS PubMed.
- G. Ménard and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4409–4412 CrossRef PubMed.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513–516 CrossRef PubMed.
- D. Roth, J. Stirn, D. W. Stephan and L. Greb, J. Am. Chem. Soc., 2021, 143, 15845–15851 CrossRef CAS PubMed.
- N. Ansmann, T. Thorwart and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202210132 CrossRef CAS PubMed.
- D. Hartmann, T. Thorwart, R. Müller, J. Thusek, J. Schwabedissen, A. Mix, J. H. Lamm, B. Neumann, N. W. Mitzel and L. Greb, J. Am. Chem. Soc., 2021, 143, 18784–18793 CrossRef CAS PubMed.
- T. Thorwart, D. Hartmann and L. Greb, Chem. – Eur. J., 2022, 28, e202202273 CrossRef CAS PubMed.
- Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868–7871 CrossRef CAS PubMed.
- K. S. Lee, D. Katsoulis and J. Choi, ACS Catal., 2016, 6, 1493–1496 CrossRef CAS.
- B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508–7510 CrossRef CAS PubMed.
- D. Y. Wang, X. Y. Chen, J. J. Wong, L. Q. Jin, M. J. Li, Y. Zhao, K. N. Houk and Z. Z. Shi, Nat. Commun., 2021, 12, 524 CrossRef CAS PubMed.
- T. Tsuchimoto, M. Fujii, Y. Iketani and M. Sekine, Adv. Synth. Catal., 2012, 354, 2959–2964 CrossRef CAS.
- Y. H. Ma, S. J. Lou, G. Luo, Y. Luo, G. Zhan, M. Nishiura, Y. Luo and Z. M. Hou, Angew. Chem., Int. Ed., 2018, 57, 15222–15226 CrossRef CAS PubMed.
- K. Yamaguchi, Y. Wang, T. Oishi, Y. Kuroda and N. Mizuno, Angew. Chem., Int. Ed., 2013, 52, 5627–5630 CrossRef CAS PubMed.
- T. Baba, A. Kato, H. Yuasa, F. Toriyama, H. Handa and Y. Ono, Catal. Today, 1998, 44, 271–276 CrossRef CAS.
- J. Ishikawa and M. Itoh, J. Catal., 1999, 185, 454–461 CrossRef CAS.
- J. Ishikawa, K. Inoue and M. Itoh, J. Organomet. Chem., 1998, 552, 303–311 CrossRef CAS.
- M. G. Voronkov, N. I. Ushakova, I. I. Tsykhanskaya and V. B. Pukhnarevich, J. Organomet. Chem., 1984, 264, 39–48 CrossRef CAS.
- A. A. Toutov, K. N. Betz, D. P. Schuman, W. B. Liu, A. Fedorov, B. M. Stoltz and R. H. Grubbs, J. Am. Chem. Soc., 2017, 139, 1668–1674 CrossRef CAS PubMed.
- A. A. Andreev, V. V. Konshin, N. V. Komarov, M. Rubin, C. Brouwer and V. Gevorgyan, Org. Lett., 2004, 6, 421–424 CrossRef CAS PubMed.
- R. Shimizu and T. Fuchikami, Tetrahedron Lett., 2000, 41, 907–910 CrossRef CAS.
- S. Bähr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52–59 CrossRef PubMed.
- R. R. Fraser, T. S. Mansour and S. Savard, Can. J. Chem., 1985, 63, 3505–3509 CrossRef CAS.
- T. A. Rokob, A. Hamza, A. Stirling, T. Soos and I. Pápai, Angew. Chem., Int. Ed., 2008, 47, 2435–2438 CrossRef CAS PubMed.
- S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed.
- D. Hartmann, S. Braner and L. Greb, Chem. Commun., 2021, 57, 8572–8575 RSC.
- G. R. Whittell, E. I. Balmond, A. P. M. Robertson, S. K. Patra, M. F. Haddow and I. Manners, Eur. J. Inorg. Chem., 2010, 3967–3975 CrossRef CAS.
- B. Waerder, M. Pieper, L. A. Korte, T. A. Kinder, A. Mix, B. Neumann, H. G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015, 54, 13416–13419 CrossRef CAS PubMed.
- P. Holtkamp, F. Friedrich, E. Stratmann, A. Mix, B. Neumann, H. G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 5114–5118 CrossRef CAS PubMed.
- P. Sarkar, S. Das and S. K. Pati, Chem.–Asian J., 2022, 17, e202200148 CrossRef CAS PubMed.
- R. T. Cooper, J. S. Sapsford, R. C. Turnell-Ritson, D. H. Hyon, A. J. P. White and A. E. Ashley, Philos. Trans. R. Soc., A, 2017, 375, 20170008 CrossRef PubMed.
- D. J. Scott, N. A. Phillips, J. S. Sapsford, A. C. Deacy, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2016, 55, 14738–14742 CrossRef CAS PubMed.
- T. J. Herrington, B. J. Ward, L. R. Doyle, J. McDermott, A. J. P. White, P. A. Hunt and A. E. Ashley, Chem. Commun., 2014, 50, 12753–12756 RSC.
- M. Reissmann, A. Schäfer, S. Jung and T. Müller, Organometallics, 2013, 32, 6736–6744 CrossRef CAS.
- A. Schäfer, M. Reissmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636–12638 CrossRef PubMed.
- F. Medici, J. Maury, G. Lemiere and L. Fensterbank, Chem. – Eur. J., 2019, 25, 9438–9442 CrossRef CAS PubMed.
- N. Ansmann, D. Hartmann, S. Sailer, P. Erdmann, R. Maskey, M. Schorpp and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202203947 CrossRef CAS PubMed.
- U. Frick and G. Simchen, Synthesis, 1984, 11, 929–930 CrossRef.
- M. W. Majchrzak and G. Simchen, Synthesis, 1986, 1986, 956–958 CrossRef.
- A. L. Liberman-Martin, R. G. Bergman and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 5328–5331 CrossRef CAS PubMed.
- D. Hartmann, M. Schädler and L. Greb, Chem. Sci., 2019, 10, 7379–7388 RSC.
- R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef CAS PubMed.
- T. Thorwart, D. Roth and L. Greb, Chem. – Eur. J., 2021, 27, 10422–10427 CrossRef CAS PubMed.
- F. S. Tschernuth, T. Thorwart, L. Greb, F. Hanusch and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 25799–25803 CrossRef CAS PubMed.
- L. T. An, J. P. Zou, L. L. Zhang and Y. Zhang, Tetrahedron Lett., 2007, 48, 4297–4300 CrossRef CAS.
- P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 6117–6134 RSC.
- H. Firouzabadi, N. Iranpoor and F. Nowrouzi, Chem. Commun., 2005, 6, 789–791 RSC.
- C. C. Lin, J. M. Hsu, M. N. V. Sastry, H. L. Fang, Z. J. Tu, J. T. Liu and C. F. Yao, Tetrahedron, 2005, 61, 11751–11757 CrossRef CAS.
- Z. G. Li, Z. J. Shi and C. He, J. Organomet. Chem., 2005, 690, 5049–5054 CrossRef CAS.
- L. M. Hodges, J. Gonzalez, J. I. Koontz, W. H. Myers and W. D. Harman, J. Org. Chem., 1993, 58, 4788–4790 CrossRef CAS.
- V. Corce, L. M. Chamoreau, E. Derat, J. P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed., 2015, 54, 11414–11418 CrossRef CAS PubMed.
- L. Chenneberg, C. Leveque, V. Corce, A. Baralle, J. P. Goddard, C. Ollivier and L. Fensterbank, Synlett, 2016, 27, 731–735 CrossRef CAS.
- M. Jouffroy, D. N. Primer and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef CAS PubMed.
- C. Leveque, L. Chenneberg, V. Corce, J. P. Goddard, C. Ollivier and L. Fensterbank, Org. Chem. Front., 2016, 3, 462–465 RSC.
- C. Leveque, L. Chenneberg, V. Corce, C. Ollivier and L. Fensterbank, Chem. Commun., 2016, 52, 9877–9880 RSC.
- E. Levernier, K. Jaouadi, H. R. Zhang, V. Corce, A. Bernard, G. Gontard, C. Troufflard, L. Grimaud, E. Derat, C. Ollivier and L. Fensterbank, Chem. – Eur. J., 2021, 27, 8782–8790 CrossRef CAS PubMed.
- S. Witzel, K. Sekine, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2018, 54, 13802–13804 RSC.
- W. M. Seganish and P. DeShong, Org. Lett., 2004, 6, 4379–4381 CrossRef CAS PubMed.
- W. M. Seganish and P. DeShong, J. Org. Chem., 2004, 69, 1137–1143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2279861. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03488g |
| This journal is © The Royal Society of Chemistry 2023 |