
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective separation of planar and non-planar hydrocarbons using an aqueous Pd6 interlocked cage†
Debsena
Chakraborty
 a,
Rupak
Saha
a,
Jack K.
Clegg
b and
Partha Sarathi
Mukherjee
*a
a,
Rupak
Saha
a,
Jack K.
Clegg
b and
Partha Sarathi
Mukherjee
*a
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: psm@iisc.ac.in
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, Queensland 4072, Australia
First published on 14th September 2022
Abstract
Polycyclic aromatic hydrocarbons (PAHs) find multiple applications ranging from fabric dyes to optoelectronic materials. Hydrogenation of PAHs is often employed for their purification or derivatization. However, separation of PAHs from their hydrogenated analogues is challenging because of their similar physical properties. An example of such is the separation of 9,10-dihydroanthracene from phenanthrene/anthracene which requires fractional distillation at high temperature (∼340 °C) to obtain pure anthracene/phenanthrene in coal industry. Herein we demonstrate a new approach for this separation at room temperature using a water-soluble interlocked cage (1) as extracting agent by host–guest chemistry. The cage was obtained by self-assembly of a triimidazole donor L·HNO3 with cis-[(tmeda)Pd(NO3)2] (M) [tmeda = N,N,N′,N′-tetramethylethane-1,2-diamine]. 1 has a triply interlocked structure with an inner cavity capable of selectively binding planar aromatic guests.
Introduction
Selective sequestration of substrates is a crucial step for many enzymatic reactions and biological processes.1 In such processes, enzymes or other proteins show highly specific binding of guests within suitable pockets using mechanisms such as Fischer's lock and key2 or Koshland's induced fit.3 Inspired by such systems, multiple artificial analogues have been designed to mimic the binding property of naturally occurring host molecules.4 These artificial analogues can be broadly classified into macromolecular hosts and supramolecular or molecular hosts.5 Macromolecular hosts include porous covalent and coordination frameworks which often have very low solubility in common solvents. This leads to a loss of solution-processability and restricts their uses.5 Other molecular hosts, such as porous organic cages,6 mostly exhibit selective guest uptake in their solid or crystalline state. This limits their application to uptake of only gases and low-boiling liquids, and therefore, selective uptake between solid substrates is rarely achieved.7 Supramolecular hosts are formed by the self-assembly of multiple subunits. Such hosts, like the ones formed by metal–ligand coordination,8–11 have multiple advantages such as synthetic ease and solubility in common solvents, including water. Specifically, water-soluble hosts have a high affinity for guest encapsulation due to multiple hydrophobic interactions.12 Owing to these advantages, self-assembled coordination systems have been used for multiple applications ranging from light-harvesting,13 catalysis,14 sensing,15 separation,16 and stabilization of reactive intermediates.17Polycyclic aromatic hydrocarbons (PAH) are common planar compounds which are ideal candidates for fabricating electronic and optical devices because of their rigid, planar, and conjugated structure.18 Although important, such molecules are micropollutants in water18 and hence their segregation in water is environmentally beneficial.16,19 The hydrogenation of such PAHs is often employed for making derivatives to tune the optoelectronic properties of these molecules. However, such hydrogenated products have similar physical properties as their parent PAHs. For example, benzene and cyclohexane have similar boiling points of ca. 80 °C; naphthalene and 1,2,3,4-tetrahydronaphthalene have boiling points of 218 °C and 207 °C, respectively, and thus cannot be separated by simple distillation. Similarly, anthracene (A) obtained naturally in coal tar is mixed with its isomer phenanthrene (P), and carbazole. To obtain pure anthracene from this crude, first hydrogenation is carried out to predominantly form 9,10-dihydroanthracene (H2A) and unreacted phenanthrene and carbazole. Carbazole can be easily separated from the mixture because of its difference in solubility. However, to separate phenanthrene and 9,10-dihydroanthracene, high temperature (above 340 °C) fractional distillation is required, followed by recrystallization to get highly pure 9,10-dihydroanthracene which is then aromatized to obtain pure anthracene.20 This process is highly energy-intensive and inefficient. This same separation is further complicated by the fact that 9,10-dihydroanthracene and anthracene have identical Rf values in most solvents and thus chromatographic techniques are not useful specially in the milligram to gram scale range. Thus, an alternate strategy for the separation of 9,10-dihydroanthracene from its planar analogue anthracene and its isomer phenanthrene is highly desirable in coal industries.
To achieve such selective separation through host–guest chemistry, a host is required which can selectively encapsulate planar hydrocarbons. Many synthetic water-soluble host molecules are unsuitable for the selective encapsulation of planar hydrocarbons due to their large size and shape.7,16,21 Therefore, we shifted our interest towards molecular interlocked materials (MIMs) as host molecules. These compounds are formed by the interlocking or intertwining of molecular fragments so that the fragments cannot be separated from one another without breaking the whole molecule.22 However, because of the interpenetrated nature of such structures, they are often incapable of guest encapsulation.22 Furthermore, many metal–ligand coordination based interlocked systems are formed through host–guest interaction23 or by templation24 of molecular fragments and are stable only in the presence of such guests.24 Thus, these systems do not exhibit any active cavity for the uptake and release of guest molecules. Probably, it is because of this reason that most interlocked cages are revered for their difficulty in synthesis and aesthetic beauty, but their application is still underexplored.
Herein, we report the synthesis of a water-soluble triply interlocked cage 1, formed by metal–ligand coordination-driven self-assembly of guanidine-based triimidazole ligand L·HNO3 with a cis-blocked Pd(II) acceptor M (Scheme 1). Owing to the shape of the guanidine core, interlocked cage 1 had an inner cavity with dimensions suitable for guest uptake. 1 facilitated the uptake and release of planar guest molecules without disrupting the interlocked structure. Further investigations revealed that 1 showed selective uptake of planar aromatic guests over their hydrogenated non-planar analogues. Through this, efficient separation of 9,10-dihydroanthracene (H2A) from anthracene (A) and phenanthrene (P) could also be achieved. To the best of our knowledge, the present results demonstrate for the first time an example of a water-soluble interlocked molecular host capable of showing guest encapsulation and de-encapsulation, and challenging separation of phenanthrene/anthracene from non-planar 9,10-dihydroanthracene by simple aqueous extraction at ambient conditions.
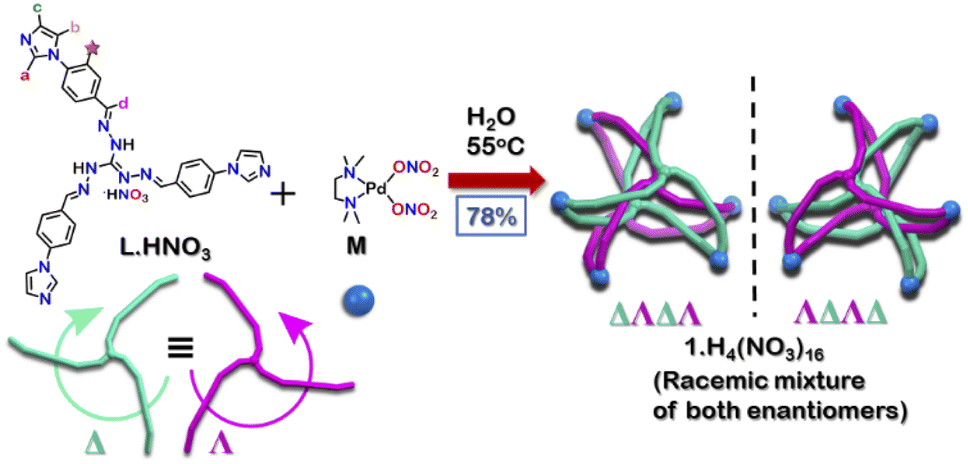 | ||
| Scheme 1 Synthesis of interlocked Pd6 cage 1 using a triimidazole donor L·HNO3. | ||
Results and discussion
Synthesis and characterization
The tri-imidazole ligand L·HNO3 was synthesized by the condensation of 4-(1H-imidazol-1-yl)benzaldehyde and triaminoguanidinium nitrate in a ethanol/water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture in good yield (Scheme S1†).25 The product was characterized by ESI-MS in methanol and multinuclear NMR in DMSO-d6 (Fig. S4–S7†). To get the interlocked cage 1, L·HNO3 was self-assembled with a cis-blocked 90° Pd(II) acceptor M in a 2:3 ratio at 55 °C in water for 24 hours (Scheme 1). It gave a turbid orange solution which was then centrifuged to obtain the aqueous solution of 1. Slow vapor diffusion of acetone into the aqueous solution of the product yielded pure crystals of 1.
:1) mixture in good yield (Scheme S1†).25 The product was characterized by ESI-MS in methanol and multinuclear NMR in DMSO-d6 (Fig. S4–S7†). To get the interlocked cage 1, L·HNO3 was self-assembled with a cis-blocked 90° Pd(II) acceptor M in a 2:3 ratio at 55 °C in water for 24 hours (Scheme 1). It gave a turbid orange solution which was then centrifuged to obtain the aqueous solution of 1. Slow vapor diffusion of acetone into the aqueous solution of the product yielded pure crystals of 1.
The 1H NMR spectrum of 1 in D2O at room temperature revealed the presence of seven distinct peaks in the aromatic region (Fig. 1 and S13†). Quite surprisingly, the ligand L·HNO3 had only six peaks (Fig. S4†) in its NMR spectrum. The presence of an extra peak in the self-assembled product suggested the formation of either an irregular structure or multiple self-assembled products. However, the 1H DOSY NMR spectrum showed that all the peaks correspond to same diffusion coefficient [D = 1.58 × 10−10 m2 s−1 (logD = −9.8)] (Fig. 1 and S20†), which confirmed the formation of a single self-assembled product.
To better understand the stoichiometry of the self-assembled product, ESI-MS spectrum of the PF6− analogue was recorded in acetonitrile. The spectrum showed multiple peaks at m/z = 1635.957, 1190.222, 923.390 due to the charged fragments [M6L4(PF6)9]3+, [M6L4(PF6)8]4+ and [M6L4(PF6)7]5+ (Fig. S21 and S22†). This confirmed a [6 + 4] self-assembly of the ditopic acceptor (M) and tridentate planar donor (L). Such a combination of the donor and acceptor may yield either an interlocked structure or a double-square or an octahedral architecture.26
To understand the exact nature of architecture formed, 1H–1H COSY, NOESY and 1H–13C HSQC NMR of 1 were recorded in D2O (Fig. S16–S19†) and compared with that for L·HNO3 in DMSO-d6 (Fig. S5–S7†). Detailed analysis showed that the two most downfield shifted proton peaks in the 1H NMR spectrum of 1 at 9.10 and 8.87 ppm are that from the α-imidazole protons in L·HNO3 (Fig. 1). The integration of these two peaks showed that they were in a 1:1 ratio (Fig. S13†). If the architecture was a double-square, the α-imidazole peaks are expected to split in a 1:2 ratio. While if the architecture is an interlocked cage, the α-imidazole peaks are expected to split in a 1:1 ratio as observed.27
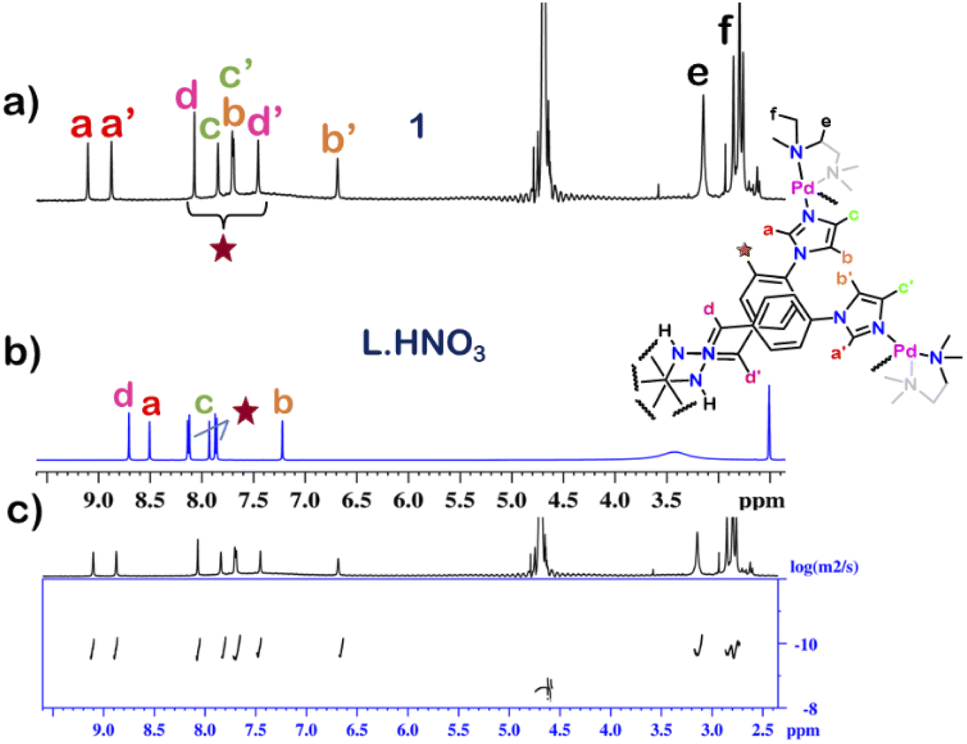 | ||
| Fig. 1 Stacked 1H NMR spectra of the (a) cage 1 in D2O, (b) ligand L·HNO3 in DMSO-d6 and (c) diffusion-ordered 1H NMR of 1 in D2O. | ||
The structure was unequivocally established by single-crystal XRD (Fig. 2 and S1–S3†). Diffraction quality single crystals of 1 were grown by the slow vapour diffusion of acetone into a concentrated aqueous solution of 1 (with NO3− as the counterion). X-ray diffraction data were collected by using a synchrotron radiation source.281 crystalized in the trigonal space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1 as a triply interlocked M6L4 cage. Unlike the known M6L4 interlocked cages,231 has an asymmetrical disposition of the ligands with an easily assessable inner cavity. The crystal structure of 1 revealed that the interplanar distances between the different ligands are 3.3 Å in the periphery and 6.3 Å in the centre. The distances between two adjacent Pd centres are 9.6 Å and 13.2 Å (Fig. 2). These dimensions indicate that 1 has an internal cavity big enough to accommodate guest molecules.27 This was unique as interlocked structures generally do not have internal cavity big enough for guest encapsulation.29
c1 as a triply interlocked M6L4 cage. Unlike the known M6L4 interlocked cages,231 has an asymmetrical disposition of the ligands with an easily assessable inner cavity. The crystal structure of 1 revealed that the interplanar distances between the different ligands are 3.3 Å in the periphery and 6.3 Å in the centre. The distances between two adjacent Pd centres are 9.6 Å and 13.2 Å (Fig. 2). These dimensions indicate that 1 has an internal cavity big enough to accommodate guest molecules.27 This was unique as interlocked structures generally do not have internal cavity big enough for guest encapsulation.29
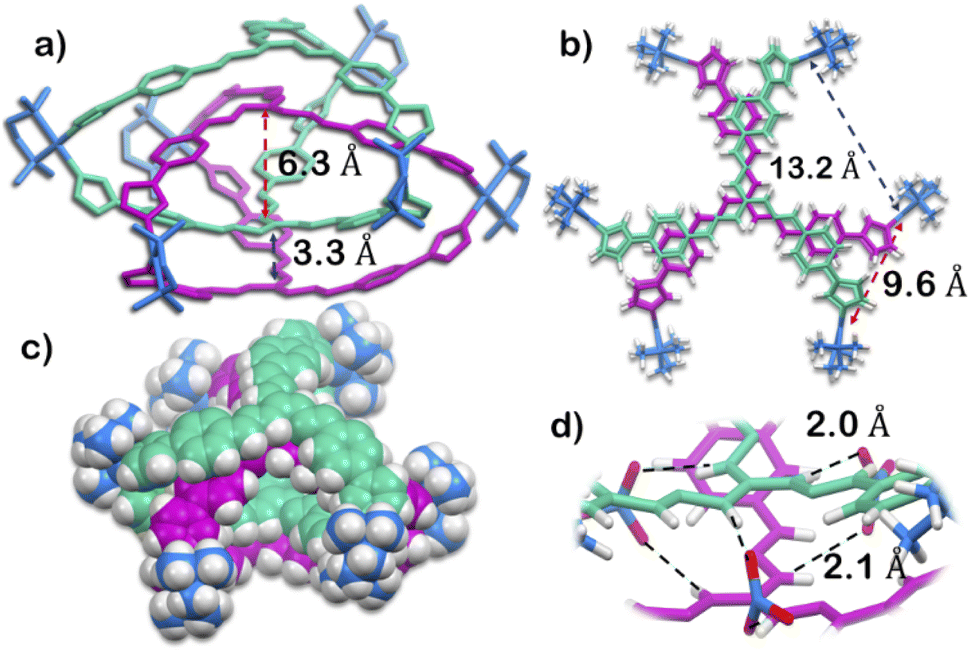 | ||
| Fig. 2 X-Ray crystal structure of 1. (a) Side view showing the inter-ligand distance of the interior cavity (H atoms and anions removed for clarity). (b) Top view showing the inter Pd distance (anions removed for clarity). (c) Side view (space fill model; anions removed for clarity). (d) Nitrate binding ability of 1 using the guanidine core. [Colour scheme: H, white; O, red; N of nitrate, blue; C, N of ligand with Δ orientation, green; C, N of ligand with Λ orientation, violet; C, N, Pd of acceptor unit, blue]. | ||
Owing to the propeller shape of the guanidine core, the interlocked cage also has a helical structure with two different orientations (Δ and Λ) (Scheme 1). The crystal structure shows that each cage is composed by the interlocking of two M3L2 cage subunits. Where one cage subunit has both ligands in the Δ orientation, and the other subunit is an isomeric cage, where both the ligands are oriented in a Λ orientation. Each subunit could thus be termed as either a ΔΔ cage or a ΛΛ cage. The M3L2 subunit (ΔΔ or ΛΛ cage) is achiral due to the presence of a plane of symmetry perpendicular to the C3 principal axis (Fig. S2†). Interestingly, due to interlocking this plane of symmetry is lost and the resultant interlocked cage 1 is chiral. Using the same notation as that of the subunits, these two enantiomers can be termed as ΔΛΔΛ or ΛΔΛΔ (Fig. 3). The crystal structure of 1 also shows that in each unit cell four molecules of 1 are present, of which two are the ΔΛΔΛ isomers and the other two are the ΛΔΛΔ isomers (Fig. S1†). This however meant that 1 is a racemic mixture containing equal amount of both the isomeric cages. The chirality of the interlocked cage 1, where the interlocking of achiral subunits resulted in the formation of a chiral interlocked structure, is noteworthy.
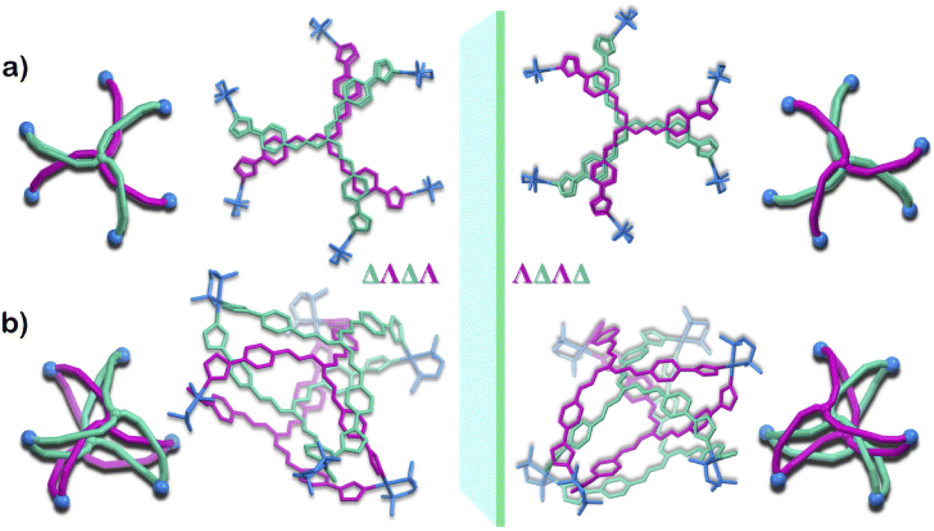 | ||
| Fig. 3 Crystal structure of 1 showing the enantiomeric relation of the two isomers ΔΛΔΛ and ΛΔΛΔ, a graphical representation is given alongside the crystal structure. (a) Top view (b) side view (H atoms and anions omitted for clarity). [Colour codes: C, N of ligand with Δ orientation, green; C, N of ligand with Λ orientation, violet; C, N, Pd of acceptor unit, blue.]. | ||
Finally, the crystal structure also showed the hydrogen bonding capability of the cage 1. Two N–H groups in the guanidine core of two ligand participate in H-bonding interaction with one nitrate ion. This nitrate ion has an orthogonal orientation to the two ligands. As there are three N–H groups in a ligand, and 1 comprised of four ligands, it participates in H-bonding interaction with six nitrate ions (Fig. 2 and S2†). Such H-bonding was also a unique consequence of the interlocked structure of 1.
Although cage 1 is robust and stable to other organic solvents. The cage formation reaction only occurs in water. In other solvents like DMSO or acetonitrile multiple polymeric or oligomeric species were formed. Similarly, use of acceptor (M) with other counterions such as PF6− or triflate led to the formation of multiple species which could not be characterised by 1H NMR or ESI-MS. Thus, the H-bonding interaction with NO3− coupled with the hydrophobic effect arising due to the use of aqueous medium, is the most probable driving force behind the formation of this triply interlocked cage (1) in water.
Guest encapsulation studies
From the crystal structure, 1 is expected to have two different guest binding sites. One is interior binding of guest into the inner cavity of 1. The other is exterior binding using H-bonding interactions as shown for nitrate ions. To investigate the guest binding abilities of 1, 4-chloro-β-nitrostyrene (Ns) was taken as a model guest molecule. Ns has a nitro group which could participate in H-bonding like the nitrate ion. It also has a hydrophobic styrene moiety which could favour encapsulation inside 1. To check this, excess (ca. 5 equivalents) of solid Ns was added to a solution of 1 in D2O. This mixture was stirred for 12 hours and then centrifuged to remove excess of Ns. This solution was then used to characterise the host–guest complex Ns⊂1. The 1H NMR of Ns⊂1 showed new peaks in 6–7 ppm region (Fig. 4 and S23†), with such a shift of the guest peaks characteristic of interior binding. Further proof of binding was obtained from the 1H DOSY spectrum of Ns⊂1 in D2O. It showed that all peaks correspond to a single diffusion band (logD = −10) (Fig. S24†). However, because of the broad nature of the guest peaks, host–guest stoichiometry could not be accurately determined. We thus investigated the host–guest behaviour with other planar guests.
It was found through 1H NMR analysis that 1 was able to encapsulate many polycyclic aromatic hydrocarbons (PAHs) like pyrene, phenanthrene (P) and anthracene (A) (Fig. 4 and S25†). Much like with Ns, it was seen that for both P⊂1 and A⊂1, encapsulation took place in the inner cavity of 1. All the 1H NMR peaks for P⊂1 had a single diffusion coefficient (logD = −9.8) (Fig. S26†) and 1H NOESY spectrum showed correlation between host and guest peaks (Fig. S27†), both characteristic for interior guest encapsulation. Similar results were also obtained for A⊂1 (Fig. S28 and S29†). However, as 1 is chiral encapsulation of the guest inside 1 led to loss of symmetry of the guest and several new peaks for the host–guest complex are observed. To accurately determine the host–guest ratio, titration experiments were carried out for A⊂1 and P⊂1.
To perform the 1H NMR titration a stock solution of 1 was prepared in 0.5 mL D2O with tetrabutylammonium nitrate [(N(Bu)4)NO3] as the internal standard. As both P and A are insoluble in water, stock solutions of P in CD3OD (0.017 M) and A in DMSO-d6 (0.017 M) were prepared. The 1H NMR titration was performed by adding aliquots of guest stock solution (10 μL) to the stock solution of 1. After each addition 1H NMR was recorded without any time delay. It was found that both the guests (A and P) were bound to 1 by slow exchange on the NMR time scale at 25 °C (Fig. S30 and S32†). Thus, the host–guest concentration [HG] could be determined by integration with respect to the internal standard. The plot of [HG] vs. equivalence of guest added showed that saturation in concentration of [HG] was achieved when ca. 1 equivalent of either A or P was added (Fig. S31 and S33†). From this we could roughly conclude that for both the systems A⊂1 and P⊂1, the host to guest stoichiometry is 1:1. From the 1H NMR titration, the apparent association constant could also be calculated using the Hill equation.30 The apparent association constants for the formation of A⊂1 and P⊂1 were found to be 9.03 × 102 M−1 (Fig. S31†) and 6.42 × 103 M−1 (Fig. S33†), respectively.31
The guest binding ability of 1 was unique and thus we wanted to check the importance of the interlocked structure towards this property. To check this we constructed a non-interlocked cage 2 using ligand L′.HNO3 with the same guanidine core (Fig. 5). Unlike L·HNO3 which had rigid imidazole donors, L′·HNO3 has flexible imidazole arms (Fig. S9–S11†). Self-assembly of L′·HNO3 with acceptor M in a 2:3 ratio in water gave an orange solution. Complex 2 was first characterized by 1H NMR (Fig. 5 and S34†), which showed the presence of six peaks in the aromatic region with an NMR pattern similar to that for L′·HNO3 in DMSO-d6. Absence of extra peak in the 1H NMR spectra indicated the formation of a non-interlocked structure. All the peaks of 2 correspond to the same diffusion coefficient (logD = −9.3) in the DOSY NMR which confirmed the formation of a single assembly.
To better understand the stoichiometry of the self-assembled product, ESI-MS spectrum of the PF6− analogue was recorded in acetonitrile. The spectrum showed multiple peaks at m/z = 1232.343, 773.240, 543.686 correspond to the charged fragments 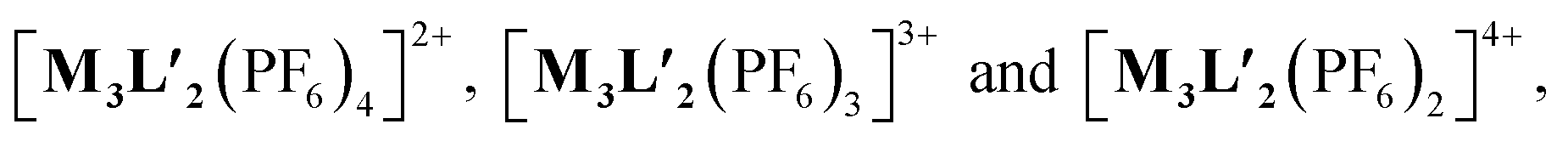 respectively (Fig. S38 and S39†). This confirmed that introduction of flexibility in the ligand backbone led to the formation of a
respectively (Fig. S38 and S39†). This confirmed that introduction of flexibility in the ligand backbone led to the formation of a 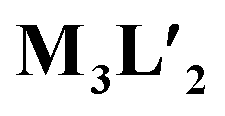 architecture. Complex 2 formed very tiny crystals upon slow vapor diffusion of acetone into a concentrated aqueous solution of the cage. These could not be used for X-ray structure elucidation, thus the structure of 2 was optimized semi-empirically using the PM6 model. The optimized structure showed that 2 has a large inner cavity with an inter ligand distance of 9.5 Å and a 21.9 Å distance between two Pd atoms (Fig. 5).
architecture. Complex 2 formed very tiny crystals upon slow vapor diffusion of acetone into a concentrated aqueous solution of the cage. These could not be used for X-ray structure elucidation, thus the structure of 2 was optimized semi-empirically using the PM6 model. The optimized structure showed that 2 has a large inner cavity with an inter ligand distance of 9.5 Å and a 21.9 Å distance between two Pd atoms (Fig. 5).
If the encapsulation behavior exhibited by 1 was a result of its ligand backbone, 2 is also expected to show same host–guest property. This however was not the case as 2 was found to show no affinity towards any guests (planar or non-planar). This proved that the interlock structure of 1 was pivotal for guest encapsulation. The inability of 2 to encapsulate any guest is probably because of its big pocket size which did not facilitate interactions strong enough to stabilize a guest inside its cavity.
Selective guest encapsulation: planar vs. non-planar
Although 1 could encapsulate planar hydrocarbons like anthracene and phenanthrene, it did not encapsulate molecules with adamantane or camphor moiety (Fig. 4). Such observation was very interesting and led to the idea that 1 could be used for the separation of planar and non-planar hydrocarbons. Such separation is highly challenging in coal industry, especially the separation of 9,10-dihydroanthracene (H2A) from its planar analogues anthracene (A) and its isomer phenanthrene (P). Further, small scale (milligram level) separation is even more difficult due to almost same Rf values of these compounds. To check this, first encapsulation of H2A by 1 was studied. Excess of solid H2A (ca. 5 equivalents) was added to a D2O solution of 1 and stirred for 12 hours. Then the solution was centrifuged to remove excess guests and the 1H NMR of the supernatant was recorded. The 1H NMR of H2A + 1 was found to be identical to that of 1 (Fig. 6). This proved that 1 did not encapsulate H2A.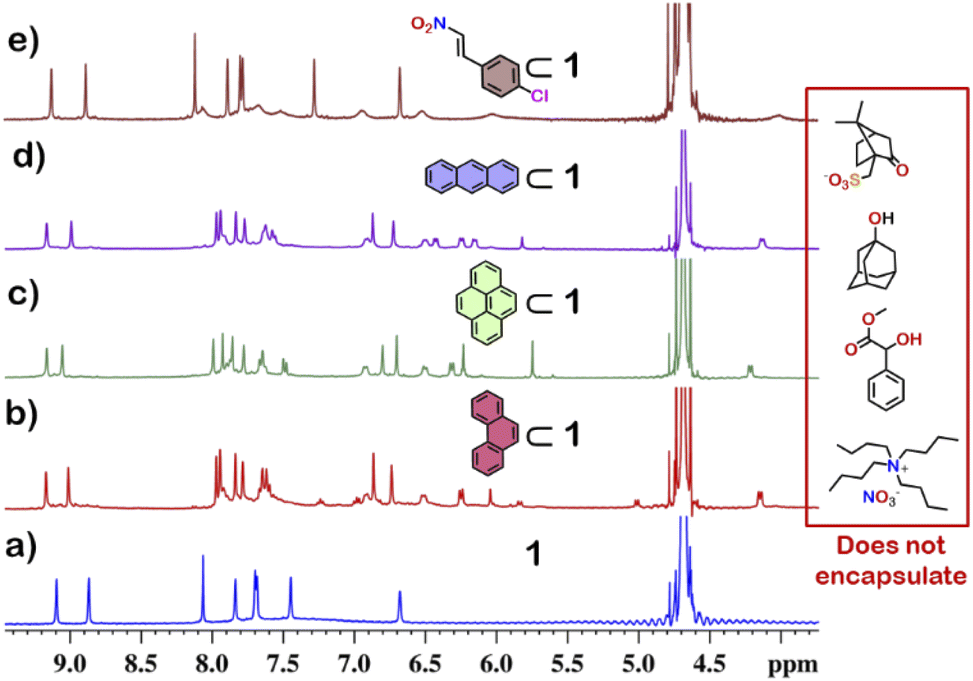 | ||
| Fig. 4 Partial 1H NMR stack plot of (a) 1, (b) P⊂1 (P: phenanthrene), (c) pyrene⊂1, (d) A⊂1 (A: anthracene) and (e) Ns⊂1 (Ns: (E)-1-chloro-4-(2-nitrovinyl)benzene) in D2O showing the change in NMR by guest encapsulation. (Inset) examples of non-planar guests that could not be encapsulated by 1. | ||
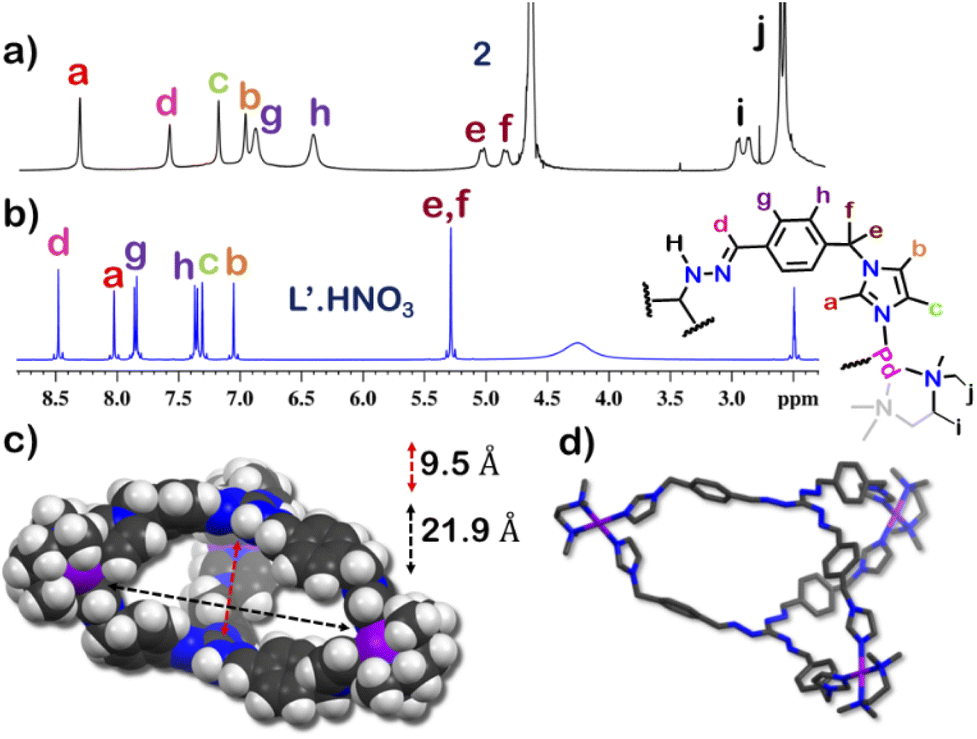 | ||
| Fig. 5 Stacked 1H NMR spectra of (a) cage 2 in D2O, (b) ligand L′.HNO3 in DMSO-d6 and (c) optimized structure of 2 (PM6 level), side view (space fill model) showing the inter ligand distance and inter Pd distance of the interior cavity. (d) Side view (capped-stick model) (H-atoms were removed for clarity) [colour scheme: H, white; C, black; N, blue; Pd, violet]. | ||
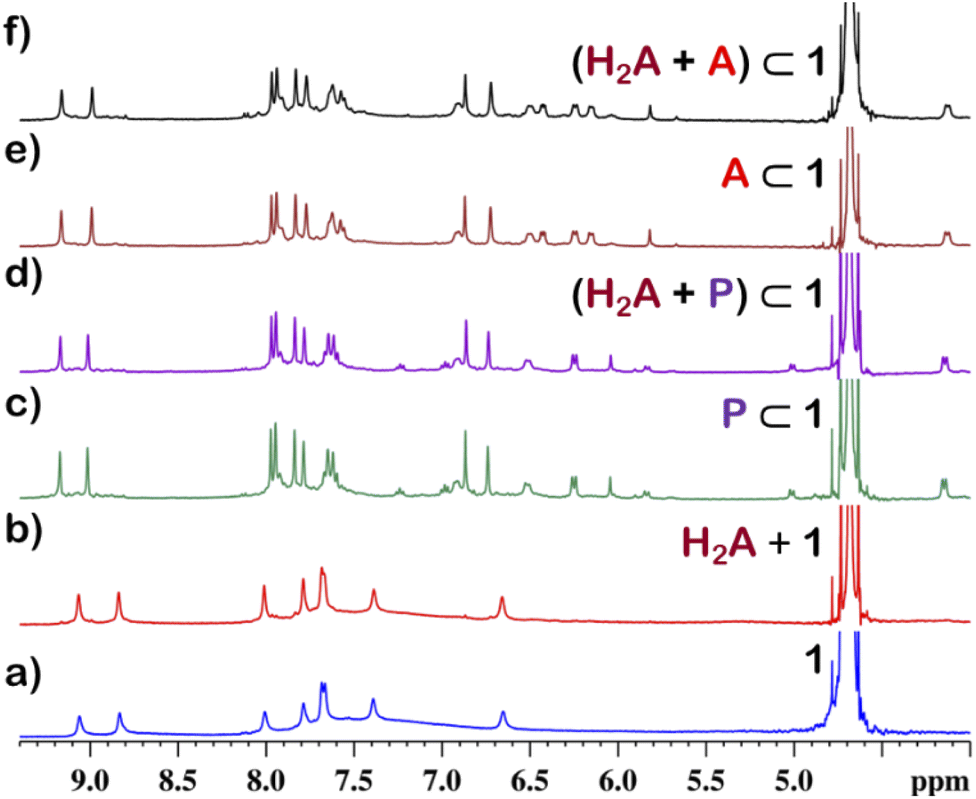 | ||
| Fig. 6 Partial 1H NMR stack plot of (a) 1, (b) (H2A + 1) (H2A = 9,10-dihydroanthracene), (c) P⊂1 (P: phenanthrene), (d) (H2A + P)⊂1, (e) A⊂1 (A: anthracene) and (f) (H2A + A)⊂1 in D2O showing the selective encapsulation of A and P. | ||
To examine the selective guest uptake, a solid mixture of equimolar H2A and A was added to a D2O solution of 1. The resultant solution was stirred for 12 hours and then centrifuged to remove excess guests (Scheme 2). The 1H NMR of the supernatant was identical to that of A⊂1 (Fig. 6 and S40†). This proved that from the mixture only anthracene was encapsulated selectively. Similar selective encapsulation of planar phenanthrene (P) was noticed when equimolar mixture of H2A and P was added to 1 (Fig. S41†). It also showed selective encapsulation of phenanthrene over 9,10-dihydroanthracene. To further verify this, the guest was extracted using chloroform. The extracted organic solvent was removed, and the resultant white solid was redissolved in 0.5 mL CDCl3. The 1H NMR of the extracted solid gave the peaks corresponding to only phenanthrene. This further verified the claim that in the host–guest complex only phenanthrene was present as guest (Fig. S42†).
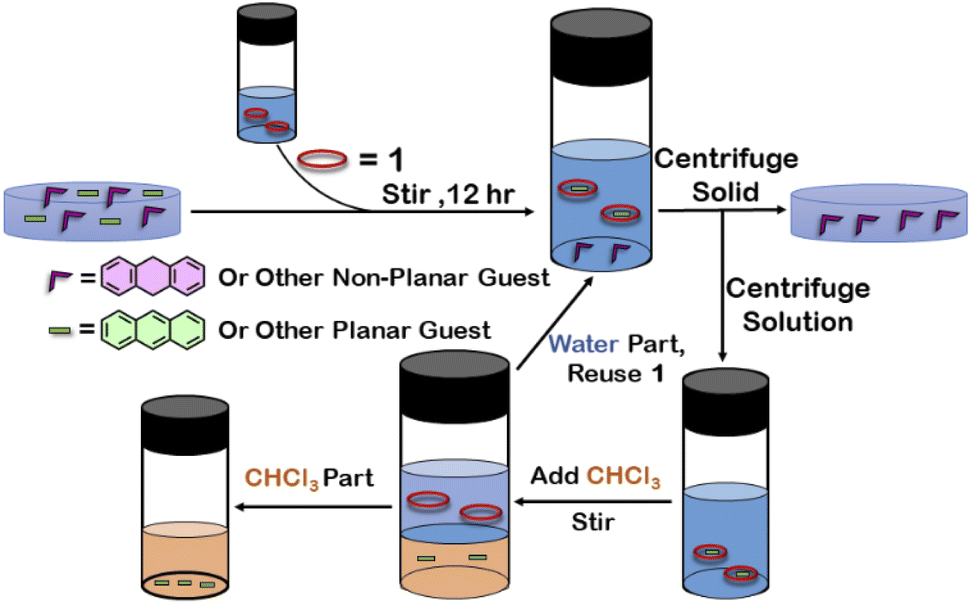 | ||
| Scheme 2 Schematic representation for the separation of planar and non-planar hydrocarbons achieved by employing host–guest interactions with 1. | ||
To check if 1 has a general tendency to separate planar and non-planar molecules. Selective encapsulation was carried out in the presence of two other non-planar guests, thianthrene (S2A) and N-methylphenothiazine (MP). Following the same experiments as employed for H2A, it was seen that both S2A and MP were not encapsulated by 1. Further selective encapsulation experiments showed that from an equimolar mixture of S2A and A, selectively only A was encapsulated (Fig. S43†). Similar results were also obtained for an equimolar mixture of MP and A (Fig. S44†). Titration of 1 with H2A, S2A and MP also showed no change in 1H NMR (Fig. S45–S47†) which further demonstrated the fact that 1 did not encapsulate these non-planar molecules. This clearly demonstrated that 1 successfully separated planar anthracene from its non-planar derivatives.
Conclusions
In conclusion, we have successfully synthesized a water-soluble interlocked cage (1) by metal–ligand coordination self-assembly of propeller shaped tridentate ligand L·HNO3 with cis-blocked Pd(II) 90° ditopic acceptor M. 1 has a triply interlocked structure with an internal cavity capable of guest uptake and release without damaging the integrity of the cage. This internal cavity was found to encapsulate planar aromatic guests like anthracene and phenanthrene. The inclusion complexes were characterized by several multinuclear NMR studies, and the host–guest ratio for the inclusion complexes was found to be 1:1. Further investigations revealed that this guest encapsulation ability was resulted from the interlocked structure, whereas a non-interlocked cage 2 with similar backbone did not show any guest encapsulation. Separation of planar polycyclic aromatic hydrocarbons from their non-planar hydrogenated analogues is a challenging problem in petrochemical industries, especially the separation of non-planar 9,10-dihydroanthracene from planar phenanthrene or anthracene by fractional distillation at high temperature. Surprisingly, the interlocked cage (1) was successful in the separation of such systems in water by selective encapsulation of planar molecules followed by their de-encapsulation using organic solvent at ambient condition. Such guest encapsulation and de-encapsulation by an interlocked cage 1 is an unusual observation as interlocked systems are either reluctant towards guests due to a lack of internal cavity or degrade if the guest is removed in the cases where the interlocked systems are formed in the presence of a guest as template. Our present investigations show a simple strategy for separating planar and non-planar hydrocarbons from their solid mixture by aqueous extraction using an interlocked cage as extracting agent at ambient condition. It also offers a new application of water-soluble interlocked cage apart from their aesthetic beauty.
Data availability
All data are provided in the ESI† and additional data can be available upon request.Author contributions
P. S. M and D. C. devised the project and designed the experiments. D. C. carried out the the experimental work together with R. S., and analyzed the data. J. K. C. collected and solved the crystallographic data. P. S. M. supervised the whole project. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. M. thanks the SERB (New Delhi, India) for financial support. D. C. is thankful to Dr A. Kumar and Dr P. Howlader for fruitful discussion, and Prime Minister Research Fellowship scheme (India) for fellowship. Part of this research was undertaken on the MX2 beamline at the Australian Synchrotron, part of ANSTO and made use of the ACRF detector. We thank the beamline staff for their support.References
- D. J. Cram and J. M. Cram, Science, 1974, 183, 803 CrossRef CAS PubMed.
- The Lock and Key Principle. The State of the Art-100 Years On, ed. J.-P. Behr, Wiley, New York, 1994 Search PubMed.
- D. E. Koshland Jr, Angew. Chem., Int. Ed. Engl., 1995, 33, 2375 CrossRef.
- (a) M. L. Merlau, M. del Pilar Mejia, S. T. Nguyen and J. T. Hupp, Angew. Chem., Int. Ed., 2001, 40, 4239 CrossRef CAS; (b) Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100 CrossRef CAS PubMed; (c) H.-J. Schneider and A. K. Yatsimirsky, Chem. Soc. Rev., 2008, 37, 263 RSC; (d) A. Chevalier, A. Osypenko, J.-M. Lehn and D. Meyer, Chem. Sci., 2020, 11, 11468 RSC; (e) J.-F. Ayme, S. Dhers and J.-M. Lehn, Angew. Chem., Int. Ed., 2020, 59, 12484 CrossRef CAS; (f) J.-F. Ayme, J.-M. Lehn, C. Bailly and L. Karmazin, J. Am. Chem. Soc., 2020, 142, 5819 CrossRef CAS PubMed; (g) M. Čonková, V. Montes-García, M. Konopka, A. Ciesielski, P. Samori and A. R. Stefankiewicz, Chem. Commun., 2022, 58, 5773 RSC.
- (a) K. Jie, Y. Zhou, E. Li and F. Huang, Acc. Chem. Res., 2018, 51, 2064 CrossRef CAS PubMed; (b) Y. Wu, J. Zhou, E. Li, M. Wang, K. Jie, H. Zhu and F. Huang, J. Am. Chem. Soc., 2020, 142, 19722 CrossRef CAS PubMed; (c) Y. Zhou, K. Jie, R. Zhao, E. Li and F. Huang, J. Am. Chem. Soc., 2020, 142, 6957 CrossRef CAS PubMed; (d) M. Kołodziejski, A. J. Brock, G. Kurpik, A. Walczak, F. Li, J. K. Clegg and A. R. Stefankiewicz, Inorg. Chem., 2021, 60, 9673 CrossRef; (e) A. Brzechwa-Chodzyńska, W. Drożdż, J. Harrowfield and A. R. Stefankiewicz, Coord. Chem. Rev., 2021, 434, 213820 CrossRef.
- (a) D. Chakraborty and P. S. Mukherjee, Chem. Commun., 2022, 58, 5558 RSC; (b) B. Hua, Y. Ding, L. O. Alimi, B. Moosa, G. Zhang, W. S. Baslyman, J. Sessler and N. M. Khashab, Chem. Sci., 2021, 12, 12286 RSC; (c) G. Zhang, Y. Ding, A. Hashem, A. Fakim and N. M. Khashab, Cell Rep. Phys. Sci., 2021, 2, 100470 CrossRef CAS; (d) Y. Ding, L. O. Alimi, B. Moosa, C. Maaliki, J. Jacquemin, F. Huang and N. M. Khashab, Chem. Sci., 2021, 12, 5315 RSC; (e) A. Dey, S. Chand, B. Maity, P. M. Bhatt, M. Ghosh, L. Cavallo, M. Eddaoudi and N. M. Khashab, J. Am. Chem. Soc., 2021, 143, 4090 CrossRef CAS PubMed.
- (a) Y. Liu, H. Wang, L. Shangguan, P. Liu, B. Shi, X. Hong and F. Huang, J. Am. Chem. Soc., 2021, 143, 3081 CrossRef CAS PubMed; (b) Y. Shi, K. Cai, H. Xiao, Z. Liu, J. Zhou, D. Shen, Y. Qiu, Q.-H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835 CrossRef CAS.
- (a) H. Wang, L.-P. Zhou, Y. Zheng, K. Wang, B. Song, X. Yan, L. Wojtas, X.-Q. Wang, X. Jiang, M. Wang, Q.-F. Sun, B. Xu, H.-B. Yang, A. C.-H. Sue, Y.-T. Chan, J. L. Sessler, Y. Jiao, P. J. Stang and X. Li, Angew. Chem., Int. Ed., 2021, 60, 1298 CrossRef CAS; (b) W. Tuo, Y. Sun, S. Lu, X. Li, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 16930 CrossRef CAS PubMed; (c) Y. Sun, C. Chen, J. Liu and P. J. Stang, Chem. Soc. Rev., 2020, 49, 3889 RSC; (d) L.-J. Chen, S. J. Humphrey, J.-L. Zhu, F.-F. Zhu, X.-Q. Wang, X. Wang, J. Wen, H.-B. Yang and P. A. Gale, J. Am. Chem. Soc., 2021, 143, 8295 CrossRef CAS PubMed; (e) W.-J. Li, X.-Q. Wang, D.-Y. Zhang, Y.-X. Hu, W.-T. Xu, L. Xu, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2021, 60, 18761 CrossRef CAS PubMed; (f) G. E. Sokolow, M. R. Crawley, D. R. Morphet, D. Asik, J. A. Spernyak, A. J. R. McGray, T. R. Cook and J. R. Morrow, Inorg. Chem., 2022, 61, 2603 CrossRef CAS PubMed.
- (a) R. Sumida, Y. Tanaka, K. Niki, Y. Sei, S. Toyota and M. Yoshizawa, Chem. Sci., 2021, 12, 9946 RSC; (b) Y. Katagiri, Y. Tsuchida, Y. Matsuo and M. Yoshizawa, J. Am. Chem. Soc., 2021, 143, 21492 CrossRef CAS; (c) Y. Domoto and M. Fujita, Coord. Chem. Rev., 2022, 466, 214605 CrossRef CAS; (d) Y. Domoto, M. Abe and M. Fujita, J. Am. Chem. Soc., 2021, 143, 8578 CrossRef CAS PubMed; (e) W. Xue, T. K. Ronson, Z. Lu and J. R. Nitschke, J. Am. Chem. Soc., 2022, 144, 6136 CrossRef CAS PubMed; (f) T. K. Ronson, J. P. Carpenter and J. R. Nitschke, Chem, 2022, 8, 557 CrossRef CAS.
- (a) C. Ngai, C. M. Sanchez-Marsetti, W. H. Harman and R. J. Hooley, Angew. Chem., Int. Ed., 2020, 59, 23505 CrossRef CAS; (b) A. J. Scott, J. Vallejo, A. Sarkar, L. Smythe, E. Regincós Martí, G. S. Nichol, W. T. Klooster, S. J. Coles, M. Murrie, G. Rajaraman, S. Piligkos, P. J. Lusby and E. K. Brechin, Chem. Sci., 2021, 12, 5134 RSC; (c) P. T. Blackburn, I. F. Mansoor, K. G. Dutton, A. M. Tyryshkin and M. C. Lipke, Chem. Commun., 2021, 57, 11342 RSC; (d) F. Sebastiani, T. A. Bender, S. Pezzotti, W.-L. Li, G. Schwaab, R. G. Bergman, K. N. Raymond, F. D. Toste, T. Head-Gordon and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 32954 CrossRef CAS PubMed; (e) S. Hasegawa, S. L. Meichsner, J. J. Holstein, A. Baksi, M. Kasanmascheff and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 9718 CrossRef CAS PubMed; (f) F. Picini, S. Schneider, O. Gavat, A. Vargas Jentzsch, J. Tan, M. Maaloum, J.-M. Strub, S. Tokunaga, J.-M. Lehn, E. Moulin and N. Giuseppone, J. Am. Chem. Soc., 2021, 143, 6498 CrossRef CAS PubMed.
- (a) X. Wang, Q. Su, Z. Zhang, J. Yang, Y. Zhang and M. Zhang, Chem. Commun., 2020, 56, 8460 RSC; (b) Y. Hou, Z. Zhang, S. Lu, J. Yuan, Q. Zhu, W.-P. Chen, S. Ling, X. Li, Y.-Z. Zheng, K. Zhu and M. Zhang, J. Am. Chem. Soc., 2020, 142, 18763 CrossRef CAS PubMed; (c) H. Wang, K. Wang, Y. Xu, W. Wang, S. Chen, M. Hart, L. Wojtas, L.-P. Zhou, L. Gan, X. Yan, Y. Li, J. Lee, X.-S. Ke, X.-Q. Wang, C.-W. Zhang, S. Zhou, T. Zhai, H.-B. Yang, M. Wang, J. He, Q.-F. Sun, B. Xu, Y. Jiao, P. J. Stang, J. L. Sessler and X. Li, J. Am. Chem. Soc., 2021, 143, 5826 CrossRef CAS; (d) G.-Q. Yin, S. Kandapal, C.-H. Liu, H. Wang, J. Huang, S.-T. Jiang, T. Ji, Y. Yan, S. Khalife, R. Zhou, L. Ye, B. Xu, H.-B. Yang, M.-P. Nieh and X. Li, Angew. Chem., Int. Ed., 2021, 60, 1281 CrossRef CAS PubMed.
- (a) E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480 CrossRef; (b) P. Howlader, S. Mondal, S. Ahmed and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 20968 CrossRef CAS PubMed; (c) B. Roy, A. K. Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916 CrossRef CAS PubMed.
- (a) K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565 CrossRef CAS PubMed; (b) Y.-X. Hu, W.-J. Li, P.-P. Jia, X.-Q. Wang, L. Xu and H.-B. Yang, Adv. Opt. Mater., 2020, 8, 2000265 CrossRef CAS; (c) T. Gorai, J. I. Lovitt, D. Umadevi, G. McManus and T. Gunnlaugsson, Chem. Sci., 2022, 13, 7805 RSC.
- (a) R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244 CrossRef CAS PubMed; (b) I. Sinha and P. S. Mukherjee, Inorg. Chem., 2018, 57, 4205 CrossRef CAS PubMed; (c) A. K. Bar, R. Chakraborty and P. S. Mukherjee, Inorg. Chem., 2009, 48, 10880 CrossRef CAS PubMed.
- (a) P. C. Purba, M. Venkateswaralu, S. Bhattacharyya and P. S. Mukherjee, Inorg. Chem., 2022, 61, 713 CrossRef CAS PubMed; (b) X. Yan, H. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. Zhou, M. Zhang, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276 CrossRef CAS.
- (a) D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 18949 CrossRef CAS PubMed; (b) A. B. Sainaba, M. Venkateswarulu, P. Bhandari, K. S. A. Arachchige, J. K. Clegg and P. S. Mukherjee, J. Am. Chem. Soc., 2022, 144, 7504 CrossRef CAS PubMed.
- (a) R. Saha and P. S. Mukherjee, Dalton Trans., 2020, 49, 1716 RSC; (b) A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600 CrossRef CAS; (c) P. Bhandari, R. Modak, S. Bhattacharyya, E. Zangrando and P. S. Mukherjee, JACS Au, 2021, 1, 2242 CrossRef CAS PubMed.
- (a) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (b) C.-E. Boström, P. Gerde, A. Hanberg, B. Jernström, C. Johansson, T. Kyrklund, A. Rannug, M. Törnqvist, K. Victorin and R. Westerholm, Environ. Health Perspect., 2002, 110, 451 Search PubMed.
- (a) C. Alvariño, E. Pía, M. D. García, V. Blanco, A. Fernández, C. Peinador and J. M. Quintela, Chem.–Eur. J., 2013, 19, 15329 CrossRef; (b) S. Ibáñez and E. Peris, Angew. Chem., Int. Ed., 2019, 58, 6693 CrossRef PubMed; (c) J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183 CrossRef CAS PubMed.
- C. B. Bennett and D. R. Sherman, Crude anthracene separation, US Pat., US2438148A, 1945.
- (a) Y. Tamura, H. Takezawa and M. Fujita, J. Am. Chem. Soc., 2020, 142, 5504 CrossRef CAS; (b) E. G. Percástegui, Eur. J. Inorg. Chem., 2021, 2021, 4425 CrossRef; (c) C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef; (d) C.-L. Liu, E. O. Bobylev, Y. Fu, D. A. Poole III, K. Robeyns, C.-A. Fustin, Y. Garcia, J. N. H. Reek and M. L. Singleton, Chem.–Eur. J., 2020, 26, 11960 CrossRef CAS PubMed.
- (a) W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Chem. Rev., 2020, 120, 6288 CrossRef CAS PubMed; (b) Z. Cui, X. Gao, Y.-J. Lin and G.-X. Jin, J. Am. Chem. Soc., 2022, 144, 2379 CrossRef CAS PubMed; (c) N. Pearce, M. Tarnowska, N. J. Andersen, A. Wahrhaftig-Lewis, B. S. Pilgrim and N. R. Champness, Chem. Sci., 2022, 13, 3915 RSC; (d) P. Rajamalli, F. Rizzi, W. Li, M. A. Jinks, A. K. Gupta, B. A. Laidlaw, I. D. W. Samuel, T. J. Penfold, S. M. Goldup and E. Zysman-Colman, Angew. Chem., Int. Ed., 2021, 60, 12066 CrossRef CAS PubMed; (e) H. V. Schröder, Y. Zhang and A. J. Link, Nat. Chem., 2021, 13, 850 CrossRef; (f) Z. Ashbridge, E. Kreidt, L. Pirvu, F. Schaufelberger, J. H. Stenlid, F. Abild-Pedersen and D. A. Leigh, Science, 2022, 375, 1035 CrossRef CAS PubMed; (g) R. L. Greenaway, V. Santolini, M. J. Bennison, B. M. Alston, C. J. Pugh, M. A. Little, M. Miklitz, E. G. B. Eden-Rump, R. Clowes, A. Shakil, H. J. Cuthbertson, H. Armstrong, M. E. Briggs, K. E. Jelfs and A. I. Cooper, Nat. Commun., 2018, 9, 2849 CrossRef CAS PubMed; (h) F. Li, J. K. Clegg, L. F. Lindoy, R. B. Macquart and G. V. Meehan, Nat. Commun., 2011, 2, 205 CrossRef.
- (a) S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016 RSC; (b) Z. Zhang, J. Zhao, Z. Guo, H. Zhang, H. Pan, Q. Wu, W. You, W. Yu and X. Yan, Nat. Commun., 2022, 13, 1393 CrossRef CAS PubMed; (c) A. Kumar and P. S. Mukherjee, Chem.–Eur. J., 2020, 26, 4842 CrossRef CAS PubMed; (d) M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52 CrossRef CAS; (e) S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060 CrossRef.
- (a) X.-Y. Chen, D. Shen, K. Cai, Y. Jiao, H. Wu, B. Song, L. Zhang, Y. Tan, Y. Wang, Y. Feng, C. L. Stern and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 20152 CrossRef CAS PubMed; (b) M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 0061 CrossRef CAS; (c) Y. Yamauchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2008, 130, 5832 CrossRef CAS.
- P. Howlader, S. Mondal, S. Ahmed and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 20968 CrossRef CAS PubMed.
- (a) D. Samanta, S. Mukherjee, Y. P. Patil and P. S. Mukherjee, Chem.–Eur. J., 2012, 18, 12322 CrossRef CAS PubMed; (b) M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52 CrossRef CAS; (c) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed.
- (a) D. Chakraborty, R. Modak, P. Howlader and P. S. Mukherjee, Chem. Commun., 2021, 57, 3995 RSC; (b) Y.-W. Zhang, R. Das, Y. Li, Y.-Y. Wang and Y.-F. Han, Chem.–Eur. J., 2019, 25, 5472 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885 CrossRef PubMed.
- Y.-W. Zhang, S. Bai, Y.-Y. Wang and Y.-F. Han, J. Am. Chem. Soc., 2020, 142, 13614 CrossRef CAS PubMed.
- D. Yang, J. L. Greenfield, T. K. Ronson, L. K. S. von Krbek, L. Yu and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 19856 CrossRef CAS PubMed.
- H. Takezawa, T. Murase, G. Resnati, P. Metrangolo and M. Fujita, J. Am. Chem. Soc., 2014, 136, 1786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, ESI-MS, calculation details, optimised structure, experimental details (PDF). CCDC 2174542. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04660a |
| This journal is © The Royal Society of Chemistry 2022 |