
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytic amide bond formation
Max
Lubberink†
 ,
William
Finnigan
and
Sabine L.
Flitsch
*
,
William
Finnigan
and
Sabine L.
Flitsch
*
Manchester Institute of Biotechnology, Department of Chemistry, The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. E-mail: Sabine.Flitsch@manchester.ac.uk
First published on 28th March 2023
Abstract
Amide bonds occur in over half of target compounds in medicinal chemistry patents, making amide bond formation the most commonly performed reaction in the pharmaceutical industry. The growing demand for green and sustainable catalytic methods for amide bond synthesis has driven the interest in biocatalytic methods. We have compiled the literature on this topic in a publicly available database (RetroBioCat) and use this database to discuss the most commonly used strategies. These include the use of low water systems for driving equilibria and kinetics in hydrolase-catalysed reactions towards amide bond formation. Alternatively, in aqueous medium, ATP-dependent enzymes have been shown to be particularly promising and can be coupled to ATP recycling systems.
Introduction
The amide bond plays a fundamental role in Nature and its prevalence in medicinal chemistry is exemplified by the fact that it occurs in over half of all compounds listed in medicinal chemistry patents and has been the most commonly performed reaction in the pharmaceutical industry for decades.1,2Accordingly, there is a wide choice for chemical methods of amide bond formations, which will not be reviewed here.3 However, many solvents and reagents commonly used for amide synthesis lack green credentials, and the development of sustainable methodologies is still of great interest.4–7 In 2018, a roundtable of pharmaceutical industry experts (GCIPR) identified amide bond formations as a top priority for the development of alternative more sustainable methods and biocatalysis was named as one of the most promising research fields to fulfil this need.8
Biocatalysis encompasses the use of enzymes for organic synthesis and has been used as a replacement for a wide variety of chemical processes for decades. Over recent years, enzymes have been increasingly utilized for the synthesis of more complex molecules such as pharmaceutical drugs, having advantages as catalysts with high chemo- and stereoselectivity.9 Limitations in reactivity and selectivity can be overcome by using large metagenomic databases as sources for new enzymes and by protein engineering.10 In particular, directed evolution, for which the 2018 Nobel Prize was awarded to Frances Arnold has transformed the efficiency and speed of development of industrial enzymes by orders of magnitude.11 Other great benefits of using biocatalysis are its economic and environmental advantages over traditional chemical synthesis. Enzymes are biodegradable, non-toxic and produced from cheap renewable resources. Enzymatic reactions are often performed under aqueous conditions using mild reaction temperatures and frequently circumvent protection/deprotection strategies or functional group activation. Therefore, biocatalytic methods generally lead to less waste, are more cost-effective than conventional organic synthesis, and are therefore seen as potentially more sustainable.12
In Nature, the amide bond is ubiquitous and therefore, biosynthesis provides a wide variety of amide bond-forming strategies which can be harnessed for use in biocatalysis. This article focuses on cell-free applications of amide bond-forming enzymes for the synthesis of industrially relevant compounds.
To accompany this review, we have also curated data from across the literature for enzymatic amide bond formation into a database linked to the biocatalytic retrosynthesis tool RetroBioCat,13 allowing this information to be interactively explored and interrogated (available through retrobiocat.com). The database is continuously updated and is a community-based effort. As of February 2023, the database comprises 388 citations, 1831 enzymes, 4659 product molecules and 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 946 reactions, the detailed discussion of which is beyond this review. We have used the data collected in this database to analyse and discuss certain aspects of the amidation topic, such as the use of different organic solvents in lipase-catalysed amidation reactions and the range of amide bond-forming enzymes available for organic synthesis (Fig. 1).
946 reactions, the detailed discussion of which is beyond this review. We have used the data collected in this database to analyse and discuss certain aspects of the amidation topic, such as the use of different organic solvents in lipase-catalysed amidation reactions and the range of amide bond-forming enzymes available for organic synthesis (Fig. 1).
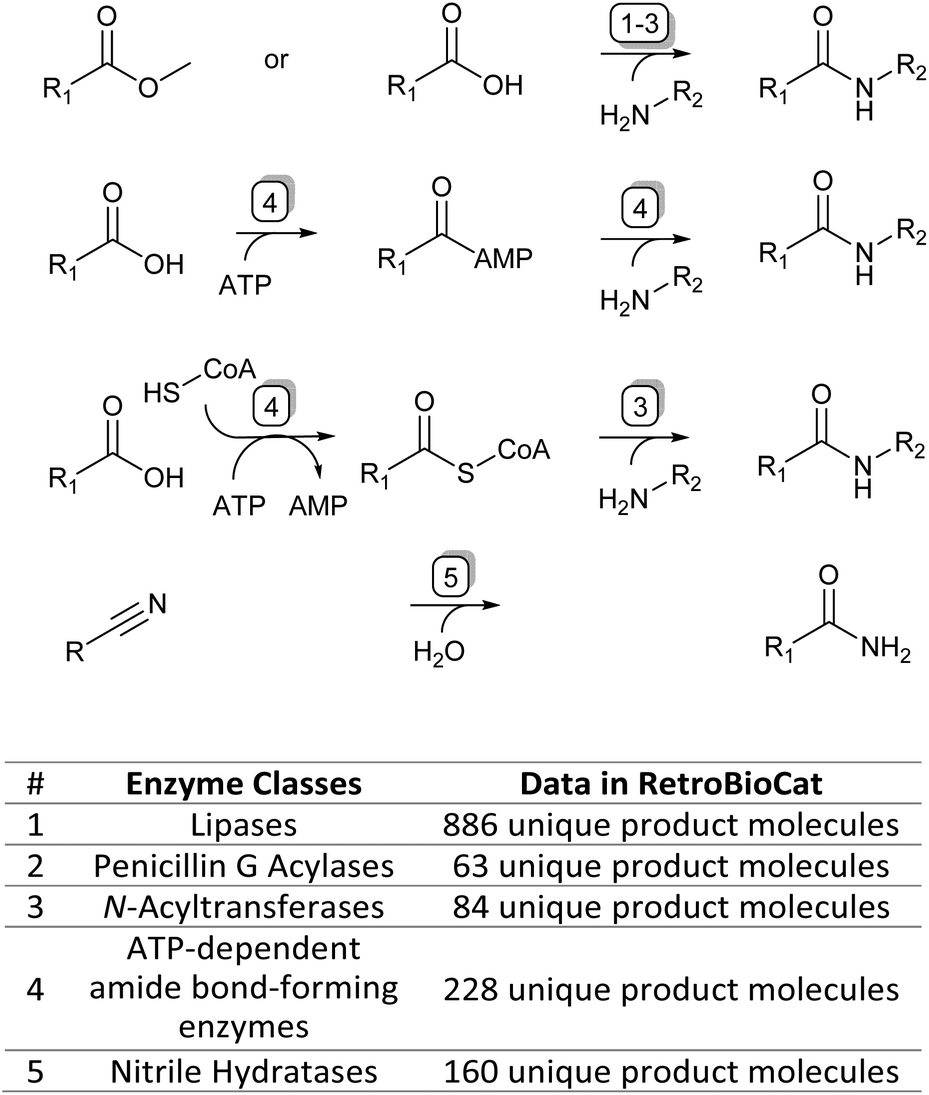 | ||
| Fig. 1 The four main enzymatic amidation strategies discussed in this review (top) and corresponding enzyme classes with their data availability in the RetroBioCat database (bottom). | ||
Aminolysis of esters
Lipases
Lipases are the most studied enzymes for amide bond formation (Fig. 1). They are members of the family of serine hydrolases that naturally catalyse the hydrolysis of triglyceride ester bonds at the oil–water interface and are therefore naturally stable in hydrophobic conditions.14 Zaks et al. reported that lipases show increased thermostability and selectivity in anhydrous conditions and that transacylation reactions are favourable over hydrolysis under these conditions, thus promoting the aminolysis of esters leading to amides.15,16 This work is often seen as a landmark discovery, showing enzymes are more widely applicable in organic solvents than previously thought possible.12 The enzymes used in these early studies were isolated from porcine pancreas (PPL)17 or Candida cylindracea (CCL),18 but over time the microbial Candida antarctica lipase B (CAL-B), in its free form or its many commercially available immobilized forms, has grown to be the most commonly used lipase for amidation reactions.19,20 Indeed, among the reactions listed in the RetroBioCat13 database of biocatalytic reactions, CAL-B is the most prevalent enzyme used for amidation reactions. CAL-B is a member of the serine hydrolase family with its three characteristic active site amino acids (aspartic acid, histidine and serine) forming the catalytic triad.19Kinetic resolutions of amines
The enantiospecific properties of lipases have been extensively exploited for the synthesis of chiral amines through kinetic resolution. Lipase-catalysed kinetic resolution processes have been used on an industrial scale, for example by BASF as shown in Scheme 1: here, a lipase is used in organic solvent to selectively acylate the R-enantiomer of racemic (R,S)-1 leaving enantiopure (S)-1 unreacted. After the separation of the reaction products, amide 3 can be hydrolysed under basic conditions yielding (R)-1 in excellent enantiopurity.21,22 As lipases are known for their exceptionally broad substrate scope, the variety of amines that can be resolved using this method is very diverse and can in some cases be done on multi-ton scales.22 A large amount of academic research has gone into the enantioselective acylation of amines following this principle, exploring the substrate scope and reaction conditions.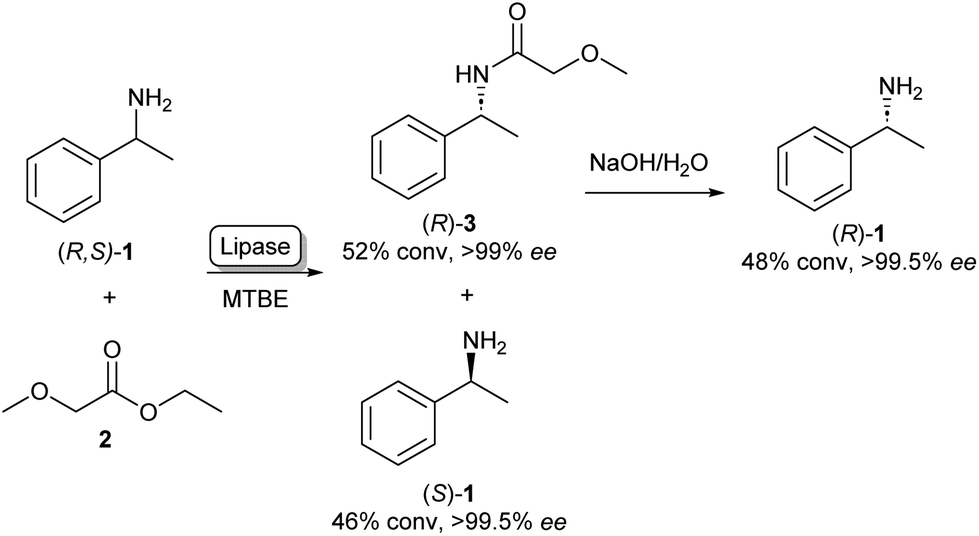 | ||
| Scheme 1 Lipase catalysed kinetic resolution of amines: selective acylation of (R,S)-1 using lipase in MTBE (methyl tert-butyl ether) yielding (S)-1. Basic hydrolysis of amide 3 yields (R)-1.21 | ||
As the kinetic resolution outlined in Scheme 1 inherently yields maximum conversion rates of 50% per enantiomer, there has been a significant interest in dynamic kinetic resolution (DKR) reactions. This method uses a racemisation agent, usually in the form of a metal or an organometallic catalyst, to convert the unreacted amine enantiomer back to the racemate, generating a cycle that allows these kinetic resolution reactions to reach full conversions to enantiopure amides (Scheme 2).23,24
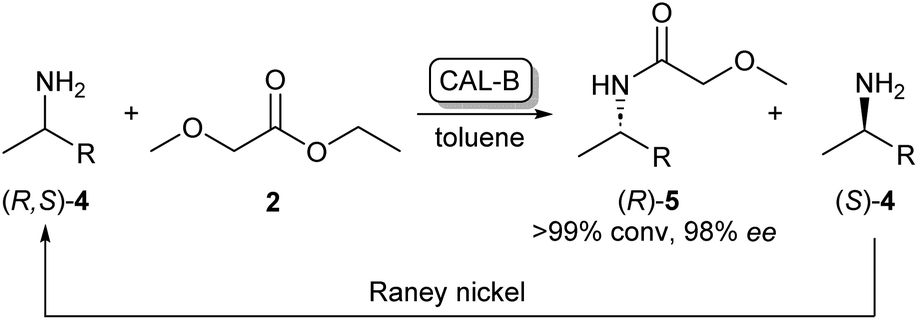 | ||
| Scheme 2 CAL-B/RANEY® nickel catalysed dynamic kinetic resolution of 4 yielding enantiopure (R)-5. CAL-B selectively acylates (R)-4 while the unreacted (S)-4 is converted back to the racemate by the RANEY® nickel catalyst.23 | ||
Use of non-aqueous solvents
The versatility of lipases for amide bond formation and other reactions has been reviewed extensively over the years.20,25 However, the need for organic solvents in lipase catalysed amidations raises the question if this is the most sustainable and safe synthesis method. Solvent selection guides such as published by CHEM21 have made efforts to rank commonly used solvents based on health, safety and environmental scores.5 The data that can be extracted from our RetroBioCat database provides a systematic overview of what organic solvents are most commonly used for lipase-catalysed amide bond formations in the scientific literature, with over 100 papers containing over 1200 entries of lipase-catalysed amide bond formations included.Fig. 2 lists the most commonly used solvents for lipase catalysed amidation reactions in the database. The colours for each solvent refer to their desirability in the CHEM21 solvent selection guide, based on criteria involving safety, health and environment.5 The four most prevalent solvents in the database are ranked “hazardous”, whereas diethyl ether and tetrachloromethane have a “highly hazardous” ranking, meaning the use of these solvents should be avoided at all costs.
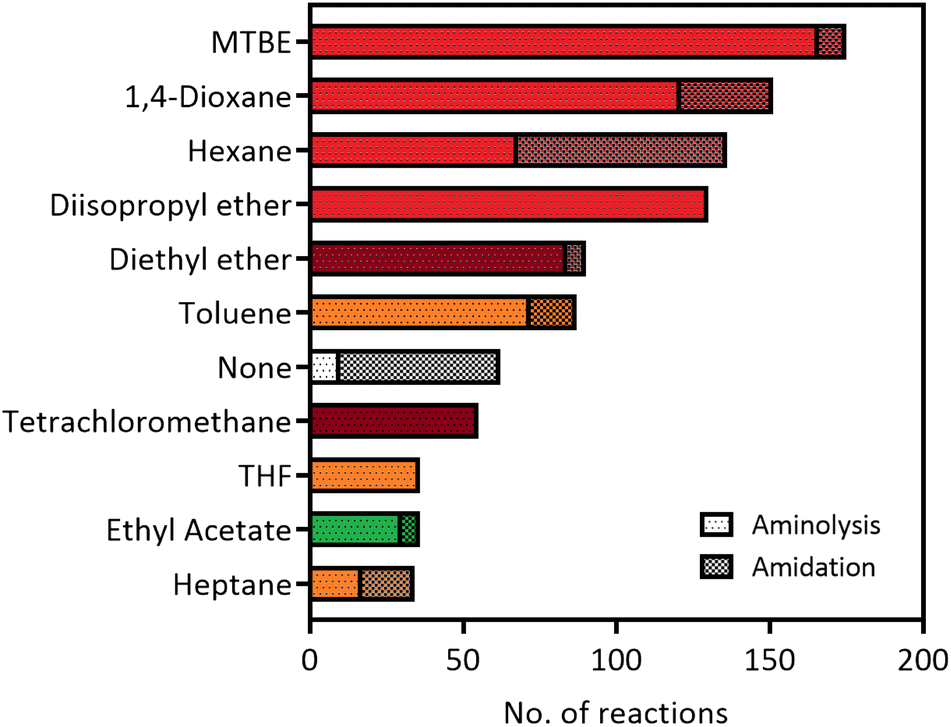 | ||
| Fig. 2 The most used solvents for lipase catalysed amidation reactions in the RetroBioCat database. The individual solvents are split up between reaction types (aminolysis of esters and direct amidation of carboxylic acids) and are colour coded by their ranking using the CHEM21 solvent selection guide (dark red: highly hazardous, red: hazardous, yellow: problematic, green: recommended).5 | ||
Overall, Fig. 2 illustrates that most solvents used in optimized lipase amide-forming reactions are not suitable as green solvents within the pharmaceutical industry and academia and pose substantial occupational health hazards. In this respect, moving towards amidation reactions under aqueous conditions is highly desirable, as water is ranked as the most recommended solvent in this selection guide. In some cases, lipases are able to catalyse amide bond formation in aqueous environments. In addition, a number of other enzyme classes have been shown to catalyse this reaction in water using a variety of mechanisms. These aqueous amidation strategies will be covered in the remaining part of this review.
Penicillin G acylases
A further group of hydrolase enzymes that have been exploited for amide bond formation are the penicillin G acylases (PGAs). PGAs natively catalyse the reversible hydrolysis of the amide bond in penicillin antibiotics (Scheme 3A). This characteristic has been exploited for the synthesis of semi-synthetic penicillins and cephalosporins by hydrolysing the amide bond of penicillin-like structures and subsequent reinstallation of an alternative acyl side chain to the beta-lactam core structure (Scheme 3B).26–28 PGAs have been used on industrial scales for processes like these for decades.29 Exploiting the substrate promiscuity, this enzyme class has also been utilized in the synthesis of dipeptides30,31 and the kinetic resolution of amines.32,33 For example, immobilized PGA was utilized for the resolution of the β-aminoester 11 using 7 as an acyl donor, resulting in an enantiopure precursor for the platelet aggregation inhibitor xemilofiban (S)-11 on a 70 L scale (Scheme 3C).34 Recently, PGA has been utilized by researchers at Merck for larger biomolecules. They report the selective installation of cleavable phenyl acetamide groups at either of three available primary amine positions of human insulin. Extensive directed evolution campaigns led to a toolbox of tailored PGA variants for the acylation or hydrolysis (i.e. protection/deprotection) for each of the targetable amino moieties. The selective modification of insulin led to homogeneous samples of bioconjugates that are highly valuable as biological probes or therapeutics.35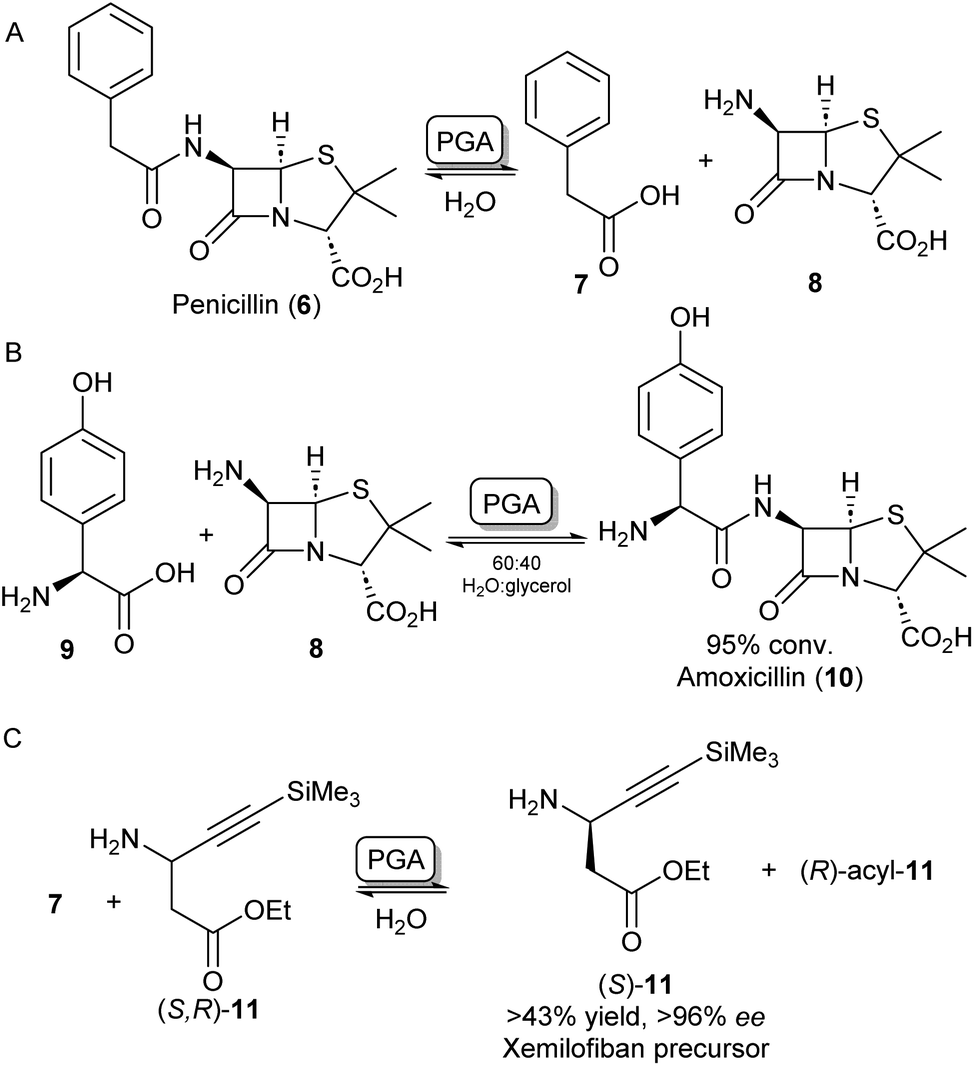 | ||
| Scheme 3 Applications of penicillin G acylase (PGA). (A) PGA catalyses the hydrolysis of penicillin into 7 and 8. (B) PGA can reinstall alternative acyl side chains such as 9 to synthesize analogous antibiotics like amoxicillin 10.26 (C) Racemic β-aminoester 11 can be deracemized on an industrial scale to obtain drug precursor (S)-11 in high ee's.34 | ||
N-Acyltransferases
In some cases, the formation of esters as more reactive intermediates for biocatalytic amide bond formation is used in natural metabolism. For example, in the biosynthesis of A-503083 B (14, Scheme 4A), a capuramycin-type antibiotic, the carboxylic acid 12 is activated by an S-adenosylmethionine–dependent carboxyl methyltransferase forming a methyl ester, which subsequently undergoes N-acylation catalysed by CapW.36 This N-acetyltransferase (NAT) has been used to couple its native methyl ester substrate to a wide array of amine donors under aqueous conditions and some of these amide products showed antimicrobial activity.37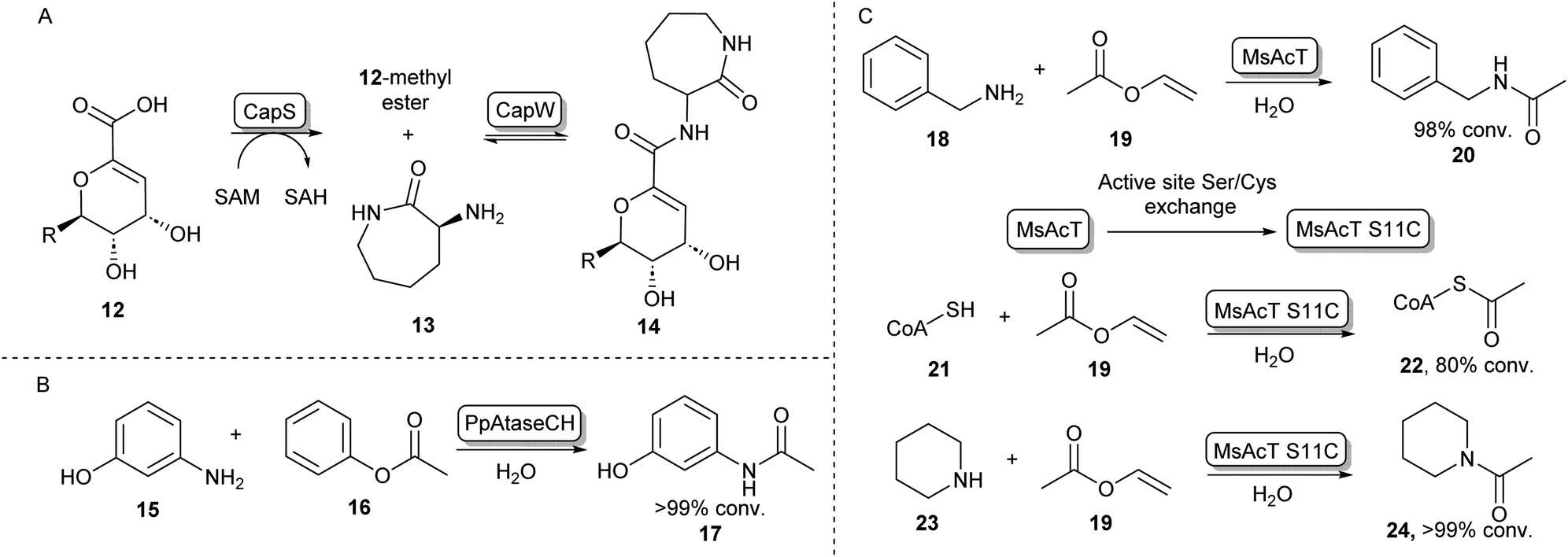 | ||
| Scheme 4 N-Acyl transferases (A) the biosynthesis of antibiotic 14 proceeds through the O-methylation of carboxylic acid substrate 12, followed by aminolysis with 13 catalysed by CapW.36 (B) C-acyl transferase PpAtaseCH has been shown to catalyse amide bond formations with aniline derivatives such as 15.39 (C) wild-type MsAcT acetylates a range of primary amines. Mutating the active site serine to a cysteine improves activity and allows acetylation of thiols including co-enzyme A and the acetylation of secondary amines. These reactions were not accessible using the wild-type enzyme.43,45 | ||
In the search for more biocatalysts for amide bond formation, the acyltransferase from Pseudomonas protegens (PpATaseCH) which previously had been reported to catalyse C–C bond formation,38 was shown to possess chemical promiscuity when 15 was introduced to the reaction (Scheme 4B). Instead of the expected C–C bond formation, C–N bond formation was observed, and a variety of anilines could be acetylated using a phenyl or an enol ester as acyl donors under aqueous conditions, with no requirement of co-factors.39 Similarly, promiscuous acyltransferase activity has been observed in the family VIII carboxylesterase EstCE1, with the enzyme enabling synthesis of the drug moclobemide (for structure see Scheme 8, 42) from ethyl 4-chlorobenzoate and 4-(2-aminoethyl)morpholine with 20% conversion. Guided by the enzyme crystal structure, the three amino acid motif responsible for acyltransferase activity was introduced using protein engineering to a hydrolase enzyme, transforming it into an acyltransferase enzyme.40
Furthermore, an acetyltransferase from Mycobacterium smegmatis (MsAcT) had previously been shown to catalyse ester formation under aqueous conditions.41 This enzyme too was shown to possess reaction promiscuity towards amide formation, as was initially shown by acetylating a variety of primary amines that were in turn formed by a transaminase, thus shifting the equilibrium towards amide bond formation.42 In further studies, Contente et al. have shown that MsAcT catalyzes the acetylation of a wide range of aliphatic and aromatic amines under aqueous conditions and that this system is capable of producing melatonin on a multi-gram scale using immobilized enzyme in flow.43,44 Notably, using mechanistic insights into the enzymes’ catalytic triad mechanism, the group has mutated the catalytic serine residue in the active site to cysteine, thereby allowing the formation of an enzyme-bound thioester intermediate, rather than an ester intermediate. This new reactivity was exploited by introducing thiol nucleophiles allowing the formation of a range of thioesters. Interestingly, the biologically important acetyl-CoA (22) could be formed in large amounts using 19 and 21 as substrates (Scheme 4C). Further exploiting the S11C MsAcT mutant, a range of secondary amines were acetylated forming tertiary amides such as 24, in transformations that were previously inaccessible by this enzymatic system.45
Amidation of carboxylic acids
Amidation of carboxylic acids by lipases
Carboxylic acid activation in the form of an ester is crucial for the activity of most lipase and NAT-catalysed aminolysis reactions.46 However, to avoid this activation step, some research has gone into the direct amidation of carboxylic acids using lipases. For example, Baldessari et al. showed that when a carboxylic acid substrate is incubated with CAL-B in ethanol, the ethanol will function as both a solvent and a nucleophile forming the ethyl ester. Upon addition of an amine nucleophile, the in situ formed ester will undergo aminolysis forming the amide.47,48 In a different strategy, CAL-B was able to directly couple carboxylic acids with amines by shifting the reaction equilibrium. Using either ionic liquids or a solvent-free system under reduced pressure to remove by-product water, chiral amides were obtained in high enantiomeric ratios.49 Another strategy of low-water systems allowed direct CAL-B catalysed amidation of carboxylic acids by using molecular sieves, thus preventing hydrolysis.50 As a rare example, Zeng et al. have described the intracellular lipase SpL, which is able to catalyse amidation of both esters and carboxylic acids. As this lipase acts intracellularly rather than extracellularly as previously discovered lipases, SpL was used in whole E. coli cells in hexane yielding a range of amides on preparative scale.51 In another recent effort, reaction and enzyme engineering of CAL-B allowed the direct amidation of unprotected L-proline with ammonia.52 Amidation of unprotected amino acids is an otherwise highly challenging reaction for lipases.ATP-dependent amide bond-forming enzymes
Because of the high prevalence of amide bond formations in the chemical and pharmaceutical industry, there is a significant interest in more sustainable amidation methods. Moving from organic solvents to aqueous reaction conditions is a crucial step in achieving this sustainability.Nature has evolved a wide array of enzymes able to perform amide bond formation in water. The majority of these enzymes activate the carboxylic acid substrate using the co-factor adenosine-triphosphate (ATP), forming highly reactive acyl-phosphate intermediates such as acyl-adenylate intermediate (25, Scheme 5).54
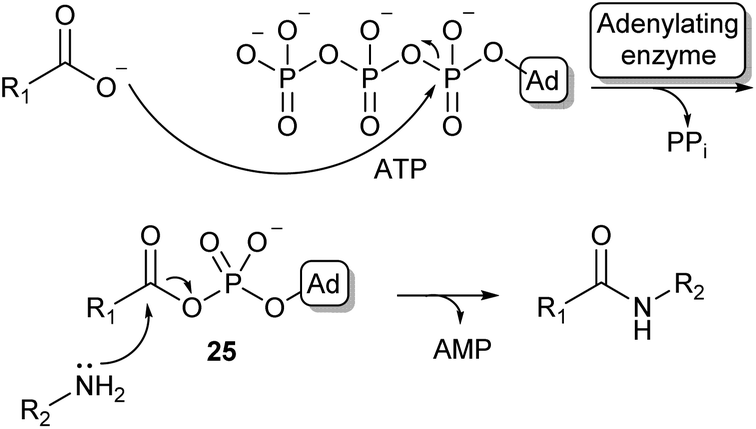 | ||
| Scheme 5 Carboxylic acid activation by adenylation via an acyl adenylate intermediate (25) and subsequent amidation (Ad = adenosine). | ||
Most amide bonds in living cells are formed within the ribosome where mRNA is used as a template to incorporate one of the 20 proteinogenic L-amino acids into a growing peptide chain.55 In the fungi and bacteria kingdoms, peptides are also synthesized outside of the ribosome by large multimodular enzyme complexes known as non-ribosomal peptide synthases (NRPSs). These enzymes act independently from mRNA and each NRPS specifically synthesizes one type of peptide product. Non-ribosomal peptides (NRPs) display a large structural diversity and often have a macrocyclic structure (26, Scheme 6). They are classified as secondary metabolites in nature and some examples have become commercially available as antibiotics or anti-cancer drugs. NRPs often contain a larger diversity of chemical groups compared to ribosomal peptides including a wide variety of non-proteinogenic amino acids.55,56 NRPSs typically consist of multiple modules that are responsible for incorporating a single amino acid into the growing peptide chain. Many structurally diverse exceptions aside, a typical NPRS module consists of three distinct domains: the adenylation (A-) domain selectively binds an amino acid and activates the acyl group using ATP forming an aminoacyl-AMP intermediate. The thiolation (T) domain contains a phosphopantetheine prosthetic group which intercepts the aminoacyl-AMP intermediate in the form of a thioester intermediate that is covalently bound to the enzyme. Finally, this active thioester intermediate is coupled to the growing peptide chain in the condensation (C) domain (Scheme 6).56 Significant amounts of research have gone into the engineering of these NRPS assembly lines to produce novel NRP products. Whilst there have been some very impressive examples of re-engineering NRPSs to broaden substrate scope, these enzymes are highly complex and evolved to specific targets. Using them as broad amidation catalysts has been very challenging because of the importance of the integrity of the linker regions and the selectivity of the individual modules.53
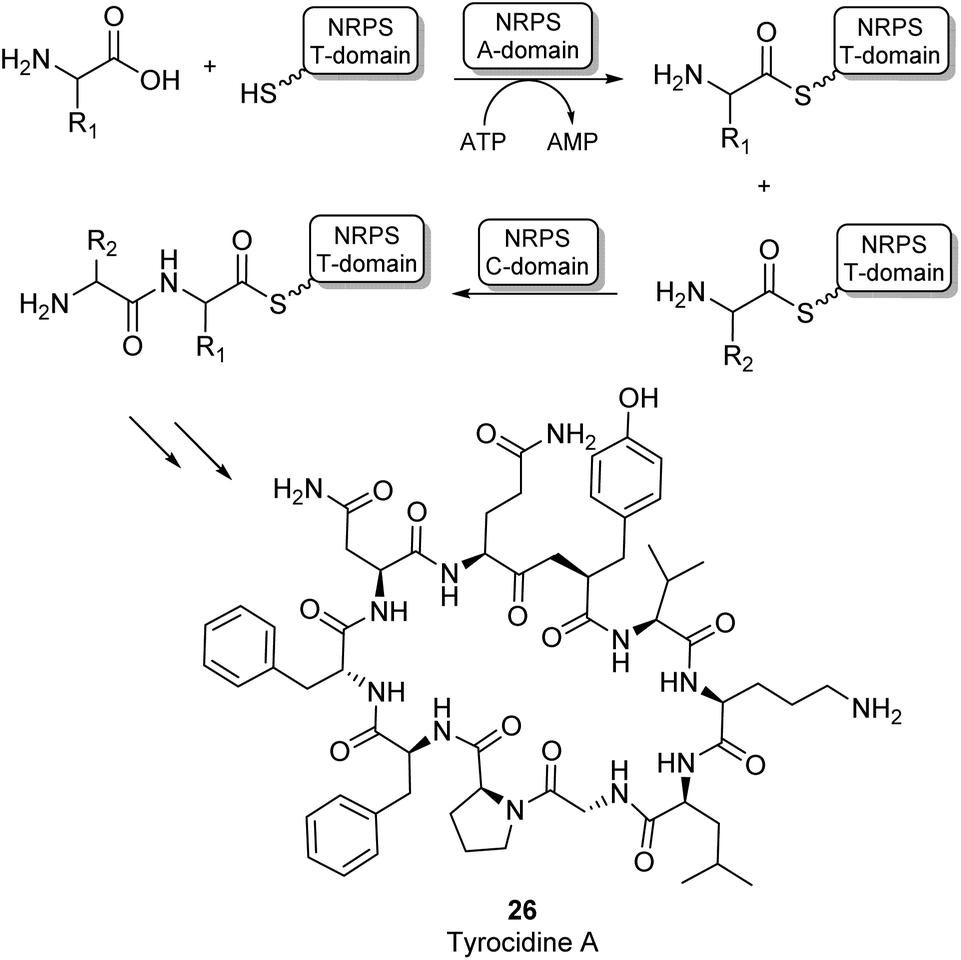 | ||
| Scheme 6 NRPS catalysed amino acid chain elongation: amino acid substrates are activated by the A-domain, allowing subsequent capture onto a T-domain in the form of a thioester. The C-domain catalyses peptide bond formation between two thioester intermediates, adding the next amino acid to the growing peptide chain.53 As an example, the structure of the NRP tyrocidine A is shown (26). | ||
Amidation of acyl adenylates
Some standalone NRPS A-domains are able to adenylate their carboxylic acid substrate and this acid activation can be used to perform amidation reactions when an amine nucleophile is introduced. Examples include the NRPS A-domain DhbA, which was shown to couple 2,3-dihydroxybenzoic acid (DHB) to L-cysteine forming N-DHB-L-cysteine,57 and the A-domain of tyrocidin synthase (TycA-A), coupling tryptophan to a range of primary and secondary amines (Scheme 7A).58 Although these A-domains are usually very specific for their carboxylic acid substrate, Zhu et al. have recently shown the 3-isocyanobutanoic acid adenylating enzyme SfaB to be able to couple a relatively broad range of aliphatic acids to a variety of primary and secondary amines.61 Adenylating enzymes involved in other natural product synthesis pathways have also been used for synthetic amide bond formation. For example, the standalone adenylating enzyme TamA, involved in the biosynthesis of tambjamine YP1, was used to couple a range of fatty acids to some α-amino acids and primary amines.62 Furthermore, pantothenate synthase (PS), which is involved in the biosynthesis of coenzyme A (CoA) was found to accept a range of amine substrates other than its native β-alanine substrate and has been exploited for the preparative synthesis of a range of pharmaceutically relevant amides like 30 (Scheme 7B).59 In a similar fashion, the adenylating activity of CoA-ligases can also be used for amide bond formation. In their native function, these enzymes adenylate their carboxylic acid substrates for the subsequent coupling with CoA forming a CoA-thioester. For example, a thermostable CoA ligase isolated from Geobacillus thermodenitrificans has been used for the preparative synthesis of 33 using a two-enzyme ATP recycling system (Scheme 7C).60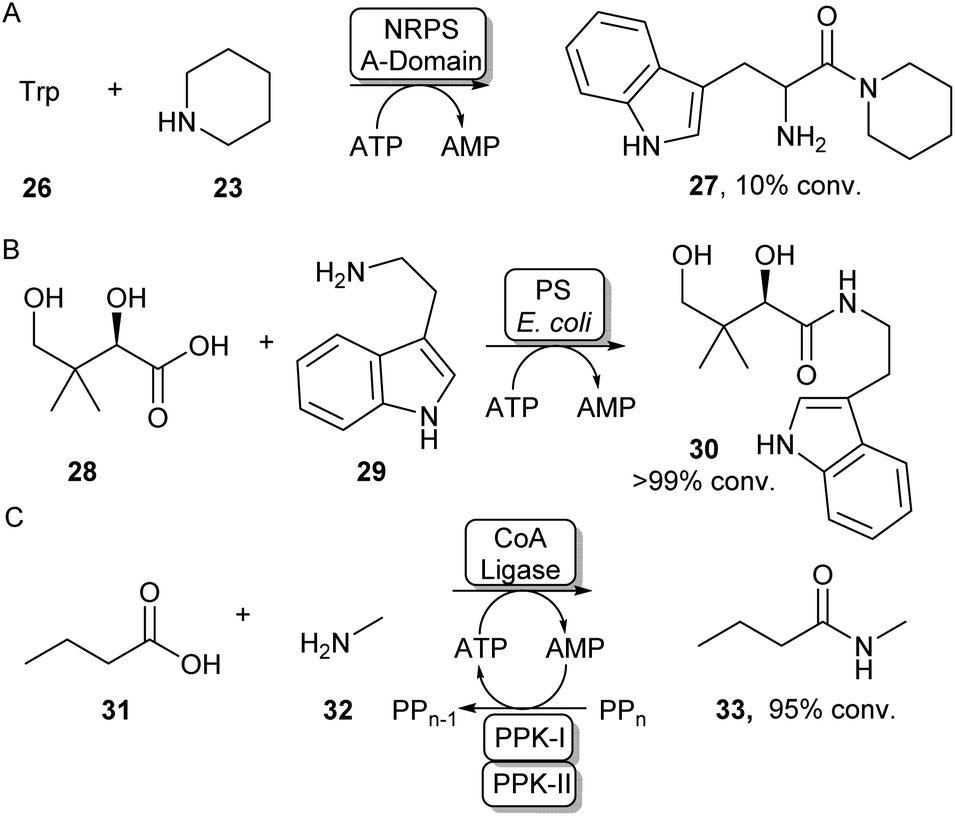 | ||
| Scheme 7 Utilising adenylating enzymes for amide bond formation. (A) The truncated A-domain from the NRPS TycA was able to adenylate tryptophan to a range of amines.58 (B) PS from E. coli has a promiscuous amine scope allowing the synthesis of a range of vitamin B5 antimetabolites59 (C) Adenylation activity from a thermostable CoA ligase allowed the preparative synthesis of 33 using a two-enzyme ATP recycling system.60 | ||
A different protein engineering strategy starts with Carboxylic acid reductases (CARs), which are multidomain proteins whose architecture resembles a single NPRS module: the A-domain activates a carboxylic acid substrate using ATP, which subsequently becomes covalently attached to the T-domain in the form of a thioester. In the final step of the CAR-catalysed reaction, this thioester intermediate is then transported to the reductase (R) domain where it is reduced to the aldehyde using the cofactor NADPH.63 CARs are known to have a very broad carboxylic acid scope due to their relatively large and hydrophobic active site pocket.64 In order to use this system for amide bond formations, it was shown that by addition of an amine nucleophile, the acyl adenylate could be intercepted forming a variety of primary, secondary or tertiary amides.65 Given that solely the adenylation activity of CAR was shown to be required for amidation, the CAR gene was truncated so that only the CAR A-domain would be expressed. It was found that the truncated CAR enzyme from Mycobacterium marinum (CARmm-A) had improved amidation activity compared to the full-length enzyme. After further optimisation by introducing a polyphosphate kinase from Cytophaga hutchinsonii (CHU) to regenerate ATP from AMP using cheap polyphosphate, this amidation method was used for the mono-acylation of symmetrical diamines. As solely the mono-acylated product was obtained using CAR-A, this method circumvented the protection/deprotection strategies used in traditional chemical methods towards these motifs. The pharmaceutical relevance was demonstrated by synthesising a range of drugs and drug precursors, for example, the antiparkinsonian agent lazabemide 36 was synthesized in a single biocatalytic step (Scheme 8A).66
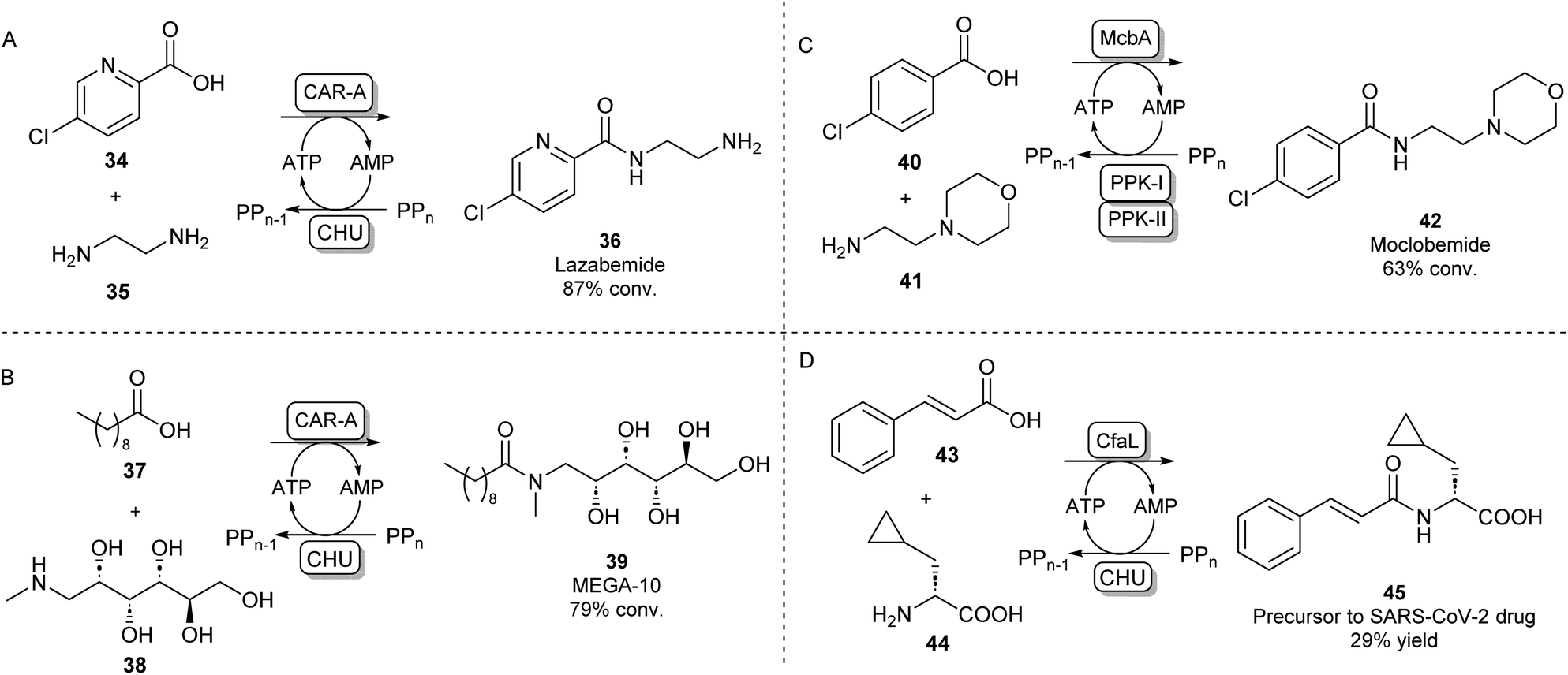 | ||
| Scheme 8 Amidation of acyl adenylates. CAR-A has been used for the mono-acylation of diamines66 (A) and for the selective N-acylation of aminoalcohols67 (B). ABSs McbA and CfaL have been used for the synthesis of pharmaceutically relevant amide products like moclobemide68 (C) and a SARS-CoV-2 drug precursor (D) respectively.69 | ||
In further studies, the promiscuity of the CAR-A system was exploited for the amide coupling of amino alcohols to fatty acids showing a very high selectivity of amide bond formation over ester formation. This allowed the one-step synthesis of commercially relevant glucamide surfactants like 39 with advantages over lipase-catalysed syntheses where mixtures of amide and ester products are obtained (Scheme 8B).67 It has been hypothesised that the acyl adenylate intermediate in this system might undergo nucleophilic attack independent from the enzyme, so, the N over O-selectivity observed in this reaction is likely due to the intrinsic reactivity of acyl phosphates in water.67 This feature might also explain the exceptionally large amine scope of the CAR-A system.
Indeed, it is not uncommon in Nature to use the adenylation strategy for direct amidation with an amine nucleophile. ATP-dependent amide bond synthases (ABSs) have been identified to catalyse amide bond formation in secondary metabolite pathways by accepting an amine directly into the active site of the enzyme, not requiring the involvement of a thioester intermediate. For example, the ABS isolated from Marinactinospora thermotolerans (McbA) has been shown to couple its native β-carboline-derived acid substrate with a range of primary amines.70 Structural studies of McbA revealed that the enzyme also displayed promiscuity for its carboxylic acid substrate and that the enzyme catalyses both half-reactions of the adenylation-amidation sequence.71 The enzyme was subsequently exploited for the synthesis of the antidepressant drug 42 in a preparative scale using a two-enzyme ATP recycling system (Scheme 8C).68 Recently, another ABS, named coronafacic acid ligase (CfaL), was discovered to possess great selectivity towards aliphatic and aromatic acids. The enzyme was shown to possess excellent enantioselectivity and was able to couple acid substrates to a wide range of natural and unnatural α-amino acids. The enzyme structure was elucidated and engineered to synthesise a variety of APIs on a preparative scale (45, Scheme 8D).69 ABSs have shown tremendous potential for aqueous amide bond synthesis as they catalyse the direct coupling of acid and amine substrates and are amenable to enzyme engineering to increase their efficiencies and substrate scopes.
Amidations involving coenzyme A
A second strategy toward amide bond formation prevalent in biosynthetic pathways involves adenylation of carboxylic acid substrates and subsequent conversion to a CoA-thioester by a CoA ligase. The CoA-thioester is then coupled to an amine by a further N-acyltransferase (NAT). Because this two-enzyme system for amide bond formation uses two cofactors (ATP and CoA), some research has gone into in vitro regeneration systems to make this strategy a viable method for synthetic amide bond formation. Mordhorst et al. designed a cascade consisting of a CoA ligase and a NAT coupled to a two-enzyme ATP recycling system. Using different combinations of ligases and NATs moderate to good conversions were observed to acetyl para-aminobenzoic acid and hippuric acid products.72Even though some NATs display moderately broad substrate scopes, CoA ligases are known to be very selective for their native carboxylic acid substrate.54 This necessitates the selection of a two-enzyme combination with complementary substrate profiles to obtain the desired amide product. Using high-throughput screening of a library of CoA-ligases with a range of carboxylic acid substrates, and a second screening of a library of NATs with a combination of CoA-esters and amines, scientists at GSK have demonstrated that this system is suitable for the synthesis of a wide variety of amide products, as long as the right combination of two complementary enzymes is being used. A precursor of the kinase inhibitor 49 was obtained in high yields on a preparative scale using a two-enzyme combination in a whole cell system (Scheme 9A).73
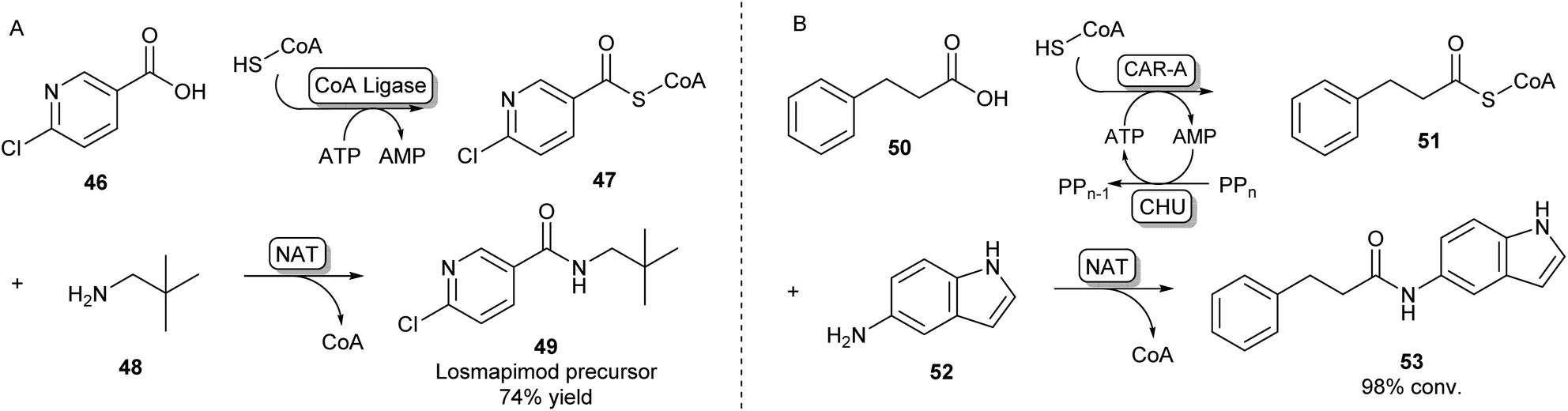 | ||
| Scheme 9 Amidations involving SCoA-esters. (A) CoA Ligase enzymes can adenylate and subsequently couple CoA to their carboxylic acid substrate. By selecting complementary CoA Ligases and NATs based on their substrate specificities, amides can be formed via an acyl-CoA intermediate.73 (B) CAR-A has been shown to accept CoA as a nucleophile and, because of its wide substrate scope, is able to serve as a universal CoA ligase. Therefore, CAR-A can be utilized in a comparable strategy when coupled to a NAT, forming a variety of amides.74 | ||
Recently, it was found that besides using an amine nucleophile to intercept the adenylate intermediate in the CAR-A reactions discussed previously, CAR-A was able to couple a wide variety of carboxylic acids to thiols, in particular CoA-SH, generating a range of CoA-thioesters and thus serving as a universal CoA ligase.74 This general acyl-S-CoA recycling system can be used in situ with substoichiometric amounts of thiol and is an attractive alternative to chemical ligation strategies.45,74 Coupling the CAR-A mediated CoA-ester synthesis to a complementary NAT enzyme, the system was exploited to synthesize an exceptionally wide range of amide products, including a set of amides derived from a variety of anilines like 52, which are usually regarded as more challenging substrates because of their low nucleophilicity and are not accessible through the direct CAR-A amidation method (Scheme 9B). Selected examples were performed on a preparative scale, demonstrating the value of this approach for synthesis. Further exploiting CAR-A as a universal CoA-thioester synthetase, the system was coupled to a lysine acetyltransferase to achieve acylation of histone-derived peptide H4–20 including non-natural bio-orthogonal groups amenable to further ‘click chemistry’.74
Amidation of acyl phosphates
Another method of carboxylic acid activation by ATP is seen in a family of enzymes called ATP-grasp enzymes (Scheme 10). This family gets its name from the common structural feature they share between them, responsible for “grasping” or holding a molecule of ATP in the active site.19 The acid substrate 54 is reacted with ATP forming an acyl-phosphate intermediate (55) which subsequently undergoes nucleophilic attack by an amine leading to amide 56. A great variety of ATP-grasp enzyme-catalysed amide bond-forming reactions have been described, involved in either primary or secondary metabolism.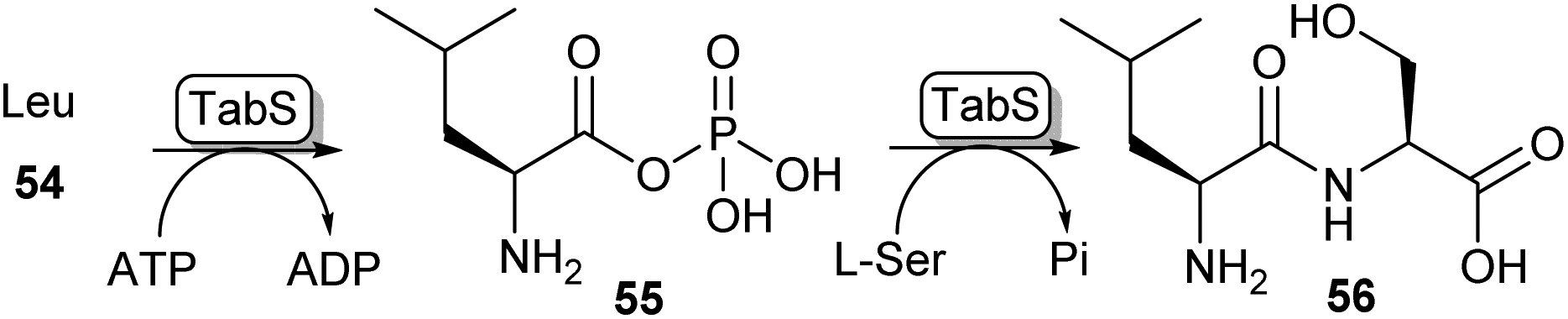 | ||
| Scheme 10 ATP grasp enzymes form amide bonds via an acyl phosphate intermediate (55). The ATP grasp enzyme isolated from Pseudomonas syringae (TabS) has been shown to synthesise a variety of dipeptides such as 56.76 | ||
Carboxylic acids accepted by the ATP-grasp family range from small acids like formic acid to large proteins being accepted as substrates, while for the amine component, a range from ammonium ions to the amine of a biotin prosthetic group of a carrier protein have been described as substrates. Even though the property of this enzyme class has been described as highly promising for use in synthetic chemistry,19 they have not been implemented as such, most likely due to the high specificity for both the acid and the amine substrates. To date, ATP-grasp enzymes have mainly been used for the synthesis of di- or oligopeptides where some promiscuity has been observed.75
For example, ywfE from Bacillus subtilis was shown to accept a handful of proteinogenic amino acids as the carboxylic acid donor, and accept up to 15 amino acids as the amine donor.77 The L-amino acid ligase TabS from Pseudomonas syringae has shown the broadest substrate promiscuity thus far, being able to accept all 20 proteinogenic L-amino acids and β-alanine as either an acid or an amine substrate (Scheme 10).76
Nitrile hydration
Nitrile hydratases
Although amine acylation is the most common synthetic route toward amides, primary amides can also be obtained by the hydration of nitrile compounds. Chemical nitrile hydration methods rely on harsh reaction conditions such as high temperatures (200–300 °C) and high pressure usually obtaining low yields without chemo- or regioselectivity. Therefore, the cobalt- or iron-dependent enzyme class of nitrile hydratase (NHase) has been used in several industrial-scale processes for decades as a more eco-friendly alternative.78 In fact, NHase catalysed industrial-scale synthesis of acrylamide (58) from acrylonitrile 57 is the first example of the use of biocatalysis for the production of commodity chemicals and the first example of the use of biotechnology in the petrochemical industry (Scheme 11A).79 NHase has also been employed as an industrial catalyst for the manufacturing of nicotinamide and 5-cyanovaleramide.80 Much research has gone into the expression,81 structure,82 mechanism,83 stability,84 selectivity85 and substrate scope86 of NHases and has been extensively reviewed elsewhere.78–80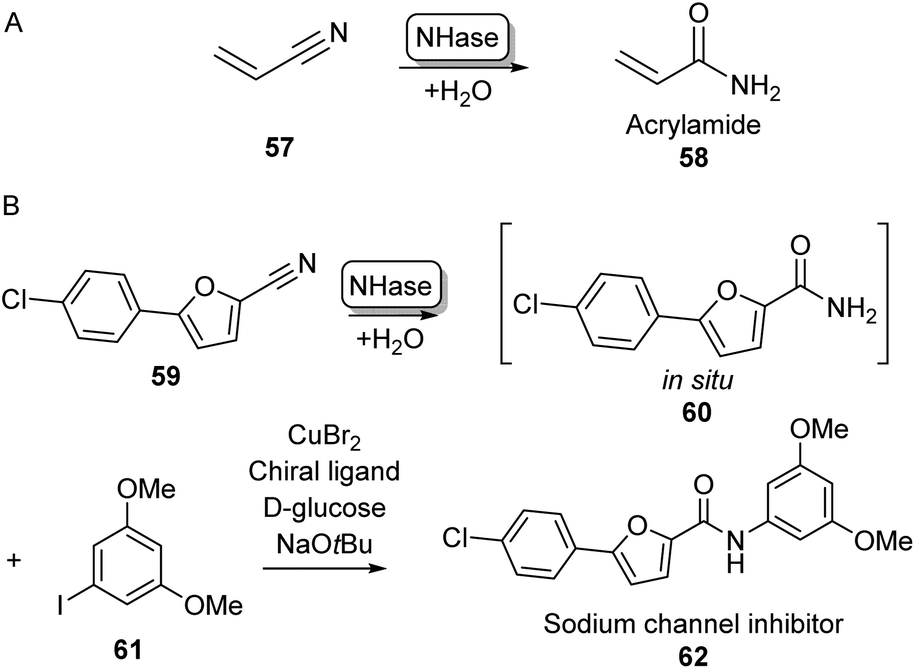 | ||
| Scheme 11 Nitrile Hydratase (NH) catalysed amide bond formation. (A) NHase catalysed acrylamide synthesis. (B) Chemoenzymatic strategy for the expansion of amide scopes using copper catalysis.87 | ||
In a notable recent example, NHases have been used in combination with copper catalysis to extend the product scope of this amidation method beyond primary amides. Using two complementary NHase subtypes (from Rhodopseudomanas palustris and Rhodococcus erythropolis respectively) in whole E. coli cells to perform nitrile hydration, the obtained primary amide was then coupled in situ to iodobenzene derivatives like 61 using CuBr2 and a diamine ligand in water, obtaining a broad range of aliphatic and aromatic amide products such as 62 on gram scales using substrate loadings in the molar range (Scheme 11B).87
Lactamisation
Cyclic amides (lactams) are found as motifs in many natural products and APIs and are important precursors in the synthesis of commercial polymers.88 A well-known example from nature is the four-membered β-lactam ring present in the most important class of currently approved antibiotics, first observed in penicillin.89 The biosynthetic pathway of the well-studied penicillin N (64) proceeds via a tripeptide precursor produced by an NRPS. This peptide is cyclized by isopenicillin N synthase (IPNS), a non-heme iron protein making use of a ferrous ion and molecular oxygen, yielding the β-lactam ring in 64 (Scheme 12A).90,91 An alternative pathway toward β-lactams has been found in the biosynthesis of clavulanic acid, an inhibitor of β-lactamases which is used to overcome antibiotic resistance. Here, an ATP-dependent β-lactam synthetase (β-LS) catalyses the intramolecular ring closure between a terminal carboxylic acid and a secondary amine (Scheme 12B).90 β-LS has been engineered using rational design to accept methyl and ethyl substituted substrates yielding clavulanic derivatives.92 Carbanepam synthase (CarA) also utilizes an ATP-dependent mechanism to produce a lactam ring but is besides four-membered rings also able to form 5- and 6-membered lactam rings.93 This enzyme has subsequently been used in a two-enzyme cascade to prepare a range of bicyclic β-lactams in excellent diastereomeric ratios.94 (BAS) was exploited to form 5-membered lactam rings via an intramolecular Dieckmann condensation. Coupling malonyl-CoA with aminoacyl-CoA esters a range of unnatural tetramic acid derivatives were obtained with one example showing anti-leukemic activity.95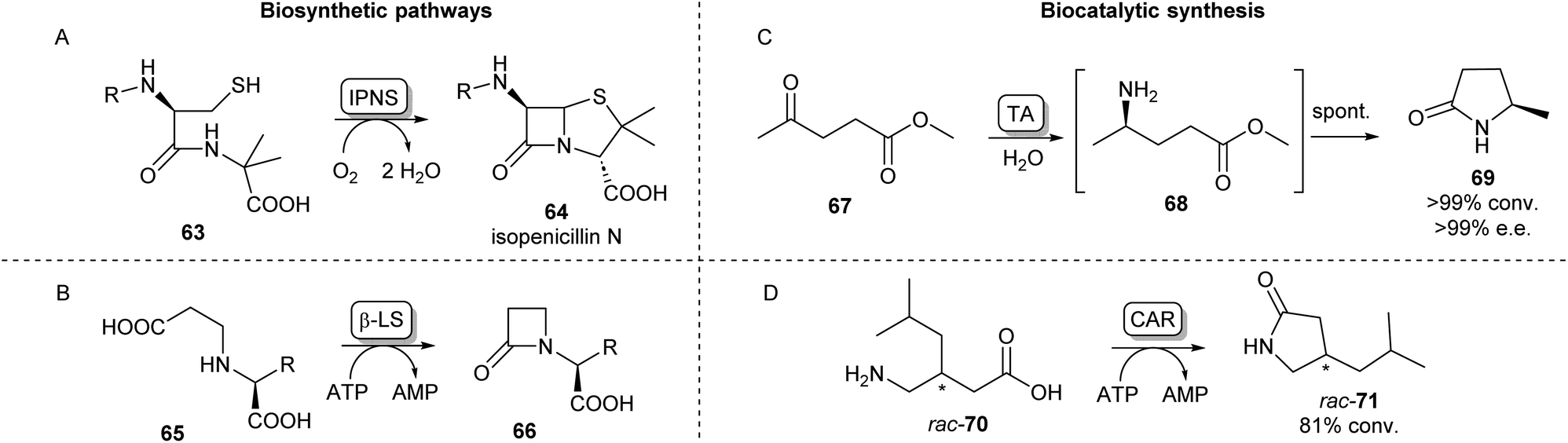 | ||
| Scheme 12 Lactam forming enzymes. Naturally occurring biosynthetic enzymes forming a beta-lactam ring (left). (A) The non-heme iron protein IPNS catalyzes the cyclisation of 63 forming isopenicillin N (64) and (B) β-LS catalyzes the ring closure of 65 forming 66, an intermediate in the clavulanic acid biosynthetic pathway.90 Enzymatic methods towards synthetic lactams (right). (C) Enantioselective TAs catalyse reductive amination of ketoesters like 67 forming intermediate 68 which undergoes spontaneous cyclisation forming lactams like 69.88 (D) Using CAR catalysed amidation a variety of amino acids have been converted into lactams.103 | ||
In a different lactamisation strategy, the wide substrate promiscuity of the polyketide synthase benzalacetone synthase.
Synthetically, lactams can be formed using an intramolecular cyclisation reaction of amino esters or amino acids catalysed by lipases.46 PPL has been shown to catalyse the lactamisation of a range of amino esters in tert-amyl alcohol,96 whereas CAL-B has been used for the cyclisation of 4-, 5-, 6-, and 8-aminooctanoic acid to their corresponding lactam products.97 In a scaled-up example, Willis et al. have utilized CAL-B for the lactamisation of a fluorinated diester in aqueous conditions on a 10 g scale.98
An alternative biocatalytic method towards lactam formation is via the reductive amination of keto- or aldehyde esters, the products of which have been shown to spontaneously cyclise to the corresponding lactams. This was first shown using the (R)-selective transaminase ATA-117 synthesising (R)-4-phenylpyrrolidin-2-one in excellent yields.99 Using similar lactamisation strategies and thorough reaction optimisations, these reactions have been applied on multi-gram scales showing excellent stereoselectivities.100,101 The versatility of this lactamisation strategy has been shown by Mourelle-Insua et al. forming a wide range of lactams from γ- and δ-keto esters by using a set of commercially available transaminases (Scheme 12C).88 Using a different reductive amination strategy, an NADPH-dependent reductive aminase has been shown to catalyse the coupling of a keto ester with propargylamine, which then formed the (R)-lactam products in high yields and ee.102 Recently, Qin et al. have exploited the previously discussed amidation activity and promiscuity of CAR for the ATP-dependent cyclisation of a range of amino acids.
Using both wild-type and mutants of CAR from Segniliparus rugosus various substituted lactams of different ring sizes were synthesised with some examples showing stereoselectivity (Scheme 12D).103 This lactamisation activity of CAR has also been applied in a one-pot biocatalytic cascade forming ε-caprolactam from cyclohexanol.104
Alternative enzymatic methods towards amides
Besides the main amidation methods described so far, there are specific examples of routes toward amides that do not fall into these categories. The NAT MsAcT which has been discussed previously has been described to catalyse the reaction between anilines and various anhydrides to form aminooxo-acids under aqueous conditions in molar substrate concentrations. Using this approach, a precursor to histone deacetylase inhibitor 74 was obtained on a multigram scale (Scheme 13A).105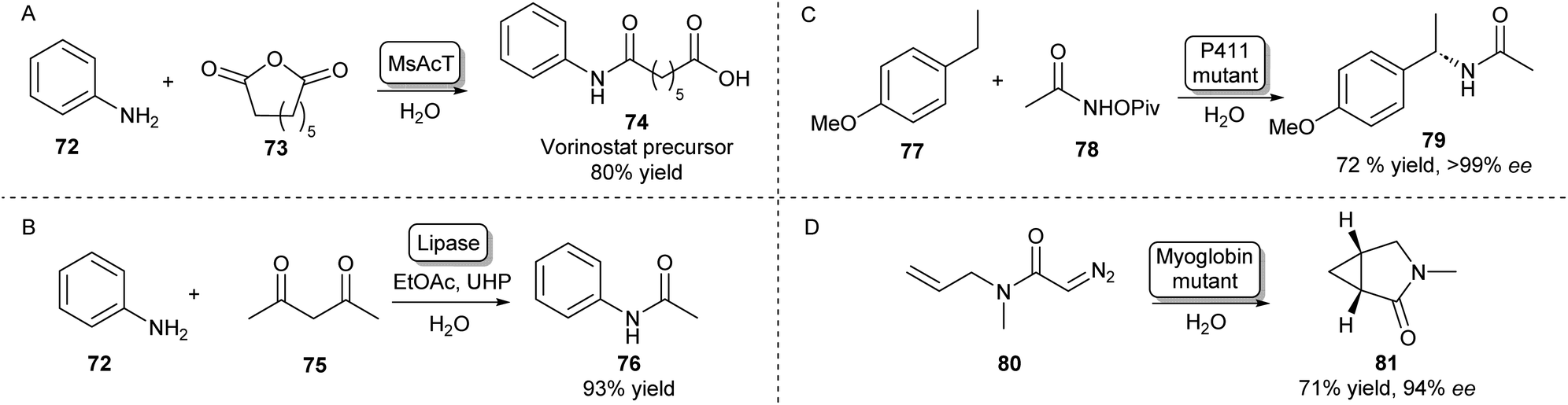 | ||
| Scheme 13 Alternative enzymatic methods towards amides. (A) The chemical promiscuity of MsAcT was exploited in the amidation of anhydrides leading to aminooxo-acids like 74.105 (B) Lipase mediated amidation of 1,3-diketones with anilines using in situ generation of a peracid.106 Access to enantioenriched amides can be achieved by heme-protein catalysed nitrene (C) or carbene (D) insertion.107,108 | ||
Amides have also been synthesised using novel biocatalytic strategies that do not rely on direct N-acylation (Scheme 13B–D). For example, a lipase was used to form amides from a range of anilines and 1,3-diketones. Mechanistically, this was achieved by the in situ generation of a peracid from ethyl acetate by the lipase, which oxidises the enaminone intermediate formed by the same lipase. This species will then rearrange to form the desired amide (Scheme 13B).106
Access to enantioenriched amides can also be achieved by nitrene or carbene transfer reactions catalysed by heme proteins, which is a very recent field that has attracted much interest.109 For example, the benzylic C–H bond positions of inert starting materials like 77 can be functionalized by highly evolved P411 enzymes (Scheme 13C). Hydroxamate precursors such as 78 form a nitrene intermediate in the enzyme active site that allows enantioselective C–H functionalisation of a wide scope of feedstock aromatic compounds with some examples performed on a preparative scale.107
Furthermore, using a carbene transfer reaction with ethyl diazoacetate, this same class of enzymes was shown to be able to catalyse the cyclopropanation of a variety of acrylamide and acrylate olefins110 allowing the synthesis of a levomilnacipran precursor111 or amide-bearing alkenes in high diastereo- and enantioselectivities.112 A similar intramolecular cyclopropanation strategy was also shown to be catalysed by myoglobin mutants leading to enantioenriched lactams (Scheme 13D).108
Conclusion
As traditional chemical methods for amide bond formation most often rely on hazardous reagents and coupling agents, scientists have been looking to develop more sustainable and greener alternatives for this very common chemical reaction for decades. Biocatalysis has played a big role in this regard. In fact, the discovery of the ability of lipases to catalyse amide bond formation has helped to kick-start the very field of biocatalysis, possessing excellent stability and substrate scopes. However, with the increasing impact of climate change, the desirability for moving away from fossil fuels and to more sustainable chemistry is becoming more and more important. As outlined in Fig. 2, lipase catalysed amidations often rely on hazardous solvents. Aqueous amidation methods are therefore highly sought after, and some highly promising enzymatic technologies have been described in this review: NATs like MsAcT and its mutants have proven very successful for the acetylation of a range of amines and even thiols in water, whereas ATP-dependent acyl adenylate forming enzymes like McbA, CfaL and CAR-A have shown to be able to couple a very diverse set of carboxylic acids and amines under aqueous conditions, forming a wide range of molecules relevant to pharmaceutical and fine chemical industries. RetroBioCat seeks to cover all papers in this area and researchers are encouraged to help populate the database as new methodologies are published. Publicly available data curation tools allow new reactions, enzymes, or papers to be suggested by anyone. Furthermore, a comprehensive data curation portal is available for the addition of enzyme sequences and activity data, with database additions to the database attributed to each user.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ML thanks CoEBio3 for funding for a studentship.References
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 2020, 4641–4651 CrossRef CAS.
- R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- S. Hazra, F. Gallou and S. Handa, ACS Sustainable Chem. Eng., 2022, 10, 5299–5306 CrossRef CAS.
- D. Procopio, C. Siciliano, S. Trombino, D. E. Dumitrescu, F. Suciu and M. L. di Gioia, Org. Biomol. Chem., 2022, 20, 1137–1149 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- R. J. Young, S. L. Flitsch, M. Grigalunas, P. D. Leeson, R. J. Quinn, N. J. Turner and H. Waldmann, JACS Au, 2022, 2, 2400–2416 CrossRef CAS PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- F. H. Arnold and F. H. Arnold– Nobel Lecture, https://www.nobelprize.org/prizes/chemistry/2018/arnold/lecture/, (accessed 23 November 2022).
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS PubMed.
- W. Finnigan, L. J. Hepworth, S. L. Flitsch and N. J. Turner, Nat. Catal., 2021, 4, 98–104 CrossRef CAS PubMed.
- M. T. Reetz, Curr. Opin. Chem. Biol., 2002, 6, 145–150 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, Science, 1984, 224, 1249–1251 CrossRef CAS PubMed.
- A. Zaks and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3192–3196 CrossRef CAS PubMed.
- A. L. Margolin and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 3802–3804 CrossRef CAS.
- V. Gotor, R. Brieva and F. Rebolledo, Tetrahedron Lett., 1988, 29, 6973–6974 CrossRef CAS.
- A. Goswami and S. G. Van Lanen, Mol. BioSyst., 2015, 11, 338–353 RSC.
- R. N. Lima, C. S. dos Anjos, E. V. M. Orozco and A. L. M. Porto, Mol. Catal., 2019, 466, 75–105 CrossRef CAS.
- F. Balkenhohl, K. Ditrich, B. Hauer and W. Ladner, J. Prakt. Chem., 1997, 339, 381–384 CrossRef CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- A. N. Parvulescu, P. A. Jacobs and D. E. De Vos, Adv. Synth. Catal., 2008, 350, 113–121 CrossRef CAS.
- R. N. Lima, C. S. dos Anjos, E. V. M. Orozco and A. L. M. Porto, Mol. Catal., 2019, 466, 75–105 CrossRef CAS.
- V. Gotor, Bioorg. Med. Chem., 1999, 7, 2189–2197 CrossRef CAS PubMed.
- X. Pan, L. Wang, J. Ye, S. Qin and B. He, Appl. Microbiol. Biotechnol., 2018, 102, 1749–1758 CrossRef CAS PubMed.
- X.-L. Zhang, M.-H. Zong and N. Li, Bioresour. Bioprocess., 2016, 3, 49 CrossRef PubMed.
- C. Fuganti, C. M. Rosell, R. Rigoni, S. Servi, A. Tagliani and M. Terreni, Biotechnol. Lett., 1992, 14, 543–546 CrossRef CAS.
- B. M. Dorr and D. E. Fuerst, Curr. Opin. Chem. Biol., 2018, 43, 127–133 CrossRef CAS PubMed.
- P. C. Pereira, I. W. C. E. Arends and R. A. Sheldon, Tetrahedron Lett., 2014, 55, 4991–4993 CrossRef CAS.
- M. Koreishi, K. Tani, Y. Ise, H. Imanaka, K. Imamura and K. Nakanishi, Biosci., Biotechnol., Biochem., 2007, 71, 1582–1586 CrossRef CAS PubMed.
- L. M. Van Langen, N. H. P. Oosthoek, D. T. Guranda, F. Van Rantwijk, V. K. Švedas and R. A. Sheldon, Tetrahedron: Asymmetry, 2000, 11, 4593–4600 CrossRef CAS.
- X. Gong, E. Su, P. Wang and D. Wei, Tetrahedron Lett., 2011, 52, 5398–5402 CrossRef CAS.
- B. H. Landis, P. B. Mullins, K. E. Mullins and P. T. Wang, Org. Process Res. Dev., 2002, 6, 539–546 CrossRef CAS.
- A. Fryszkowska, C. An, O. Alvizo, G. Banerjee, K. A. Canada, Y. Cao, D. DeMong, P. N. Devine, D. Duan, D. M. Elgart, I. Farasat, D. R. Gauthier, E. N. Guidry, X. Jia, J. Kong, N. Kruse, K. W. Lexa, A. A. Makarov, B. F. Mann, E. M. Milczek, V. Mitchell, J. Nazor, C. Neri, R. K. Orr, P. Orth, E. M. Phillips, J. N. Riggins, W. A. Schafer, S. M. Silverman, C. A. Strulson, N. Subramanian, R. Voladri, H. Yang, J. Yang, X. Yi, X. Zhang and W. Zhong, Science, 2022, 376, 1321–1327 CrossRef CAS PubMed.
- M. Funabashi, Z. Yang, K. Nonaka, M. Hosobuchi, Y. Fujita, T. Shibata, X. Chi and S. G. Van Lanen, Nat. Chem. Biol., 2010, 6, 581–586 CrossRef CAS PubMed.
- X. Liu, Y. Jin, W. Cai, K. D. Green, A. Goswami, S. Garneau-Tsodikova, K. Nonaka, S. Baba, M. Funabashi, Z. Yang and S. G. Van Lanen, Org. Biomol. Chem., 2016, 14, 3956–3962 RSC.
- N. G. Schmidt, T. Pavkov-Keller, N. Richter, B. Wiltschi, K. Gruber and W. Kroutil, Angew. Chem., Int. Ed., 2017, 56, 7615–7619 CrossRef CAS PubMed.
- A. Zadło-Dobrowolska, N. G. Schmidt and W. Kroutil, Chem. Commun., 2018, 54, 3387–3390 RSC.
- H. Müller, S. P. Godehard, G. J. Palm, L. Berndt, C. P. S. Badenhorst, A. Becker, M. Lammers and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 2013–2017 CrossRef PubMed.
- I. Mathews, M. Soltis, M. Saldajeno, G. Ganshaw, R. Sala, W. Weyler, M. A. Cervin, G. Whited and R. Bott, Biochemistry, 2007, 46, 8969–8979 CrossRef CAS PubMed.
- H. Land, P. Hendil-Forssell, M. Martinelle and P. Berglund, Catal. Sci. Technol., 2016, 6, 2897–2900 RSC.
- M. L. Contente, A. Pinto, F. Molinari and F. Paradisi, Adv. Synth. Catal., 2018, 360, 4814–4819 CrossRef CAS.
- M. L. Contente, S. Farris, L. Tamborini, F. Molinari and F. Paradisi, Green Chem., 2019, 21, 3263–3266 RSC.
- M. L. Contente, D. Roura-Padrosa, F. Molinari and F. Paradisi, Nat. Catal., 2020, 3, 1020–1026 CrossRef CAS.
- M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS.
- A. Baldessari and C. P. Mangone, J. Mol. Catal. B: Enzym., 2001, 11, 335–341 CrossRef CAS.
- P. G. Quintana, G. García Liñares, S. N. Chanquia, R. M. Gorojod, M. L. Kotler and A. Baldessari, Eur. J. Org. Chem., 2016, 2016, 518–528 CrossRef CAS.
- R. Irimescu and K. Kato, Tetrahedron Lett., 2004, 45, 523–525 CrossRef CAS.
- D. Manova, F. Gallier, L. Tak-Tak, L. Yotava and N. Lubin-Germain, Tetrahedron Lett., 2018, 59, 2086–2090 CrossRef CAS.
- S. Zeng, J. Liu, S. Anankanbil, M. Chen, Z. Guo, J. P. Adams, R. Snajdrova and Z. Li, ACS Catal., 2018, 8, 8856–8865 CrossRef CAS.
- J. Pitzer, K. Steiner, C. Schmid, V. K. Schein, C. Prause, C. Kniely, M. Reif, M. Geier, E. Pietrich, T. Reiter, P. Selig, C. Stückler, P. Pöchlauer, G. Steinkellner, K. Gruber, H. Schwab, A. Glieder and W. Kroutil, Green Chem., 2022, 24, 5171–5180 RSC.
- M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- M. Winn, S. M. Richardson, D. J. Campopiano and J. Micklefield, Curr. Opin. Chem. Biol., 2020, 55, 77–85 CrossRef CAS PubMed.
- J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32–46 CrossRef CAS PubMed.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- T. Abe, Y. Hashimoto, S. Sugimoto, K. Kobayashi, T. Kumano and M. Kobayashi, J. Antibiot., 2017, 70, 435–442 CrossRef CAS PubMed.
- R. Hara, K. Hirai, S. Suzuki and K. Kino, Sci. Rep., 2018, 8, 1–8 Search PubMed.
- M. Z. Abidin, T. Saravanan, E. Strauss and G. J. Poelarends, Org. Biomol. Chem., 2021, 19, 4515–4519 RSC.
- C. M. Lelièvre, M. Balandras, J. L. Petit, C. Vergne-Vaxelaire and A. Zaparucha, ChemCatChem, 2020, 12, 1184–1189 CrossRef.
- M. Zhu, L. Wang and J. He, Angew. Chem., Int. Ed., 2021, 60, 2030–2035 CrossRef CAS PubMed.
- P. M. Marchetti, S. Richardson, N. M. K. Mohamed Abdelmegid and D. J. Campopiano, MedChemComm, 2019, 10, 1192–1196, 10.1039/C9MD00063A.
- D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner and D. Leys, Nat. Chem. Biol., 2017, 13, 975–981 CrossRef CAS PubMed.
- M. Winkler and J. G. Ling, ChemCatChem, 2022, 14(19), e202200441, DOI:10.1002/cctc.202200441.
- A. J. L. Wood, N. J. Weise, J. D. Frampton, M. S. Dunstan, M. A. Hollas, S. R. Derrington, R. C. Lloyd, D. Quaglia, F. Parmeggiani, D. Leys, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2017, 56, 14498–14501 CrossRef CAS PubMed.
- M. Lubberink, C. Schnepel, J. Citoler, S. R. Derrington, W. Finnigan, M. A. Hayes, N. J. Turner and S. L. Flitsch, ACS Catal., 2020, 10, 10005–10009 CrossRef CAS.
- M. Lubberink, W. Finnigan, C. Schnepel, C. R. Baldwin, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2022, 61, e202205054 CrossRef CAS PubMed.
- M. R. Petchey, B. Rowlinson, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, ACS Catal., 2020, 10, 4659–4663 CrossRef CAS PubMed.
- M. Winn, M. Rowlinson, F. Wang, L. Bering, D. Francis, C. Levy and J. Micklefield, Nature, 2021, 593, 391–398 CrossRef CAS PubMed.
- C. Ji, Q. Chen, Q. Li, H. Huang, Y. Song, J. Ma and J. Ju, Tetrahedron Lett., 2014, 55, 4901–4904 CrossRef CAS.
- M. Petchey, A. Cuetos, B. Rowlinson, S. Dannevald, A. Frese, P. W. Sutton, S. Lovelock, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, Angew. Chem., Int. Ed., 2018, 11584–11588 CrossRef CAS PubMed.
- S. Mordhorst, A. Maurer, D. Popadić, J. Brech and J. N. Andexer, ChemCatChem, 2017, 9, 4164–4168 CrossRef CAS.
- H. K. Philpott, P. J. Thomas, D. Tew, D. E. Fuerst and S. L. Lovelock, Green Chem., 2018, 20, 3426–3431 RSC.
- C. Schnepel, L. R. Pérez, Y. Yu, A. Angelastro, R. S. Heath, M. Lubberink, F. Falcioni, K. Mulholland, M. A. Hayes, N. J. Turner and S. L. Flitsch, Nat. Catal., 2023, 6, 89–99, DOI:10.1038/s41929-022-00889-x.
- Y. Ogasawara and T. Dairi, Chem. – Eur. J., 2017, 23, 10714–10724 CrossRef CAS PubMed.
- T. Arai, Y. Arimura, S. Ishikura and K. Kino, Appl. Environ. Microbiol., 2013, 79, 5023–5029 CrossRef CAS PubMed.
- K. Tabata, H. Ikeda and S. Hashimoto, J. Bacteriol., 2005, 187, 5195–5202 CrossRef CAS PubMed.
- S. Prasad and T. C. Bhalla, Biotechnol. Adv., 2010, 28, 725–741 CrossRef CAS PubMed.
- M. Kobayashi and S. Shimizu, Nat. Biotechnol., 1998, 16, 733–736 CrossRef CAS PubMed.
- Z. Cheng, Y. Xia and Z. Zhou, Front. Bioeng. Biotechnol., 2020, 8, 1–18 CrossRef PubMed.
- Y. Tian, J. Chen, H. Yu and Z. Shen, J. Microbiol. Biotechnol., 2016, 26, 337–346 CrossRef CAS PubMed.
- S. Hourai, M. Miki, Y. Takashima, S. Mitsuda and K. Yanagi, Biochem. Biophys. Res. Commun., 2003, 312, 340–345 CrossRef CAS PubMed.
- Y. Yamanaka, Y. Kato, K. Hashimoto, K. Iida, K. Nagasawa, H. Nakayama, N. Dohmae, K. Noguchi, T. Noguchi, M. Yohda and M. Odaka, Angew. Chem., Int. Ed., 2015, 54, 10763–10767 CrossRef CAS PubMed.
- J. Liu, H. Yu and Z. Shen, J. Mol. Graphics Modell., 2008, 27, 529–535 CrossRef CAS PubMed.
- M.-X. Wang, Acc. Chem. Res., 2015, 48, 602–611 CrossRef CAS PubMed.
- A. R. Mashweu, V. P. Chhiba-Govindjee, M. L. Bode and D. Brady, Molecules, 2020, 25, 1–15 CrossRef PubMed.
- L. Bering, E. J. Craven, S. A. Sowerby Thomas, S. A. Shepherd and J. Micklefield, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Á. Mourelle-Insua, L. A. Zampieri, I. Lavandera and V. Gotor-Fernández, Adv. Synth. Catal., 2018, 360, 686–695 CrossRef.
- L. M. Lima, B. N. M. da Silva, G. Barbosa and E. J. Barreiro, Eur. J. Med. Chem., 2020, 208, 112829 CrossRef CAS PubMed.
- B. O. Bachmann, R. Li and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9082–9086 CrossRef CAS PubMed.
- P. L. Roach, I. J. Clifton, C. M. H. Hensgens, N. Shibata, C. J. Schofield, J. Hajdu and J. E. Baldwin, Nature, 1997, 387, 827–830 CrossRef CAS PubMed.
- J. W. Labonte, F. Kudo, M. F. Freeman, M. L. Raber and C. A. Townsend, MedChemComm, 2012, 3, 960–966 RSC.
- B. Gerratana, A. Stapon and C. A. Townsend, Biochemistry, 2003, 42, 7836–7847 CrossRef CAS PubMed.
- R. B. Hamed, J. R. Gomez-Castellanos, L. Henry, S. Warhaut, T. D. W. Claridge and C. J. Schofield, Commun. Chem., 2019, 2, 1–10 CrossRef CAS.
- T. Wakimoto, T. Mori, H. Morita and I. Abe, J. Am. Chem. Soc., 2011, 133, 4746–4749 CrossRef CAS PubMed.
- A. L. Gutman, E. Meyer, X. Yue and C. Abell, Tetrahedron Lett., 1992, 33, 3943–3946 CrossRef CAS.
- E. Stavila and K. Loos, Tetrahedron Lett., 2013, 54, 370–372 CrossRef CAS.
- N. J. Willis, C. A. Fisher, C. M. Alder, A. Harsanyi, L. Shukla, J. P. Adams and G. Sandford, Green Chem., 2016, 18, 1313–1318 RSC.
- D. Koszelewski, D. Clay, K. Faber and W. Kroutil, J. Mol. Catal. B: Enzym., 2009, 60, 191–194 CrossRef CAS.
- C. K. Chung, P. G. Bulger, B. Kosjek, K. M. Belyk, N. Rivera, M. E. Scott, G. R. Humphrey, J. Limanto, D. C. Bachert and K. M. Emerson, Org. Process Res. Dev., 2014, 18, 215–227 CrossRef CAS.
- M. D. Truppo, J. David Rozzell and N. J. Turner, Org. Process Res. Dev., 2010, 14, 234–237 CrossRef CAS.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS PubMed.
- Z. Qin, X. Zhang, X. Sang, W. Zhang, G. Qu and Z. Sun, Green Synth. Catal., 2022, 3(3), 294–297, DOI:10.1016/j.gresc.2022.05.009.
- S. Sarak, T. P. Khobragade, H. Jeon, A. D. Pagar, P. Giri, S. Lee and H. Yun, Green Chem., 2021, 23, 9447–9453 RSC.
- M. S. Christodoulou, M. L. Contente, S. Dallavalle and A. Pinto, Green Chem., 2022, 24, 4432–4436 RSC.
- L. Zhang, F. Li, C. Wang, L. Zheng, Z. Wang, R. Zhao and L. Wang, Catalysts, 2017, 7, 115 CrossRef.
- S. V. Athavale, S. Gao, Z. Liu, S. C. Mallojjala, J. S. Hirschi and F. H. Arnold, Angew. Chem., Int. Ed., 2021, 60, 24864–24869 CrossRef CAS PubMed.
- X. Ren, A. L. Chandgude and R. Fasan, ACS Catal., 2020, 10, 2308–2313 CrossRef CAS PubMed.
- Y. Yang and F. H. Arnold, Acc. Chem. Res., 2021, 54, 1209–1225 CrossRef CAS PubMed.
- H. Renata, Z. J. Wang, R. Z. Kitto and F. H. Arnold, Catal. Sci. Technol., 2014, 4, 3640–3643 RSC.
- Z. J. Wang, H. Renata, N. E. Peck, C. C. Farwell, P. S. Coelho and F. H. Arnold, Angew. Chem., Int. Ed., 2014, 53, 6810–6813 CrossRef CAS PubMed.
- O. F. Brandenberg, C. K. Prier, K. Chen, A. M. Knight, Z. Wu and F. H. Arnold, ACS Catal., 2018, 8, 2629–2634 CrossRef CAS.
Footnote |
| † Present address: Wageningen Food & Biobased Research, Wageningen, P. O. Box 17, 6700 AA, The Netherlands. |
| This journal is © The Royal Society of Chemistry 2023 |