
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhabdastrenones A–D from the sponge Rhabdastrella globostellata†
Do Thi Trangab,
Dan Thi Thuy Hanga,
Duong Thi Dunga,
Nguyen Thi Cuca,
Pham Hai Yena,
Phan Thi Thanh Huonga,
Le Thi Huyen c,
Nguyen Thi Maid,
Nguyen Xuan Nhiemab,
Bui Huu Tai*ab and
Phan Van Kiem*ab
c,
Nguyen Thi Maid,
Nguyen Xuan Nhiemab,
Bui Huu Tai*ab and
Phan Van Kiem*ab
aInstitute of Marine Biochemistry, Vietnam Academy of Science and Technology (VAST), 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. E-mail: bhtaiich@gmail.com; phankiem@yahoo.com
bGraduate University of Science and Technology, VAST, 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam
cVNU University of Science, Vietnam National University, Hanoi, 334 Nguyen Trai, Thanh Xuan, Hanoi, Vietnam
dUniversity of Transport and Communications, 3 Cau Giay, Dong Da, Hanoi, Vietnam
First published on 6th April 2022
Abstract
Three new isomalabaricanes (1–3), a new α-pyrone derivative (4), together with four known isomalabaricane analogs rhabdastrellin G (5), isogeoditin A (6), stelliferin A (7), and (13E)-isogeoditin A (8) were isolated from the marine sponge Rhabdastrella globostellata. Their chemical structures were determined by HR-ESI-MS, 1D and 2D-NMR spectroscopic data analysis. The absolute configurations were identified by Mo2(OAc)4 induced ECD spectra and TD-DFT theoretical calculated ECD spectra. Compound 6 exhibited weak cytotoxic effects against HepG2 and SKMel2 cell lines with the IC50 values of 7.53 ± 0.70 and 9.93 ± 0.95 μM, respectively.
Introduction
Sponge natural products have been highlighted since the 1950s by the discovery of unusual nucleoside derivatives, which were successfully developed to be anti-leukemia drug, cytarabine. Studies on secondary metabolites from sponges were then speedily accelerated and hundreds of new compounds are being discovered annually in recent decades.1,2 The sponge Rhabdastrella globostellata has been attracting a lot of attention for its production of a rare terpenoid group, called isomalabaricane analogs. These types of compounds display unique chemical structures with a trans-syn-trans 6/6/5 tricyclic nucleus and a polyunsaturated side chain.3 Additionally, a series of isomalabaricanes exhibited extremely high cytotoxic activity against various cancer cell lines with IC50 values in the range of nanomolar concentrations.4–7 Of these, rhabdastrellic acid A, stelletin A, and stelletin E have been accomplished through total syntheses, providing materials for studying biological mechanisms.8,9 To date, over 70 isomalabaricane analogs have been isolated from R. globostellata.6,7,10–18 It has been shown that chemical structures of isomalabaricanes mostly differ at the side chain by the oxidative degeneration, cyclization, and isomerization of double bonds.3 In the present work, continuing investigation on chemical constituents of R. globostellata, we describe herein the isolation and determination of three new isomalabaricanes (1–3), a new α-pyrone derivative (4), together with four known isomalabaricane analogs (5–8) (Fig. 1). Cytotoxic activity of compounds (1–8) was screened on HepG2, LU1, SKMel2, and MCF7 human cancerous cells using the sulforhodamine B colorimetric assay.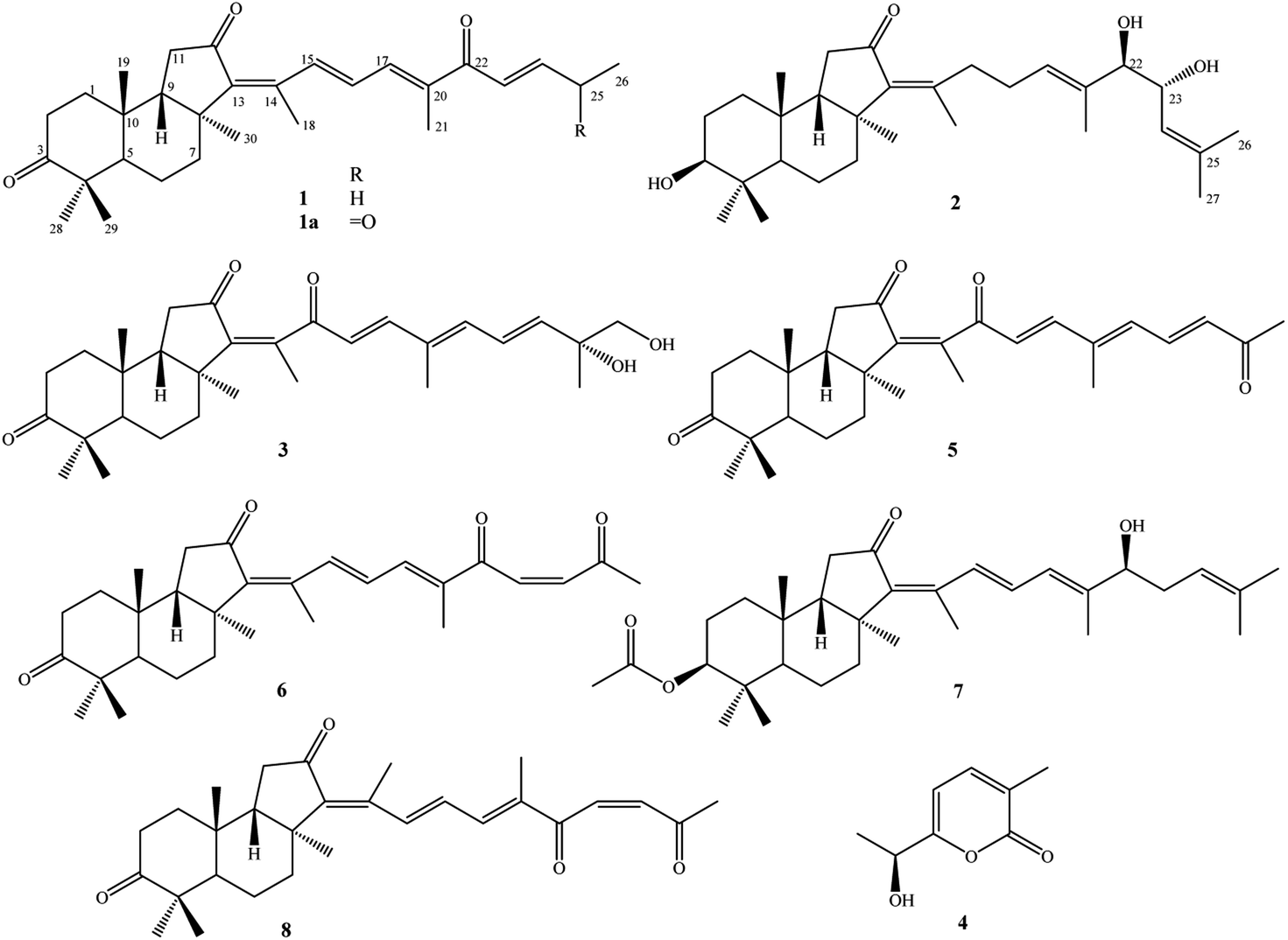 | ||
| Fig. 1 Chemical structures of 1–8 isolated from the marine sponge R. globostellata. | ||
Experimental
General experimental procedures
ECD spectra were acquired on an Applied Photophysics Chirascan spectrometer. Optical rotations were taken on a Jasco P2000 polarimeter. NMR spectra were recorded on a Bruker AVANCE III 500 MHz spectrometer. HR-ESI-MS were acquired on an Agilent 6530 Accurate-Mass QTOF LC/MS system. Semi-preparative HPLC was acquired on an Agilent 1260 Infinity II system (binary pump, autosampler, and DAD detector) using a YMC J'sphere ODS-H80 (20 × 250 mm, 4 μm) HPLC column and an isocratic mobile phase with a flow rate of 3 mL min−1. Column chromatography was performed using silica gel (40–63 μm) or ODS (150 μm) as adsorbents. Thin-layer chromatography (TLC) was carried out on pre-coated plates (silica gel 60 F254 or RP-18 F254S).Sponge material
Sponge samples were collected at the Van Phong Bay, Nha Trang, Vietnam in May 2020 and taxonomically identified to be Rhabdastrella globostellata (Carter, 1883) by Dr Tran My Linh at the Institute of Marine Biochemistry, VAST. The voucher specimen (no. NCCT-B139) was kept at the Institute of Marine Biochemistry, VAST.Extraction and isolation
The fresh sponge (40 kg) was cut into small pieces and macerated in MeOH three times (50 L, 2 h in an ultrasonic bath, at room temperature each time) to produce methanol extract (420 g). This extract was suspended with water (3 L) and separated with dichloromethane to give dichloromethane extract (320 g). The dichloromethane extract was loaded on a silica gel column and then eluted with dichloromethane/methanol (0–100% volume of methanol) to give four fractions, D1–D4. Fraction D2 (200 g) was repeatedly subjected to a silica gel column and eluted with n-hexane/acetone (0–100% volume of acetone) to give four fractions, D2A–D2D. Fraction D2B was chromatographed on a reversed-phase C-18 column, eluting with acetone/water (2/1, v/v) to give five fractions, D2B1–D2B5. Fractions D2B2 and D2B4 were purified by semi-preparative HPLC using acetonitrile (ACN) in water (30% ACN) to obtain compounds 4 (7.3 mg, tR 22.4) and 3 (15.2 mg, tR 42.3), respectively. Fraction D2C was first separated on a reversed-phase C-18 column, eluting with acetone/water (3/2, v/v) and then purified by semi-preparative HPLC using ACN in water (60% ACN) to obtain compound 5 (6.2 mg, tR 58.8). Fraction D3 (60 g) was loaded on a reversed-phase C-18 column and eluted with methanol/water (3/1, v/v) to give four fractions, D3A–D2D. Fraction D3C was further separated on a silica gel column, eluting with n-hexane/ethyl acetate (2/1, v/v) to give four fractions, D3C1–D3C4. The fraction D3C1 was purified by semi-preparative HPLC using ACN in water (70% ACN) to obtain compounds 8 (5.4 mg, tR 48.5) and 6 (5.8 mg, tR 50.0). Fraction D3C2 was purified by semi-preparative HPLC using ACN in water (95% ACN) to obtain compound 7 (5.7 mg, tR 51.5). Fraction D3C3 was purified by semi-preparative HPLC using ACN in water (85% ACN) to obtain compound 1 (10.0 mg, tR 59.4). Finally, fraction D3C4 was purified by semi-preparative HPLC using ACN in water (70% ACN) to obtain compound 2 (19.5 mg, tR 43.7).Rhabdastrenone A (1)
Pale yellow oil; [α]25D: −32.4 (c 0.1, MeOH); UV (MeOH): λmax(log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 265(3.0), 354(4.4) nm; HR-ESI-MS: m/z 437.3040 [M + H]+ (calcd. for C29H41O3, 437.3056); 1H- and 13C-NMR data are shown in Table 1.
ε) 265(3.0), 354(4.4) nm; HR-ESI-MS: m/z 437.3040 [M + H]+ (calcd. for C29H41O3, 437.3056); 1H- and 13C-NMR data are shown in Table 1.
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δCa,b | δHa,c (mult., J in Hz) | δCa,b | δHa,c (mult., J in Hz) | δCa,b | δHa,c (mult., J in Hz) | |
| a Measured in CDCl3.b Measured in 125 MHz.c Measured in 500 MHz. | ||||||
| 1 | 31.4 | 1.52 (m)/2.15 (m) | 33.3 | 1.37 (m)/1.52 (m) | 31.3 | 1.50 (m)/2.13 (m) |
| 2 | 33.4 | 2.38 (m)/2.73 (m) | 29.0 | 1.68 (m)/1.79 (m) | 33.6 | 2.38 (m)/2.72 (m) |
| 3 | 218.9 | — | 79.3 | 3.29 (dd, 5.0, 11.5) | 218.8 | — |
| 4 | 46.9 | — | 39.1 | — | 46.9 | — |
| 5 | 45.4 | 2.40 (dd, 2.0, 13.0) | 46.4 | 1.66 (br d, 13.0) | 45.4 | 2.40 (dd, 2.0, 13.0) |
| 6 | 19.6 | 1.56 (m)/1.68 (m) | 18.2 | 1.45 (m)/1.68 (m) | 19.2 | 1.56 (m)/1.65 (m) |
| 7 | 36.9 | 2.20 (m)/2.25 (m) | 37.9 | 1.98 (m)/2.03 (m) | 34.7 | 1.95 (m)/2.15 (m) |
| 8 | 45.0 | — | 43.8 | — | 43.1 | — |
| 9 | 47.9 | 1.89 (m) | 50.5 | 1.75 (m) | 48.9 | 1.94 (m) |
| 10 | 34.8 | — | 35.4 | — | 34.9 | — |
| 11 | 37.1 | 2.12 (m)/2.26 (m) | 36.6 | 2.13 (m)/2.21 (m) | 35.6 | 2.08 (m)/2.28 (m) |
| 12 | 206.4 | — | 206.5 | — | 203.6 | — |
| 13 | 147.7 | — | 145.1 | — | 145.7 | — |
| 14 | 141.7 | — | 149.8 | — | 143.7 | — |
| 15 | 138.5 | 8.32 (d, 15.5) | 34.7 | 2.51 (m)/2.67 (m) | 200.8 | — |
| 16 | 129.8 | 6.97 (dd, 15.5, 11.0) | 26.5 | 2.07 (m)/2.12 (m) | 124.8 | 6.18 (d, 16.0) |
| 17 | 138.6 | 7.20 (d, 11.0) | 128.2 | 5.46 (t, 7.0) | 147.8 | 6.93 (d, 16.0) |
| 18 | 16.0 | 2.10 (s) | 21.6 | 1.84 (s) | 17.3 | 1.99 (s) |
| 19 | 23.5 | 0.88 (s) | 22.3 | 0.98 (s) | 23.4 | 0.87 (s) |
| 20 | 139.2 | — | 134.2 | — | 134.4 | — |
| 21 | 12.4 | 2.00 (s) | 12.4 | 1.63 (s) | 12.6 | 1.92 (s) |
| 22 | 192.1 | — | 80.9 | 3.80 (d, 7.5) | 138.3 | 6.33 (d, 11.0) |
| 23 | 124.1 | 6.73 (d, 15.5) | 70.1 | 4.29 (dd, 7.5, 8.5) | 125.4 | 6.72 (dd, 11.0, 15.0) |
| 24 | 149.2 | 6.92 (dt, 6.5, 15.5) | 123.8 | 5.13 (d, 8.5) | 142.3 | 5.96 (d, 15.0) |
| 25 | 25.8 | 2.30 (m) | 137.5 | — | 73.6 | — |
| 26 | 12.5 | 1.13 (t, 6.5) | 18.5 | 1.68 (s) | 69.9 | 3.49 (d, 10.5), 3.54 (d, 10.5) |
| 27 | — | — | 25.9 | 1.71 (s) | 24.4 | 1.32 (s) |
| 28 | 29.2 | 1.12 (s) | 29.2 | 1.02 (s) | 29.2 | 1.13 (s) |
| 29 | 19.4 | 1.05 (s) | 15.9 | 0.82 (s) | 19.4 | 1.07 (s) |
| 30 | 24.6 | 1.42 (s) | 24.8 | 1.30 (s) | 24.9 | 1.46 (s) |
Rhabdastrenone B (2)
Pale yellow oil; [α]25D: −25.2 (c 0.1, MeOH); UV (MeOH): λmax(logε) 232(3.4), 339(4.1) nm; HR-ESI-MS: m/z 507.3219 [M + 35Cl]− (calcd. for C30H48O435Cl, 507.3241) and m/z 509.3228 [M + 37Cl]− (calcd. for C30H48O437Cl, 509.3212); 1H- and 13C-NMR data are shown in the Table 1.
Rhabdastrenone C (3)
Pale yellow oil; [α]25D: −27.0 (c 0.1, MeOH); UV (MeOH): λmax(logε) 223(3.3), 328(4.0) nm; HR-ESI-MS: m/z 483.3093 [M + H]+ (calcd. for C30H43O5: 483.3110); 1H- and 13C-NMR data are shown in the Table 1.
Rhabdastrenone D (4)
Colorless oil; [α]25D: −79.7 (c 0.1, MeOH); UV (MeOH): λmax(logε) 221(3.4), 296(4.1) nm; ECD (MeOH) θ(λ nm): −12.0(299) mdeg; HR-ESI-MS: m/z 155.0706 [M + H]+ (calcd. for C8H11O3, 155.0708); 1H-NMR (CD3OD, 500 MHz) δH (ppm): 7.35 (d, J = 6.5 Hz, H-4), 6.33 (d, J = 6.5 Hz, H-5), 4.53 (q, J = 6.5 Hz, H-7), 2.05 (s, H-9), 1.44 (d, J = 6.5 Hz, H-8); 13C-NMR (CDCl3, 125 MHz) δH (ppm): 166.7 (C-6), 165.5 (C-2), 142.0 (C-4), 124.4 (C-3), 102.3 (C-5), 67.2 (C-7), 21.5 (C-8), 16.5 (C-9).
ECD measurement of the molybdenum complex of compounds 2 and 3
Compounds 2 and 3 (each 0.5 mg) and Mo2(OAc)4 (1.0 mg) were dissolved in 1.0 mL anhydrous DMSO. The ECD spectrum of the obtained solution was measured immediately under wavelengths 250–500 nm. After 60 minutes, the stationary complex was formed, and the ECD spectrum of the solution was measured again and used to subtract the first ECD spectrum to obtain the Mo2(OAc)4 induced ECD spectrum of the compound.Theoretical calculation of ECD spectra
Conformational searches and geometric equilibrium were carried out using the Spartan 18 program (Wavefunction Inc., Irvine, CA, USA). Possible conformations were optimized and subjected to TDDFT calculation on Gaussian 16 program (Gaussian Inc., Wallingford, CT, USA). The calculated ECD spectra were composed after correction based on the Boltzmann distribution of the stable conformers using the SpecDis v1.71 software (University of Wuerzburg, Wuerzburg, Germany). Stereoisomer 4a (7R) was submitted to conformational searches at the molecular mechanics MMFF and equilibrium geometry was performed using a semi-empirical PM3 set. The initial stable conformers with relative energy lower than 40 kJ mol−1 were further optimized by DFT calculations at the B3LYP/6-31G(d,p) basic set. The solvent effects were taken by a polarizable continuum model (PCM) calculation with methanol as the solvent. The optimized conformers were then subjected to TDDFT calculations at the CAM-B3LYP/6-31G(d,p) level in the presence of methanol with a PCM. The ECD spectra at 30 excited states for each conformer were collected and summed to obtain the theoretical ECD spectra of each stereoisomer. The half-bands were taken at ζ = 0.3 eV. ECD spectra of enantiomers 4b (7S) were composed of a mirror image, which was of 4a (7R).Cytotoxic assay
LU1, HepG2, MCF7, and SKMel2 cell lines were obtained from Milan University, Italy and Long Island University, USA. The cells were maintained and cultured in DMEM supplemented with FBS, trypsin-EDTA, L-glutamine, sodium pyruvate, NaHCO3, and penicillin/streptomycin at 37 °C in a humidified atmosphere (5% CO2 and 95% air). Cytotoxic effects of compounds were determined using the sulforhodamine B (SRB) assay as previously described.19 In brief, the cells were incubated with/without compounds for three days in a 96-well culture plate. After incubation, cells were stained with sulforhodamine B and optical density (OD) was measured at 540 nm. The difference in OD between samples and vehicle well during experiments indicated the cell situation induced by the compounds. Results are expressed as the percentage of cell death in comparison with the vehicle as well. The dose–response curves of compounds were generated to determine IC50 values of the compounds corresponding to each cell line. Ellipticine was used as a positive control throughout the experiments.Results and discussion
Compound 1 was obtained as pale yellow oil. Its molecular formula was determined to be C29H40O3 by a protonated molecule at m/z 437.3040 [M + H]+ (calcd. for C29H41O3, 437.3056) in the HR-ESI-MS, indicating 10 degrees of unsaturation. The UV chromatogram of 1 showed two maxima absorptions at wavelengths of 265 and 354 nm, suggesting the n–π highly conjugated system. The 1H-NMR spectrum of 1 contained signals of five olefinic protons [δH 8.32 (1H, d, J = 15.5 Hz), 7.20 (1H, d, J = 11.0 Hz), 6.97 (1H, dd, J = 15.5 and 11.0 Hz), 6.92 (1H, dt, J = 15.5 and 6.5 Hz), and 6.73 (1H, d, J = 15.5 Hz)], six tertiary methyl groups [δH 0.88, 1.05, 1.12, 1.42, 2.00, and 2.10 (each 3H, s)], and a primary methyl group [δH 1.13 (3H, t, J = 6.5 Hz)]. 13C-NMR and HSQC spectra of 1 indicated signals of 29 carbons, which suggested 1 to be a nor-triterpene containing nine non-protonated carbons (three carbonyl, three olefinic, and three aliphatic carbons), seven methines (two aliphatic and five olefinic methines), six aliphatic methylenes, and seven methyl groups. Moreover, the 1H- and 13C-NMR data of 1 were close similarity to those of geoditin A (1a), an isomalabaricane nortriterpene, previously isolated from the marine sponges Geodia japonica and Rhabdastrella aff. distincta.20,21 The difference with 1a was the absence of the ketone functional group at C-25 (Fig. 1). This deduction was further confirmed by consecutive COSY correlations of H-23 (δH 6.73)/H-24 (δH 6.92)/H-25 (δH 2.30)/H-26 (δH 1.13). Additionally, the planar structure of 1 was well demonstrated by HMBC and COSY analysis as shown in Fig. 2. The stereochemistry of 1 was then elucidated by J coupling constant values from the 1H-NMR and interactions between closed protons in the NOESY spectrum. Particularly, the trans-syn-trans configuration of the tricyclic system, a usual isomalabaricane feature, was demonstrated by NOESY correlations between H3-19 (δH 0.88) and H3-29 (δH 1.05)/H-9 (δH 1.89), and between H-5 (δH 2.40) and H3-28 (δH 1.12)/H3-30 (δH 1.42) (Fig. 3). A Z-geometric configuration at double bond C-13/C-14 was indicated by the NOESY correlation between H3-30 (δH 1.42) and H3-18 (δH 2.10). On the other hand, E-geometric configurations at C-15/C-16, C-17/C-20, and C-23/C-24 were confirmed by NOESY correlations between H-15 (δH 8.32) and H-17 (δH 7.20), H-16 (δH 6.97) and H3-21 (δH 2.00), and between H-23 (δH 6.73) and H2-25 (δH 2.30). The E-configurations of double bonds at C-15/C-16 and C-23/C-24 were also supported by J coupling constants of their protons (JH-15/H-16 = 15.5 Hz and JH-23/H-24 = 15.5 Hz). Consequently, the structure of 1 was established and named rhabdastrenone A.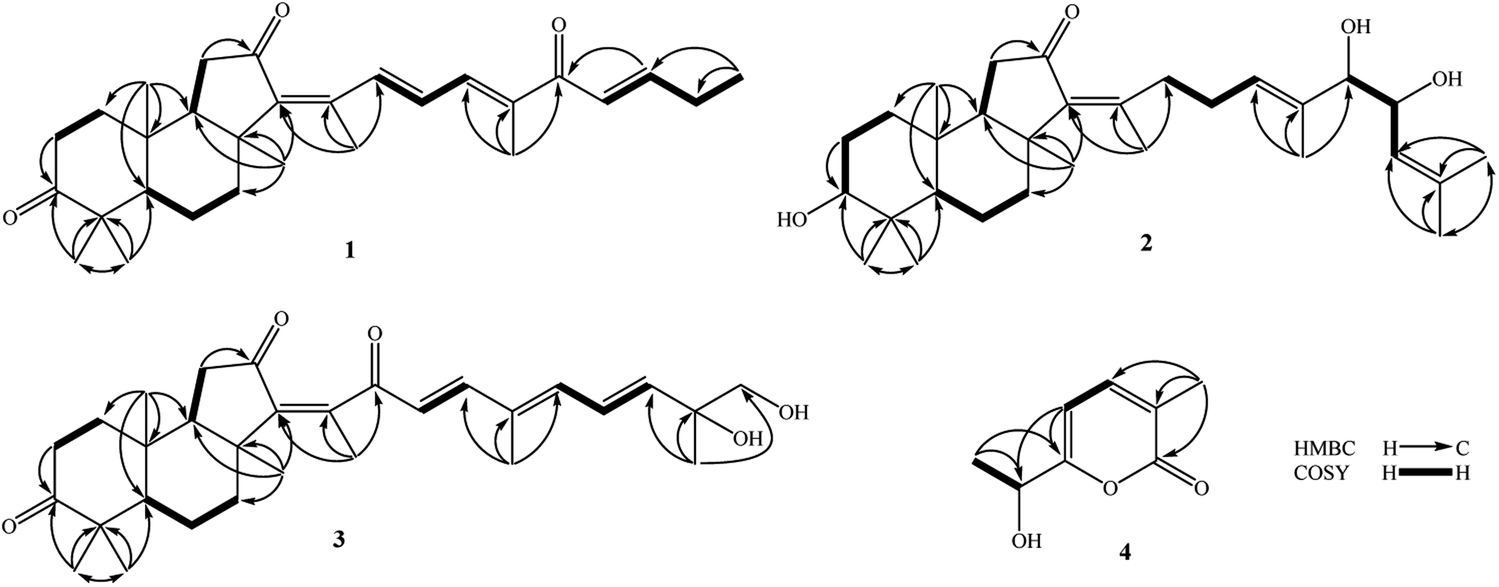 | ||
| Fig. 2 Key HMBC and COSY correlations of compounds 1–4. | ||
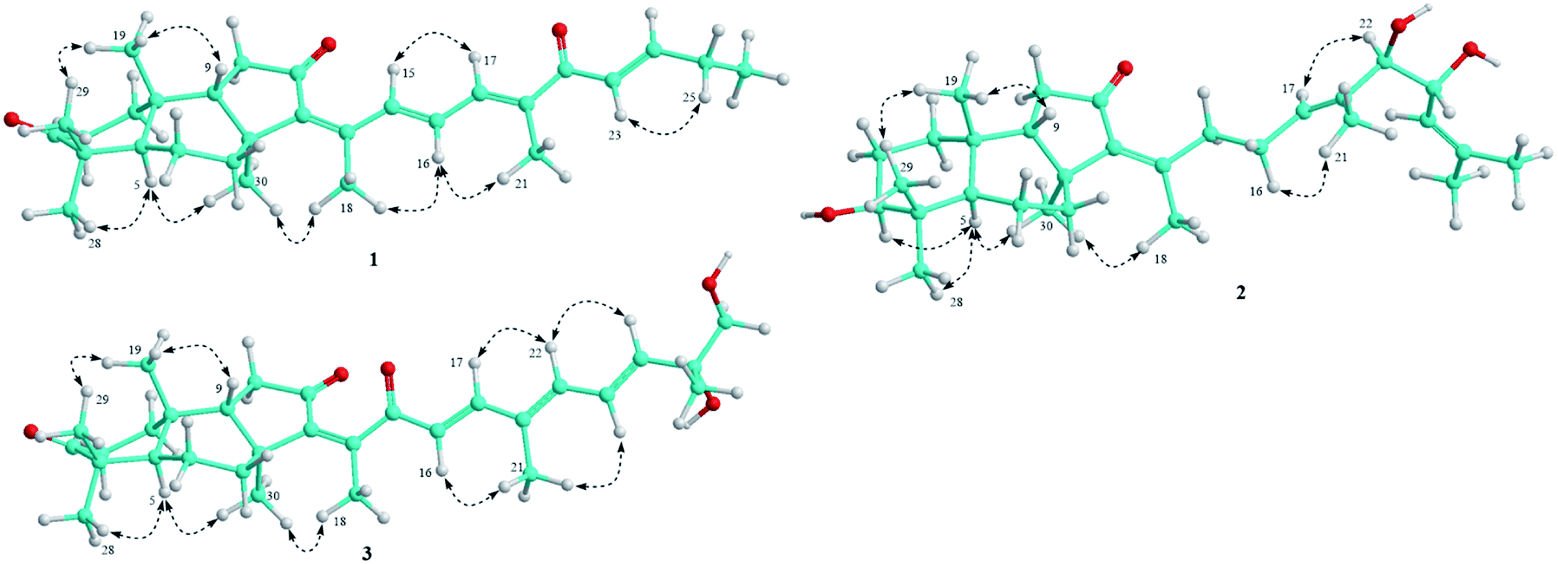 | ||
| Fig. 3 Important NOESY correlations of compounds 1–3. | ||
Compound 2 was obtained as pale yellow oil. Its molecular formula was determined to be C30H48O4 by chlorinated adduct ion at m/z 507.3219 [M + 35Cl]− (calcd. for C30H48O435Cl, 507.3241) and m/z 509.3228 [M + 37Cl]− (calcd. for C30H48O437Cl, 509.3212), showing seven degrees of unsaturation. The 1H-NMR spectra of 2 showed signals of two olefinic protons [δH 5.46 (1H, t, J = 7.0 Hz) and 5.13 (1H, d, J = 8.5 Hz)], three carbinol protons [δH 4.29 (1H, dd, J = 7.5 and 8.5 Hz), 3.80 (1H, d, J = 7.5 Hz), and 3.29 (1H, dd, J = 5.0 and 11.5 Hz)], and eight singlet methyl groups [δH 1.84, 1.71, 1.68, 1.63, 1.30, 1.02, 0.98, and 0.82 (each 3H, s)]. The 13C-NMR spectrum of 2 revealed signals of 30 carbons including one carbonyl carbon (δC 206.5), six olefinic carbons (δC 149.8, 145.1, 137.5, 134.2, 128.2, and 123.8), three oxygenated carbons (δC 80.9, 79.3, and 70.1), and others 20 sp3-hybridized carbons (δC 12.4–50.5). The aforementioned data suggested 2 to be a tricyclic isomalabaricane-type triterpene. The HMBC correlations between H3-28 (δH 1.02)/H3-29 (δH 0.82) and C-3 (δC 79.3) indicated the presence of the hydroxy group at C-3. The location of the ketone functional group at C-12 was confirmed by COSY cross-peaks of H-9 (δH 1.75)/H2-11 (δH 2.13 and 2.21), following the HMBC correlation between H2-11 and C-12 (δC 206.5). The connection between the side chain and the tricyclic system via the double bond C-13/C-14 was indicated by HMBC correlations between H3-18 (δH 1.84) and C-13 (δC 145.1)/C-14 (δC 149.8)/C-15 (δC 34.7). Continuously, COSY cross-peaks of H2-15 (δH 2.51 and 2.67)/H2-16 (δH 2.07 and 2.12)/H-17 (δH 5.46) and HMBC correlations between H3-21 (δH 1.63) and C-17 (δC 128.2)/C-20 (δC 134.2)/C-22 (δC 80.9) indicated the location of another double bond at C-17/C-20. The COSY cross-peaks of H-22 (δH 3.80)/H-23 (δH 4.29)/H-24 (δH 5.13) and HMBC correlations between H3-26 (δH 1.68)/H3-27 (δH 1.71) and C-24 (δC 123.8)/C-25 (δC 137.5) indicated the presence of hydroxy groups at C-22, C-23, and the location of the third double bond at C-24/C-25 (Fig. 2). In the NOESY spectrum of 2, correlations between H3-19 (δH 0.98) and H3-29 (δH 0.82)/H-9 (δH 1.75), and between H-5 (δH 1.66) and H3-28 (δH 1.02)/H3-30 (δH 1.30) demonstrated the trans-syn-trans configuration of the tricyclic system. The NOESY correlation between H-3 (δH 3.29) and H-5 (δH 1.66) indicated α,axial-orientation of H-3 (β,equatorial-orientation of hydroxy group) (Fig. 3). The axial orientation of H-3 was also agreed by axial–axial coupled protons between H-3 and Hax-2 (JH-3/H-2ax = 11.5 Hz). Geometric configurations of double bonds at C-13/C-14 and C-17/C-20 were determined to be 13Z,17(20)E by NOESY correlations between H3-30 (δH 1.30) and H3-18 (δH 1.84), H2-16 (δH 2.07 and 2.12) and H3-21 (δH 1.63). Relative configuration between hydroxy groups at C-22 and C-23 was determined to be threo-configuration by J coupling constant value between H-22 and H-23 (JH-22/H-23 = 7.5 Hz) as previously described for 1,2-dioxygenated compounds (erythro: J ∼ 3 Hz and threo: J ∼ 8 Hz).22 Additionally, negative chirality pattern in the Mo2(OAc)4 induced ECD spectrum of 2 [positive Cotton effect at 266 nm (+1.1 mdeg) and negative Cotton effect at 355 nm (−0.80 mdeg)] indicated the (22R,23R) absolute configurations, as previously described (ESI†).23 Consequently, the structure of 2 was established and named rhabdastrenone B.
Compound 3 was obtained as pale yellow oil. Its molecular formula was determined to be C30H42O5 by a protonated molecule at m/z 483.3093 [M + H]+ (calcd. for C30H43O5: 483.3110) in the HR-ESI-MS, indicating 10 degrees of unsaturation. The 1H-NMR and HSQC spectra of 3 showed signals of five olefinic protons [δH 6.93 (1H, d, J = 16.0 Hz), 6.72 (1H, dd, J = 11.0 and 15.0 Hz), 6.33 (1H, d, J = 11.0 Hz), 6.18 (1H, d, J = 16.0 Hz), and 5.96 (1H, d, J = 15.0 Hz)], a hydroxymethylene group [δH 3.54 and 3.49 (each 1H, d, J = 10.5 Hz)], and seven singlet methyl groups [δH 1.99, 1.92, 1.46, 1.32, 1.13, 1.07, and 0.87 (each 3H, s)]. The 13C-NMR and HSQC spectra of 3 indicated signals of 30 carbons including three carbonyl carbons (δC 218.8, 203.6, 200.8), eight olefinic carbons (δC 147.8, 145.7, 143.7, 142.3, 138.3, 134.4, 125.4, and 124.8), two oxygenated carbons (δC 73.6 and 69.9), and others 17 sp3-hybridized carbons (δC 12.6–48.9). The NMR spectral data also suggested 3 to be a tricyclic isomalabaricane-type triterpene. Similar with 1, the HMBC correlations between H3-28 (δH 1.13)/H3-29 (δH 1.07) and C-3 (δC 218.8), H2-11 (δH 2.08 and 2.28) and C-12 (δC 203.6) indicated the presence of ketone functional groups at C-3 and C-12. However, the HMBC correlation between H3-18 (δH 1.99) and C-13 (δC 145.7)/C-14 (δC 143.7)/C-15 (δC 200.8) suggested that 3 contained an additional ketone functional group at C-15. Furthermore, COSY interaction of H-16 (δH 6.18)/H-17 (δH 6.93), HMBC correlations between H3-21 (δH 1.92) and C-17 (δC 147.8)/C-20 (δC 134.4)/C-22 (δC 138.3), and COSY consecutive interactions of H-22 (δH 6.33)/H-23 (δH 6.72)/H-24 (δH 5.96) confirmed the conjugated 16,20(22),23-triene side chain. Later, the HMBC correlations between H3-27 (δH 1.32) and C-24 (δC 142.3)/C-25 (δC 73.6)/C-26 (δC 69.9) suggested the presence of hydroxy groups at C-25 and C-26 (Fig. 2). The NOESY correlations between H3-19 (δH 0.87) and H3-29 (δH 1.07)/H-9 (δH 1.94), and between H-5 (δH 2.40) and H3-28 (δH 1.13)/H3-30 (δH 1.46) demonstrated the trans-syn-trans configuration of the tricyclic system. The Z-geometric configuration at double bond C-13/C-14 was indicated by the NOESY correlation between H3-30 and H3-18. Meanwhile, E-geometric configurations at C-16/C-17, C-20/C-22, and C-23/C-24 were confirmed by NOESY correlations between H-16 (δH 6.18) and H3-21 (δH 1.92), H3-21 and H-23 (δH 6.72), and between H-22 (δH 6.33) and H-24 (δH 5.96) (Fig. 3). Absolute configuration at C-25 was determined to be 25R by negative chirality pattern in the Mo2(OAc)4 induced ECD spectrum of 3 [positive Cotton effect at 290 nm (+1.48 mdeg) and negative Cotton effect at 331 nm (−1.18 mdeg)], as previously described (ESI†).23 Consequently, the structure of 3 was established and named rhabdastrenone C.
Compound 4 was obtained as a colorless oil. Its molecular formula was determined to be C8H10O3 by a protonated molecule at m/z 155.0706 [M + H]+ (calcd. for C8H11O3, 155.0708) in the HR-ESI-MS, indicating four degrees of unsaturation. The 1H-NMR spectrum of 4 contained signals including a pair of ortho coupled olefinic protons [δH 7.35 and 6.33 (each, 1H, d, J = 6.5 Hz)], a carbinol group [δH 4.53 (1H, q, J = 6.5 Hz)], and two methyl groups [δH 2.05 (3H, s) and 1.44 (3H, d, J = 6.5 Hz)]. The 13C-NMR spectrum of 4 showed eight carbons including five sp2-hybridized carbons [one carbonyl (δC 165.5) and four olefinic carbons (δC 166.7, 142.0, 124.4, 102.3)] and three sp3-hybridized carbons [one oxygenated methine (δC 67.2) and two methyl groups (δC 21.5 and 16.5)]. The COSY spectrum of 8 revealed two pairs of coupled protons, including H3-8 (δH 1.44)/H-7 (δH 4.53) and H-4 (δH 7.35)/H-5 (δH 6.33). Continuously, a carbon backbone of 4 was established by HMBC correlations between H3-8 (δH 1.44)/H-5 (δH 6.33) and C-6 (δC 166.7)/C-7 (δC 67.2), H3-9 (δH 2.05) and C-2 (δC 165.5)/C-3 (δC 124.4)/C-4 (δC 142.0) (Fig. 2). The carbon chemical shift value of C-7 (δC 67.2) suggested the presence of a hydroxy group at C-7. Meanwhile, de-shielded carbon signals of C-2 (δC 165.5) and C-6 (δC 166.7) suggested the formation of a lactone bridge between C-2 and C-6. This deduction was also supported by the molecular formula C8H10O3 of 4. Absolute configuration at C-7 was determined to be S-configuration by ECD analysis, which showed a similar Cotton effect with that of TD-DFT calculated ECD spectrum of 7S-isomer (ESI†). Therefore, the structure of 4 was determined to be (S)-6-(α-hydroxyethyl)-3-methylpyran-2-one and named as rhabdastrenone D.
Other compounds were determined to be rhabdastrellin G (5),24 isogeoditin A (6),21 stelliferin A (7),25 and (13E)-isogeoditin A (8)21 by the consistency of their NMR spectral data with those reported in the literature (Fig. 1).
Cytotoxic activities of 1–4, 7 and 8 were evaluated against four typical human cancer cell lines including LU1, HepG2, MCF7, and SKMel2 using the SRB assay.19 With the exception of compounds 4 and 8, other compounds (1–3, 6 and 7) exhibited cytotoxic activity with IC50 values ranging from 7.53 ± 0.70 to 44.46 ± 1.47 μM (Table 2). Rhabdastrellin G (5) was previously reported to show cytotoxic effects on LU1, HepG2, MCF7, and SKMel2 with IC50 values from 75.39 ± 4.83 to 84.82 ± 6.67 μM.24 Based on IC50 values (in range of 1–10 μM),26 only compound 6 exhibited weak cytotoxic effects on HepG2 (IC50: 7.53 ± 0.70 μM) and SKMel2 (IC50: 9.93 ± 0.95 μM) cell lines. Other experiments showed inactivity (IC50 > 10 μM). Additionally, our results suggest that (13Z)-isomalabaricane (1–3, and 6) processed higher cytotoxic activity than (13E)-isomalabaricane (8).
| Comp | IC50 (μM) | |||
|---|---|---|---|---|
| LU1 | HepG2 | MCF7 | SKMel2 | |
| a Ellipticine was used as a positive control. | ||||
| 1 | 19.44 ± 1.48 | 29.35 ± 2.52 | 21.39 ± 1.04 | 12.91 ± 1.03 |
| 2 | 25.64 ± 2.34 | 21.43 ± 1.94 | 24.21 ± 1.37 | 21.70 ± 1.01 |
| 3 | 40.01 ± 1.52 | 36.82 ± 4.55 | 23.31 ± 2.43 | 34.01 ± 2.21 |
| 4 | >100 | >100 | >100 | >100 |
| 6 | 10.21 ± 1.68 | 7.53 ± 0.70 | 10.18 ± 1.60 | 9.93 ± 0.95 |
| 7 | 37.12 ± 3.46 | 44.46 ± 1.47 | 42.80 ± 1.08 | 27.63 ± 2.01 |
| 8 | >100 | >100 | >100 | >100 |
| aPos | 2.20 ± 0.20 | 2.07 ± 0.24 | 1.95 ± 0.16 | 1.18 ± 0.16 |
Conclusions
Our investigation on cytotoxic constituents of the sponge R. globostellata collected at the Van Phong Bay, Vietnam, resulted in seven isomalabaricanes (comprising three new ones) and a new α-pyrone derivative. The absolute configurations of new compounds were proved by Mo2(OAc)4 induced ECD spectra and TD-DFT theoretical calculated ECD spectra. Isogeoditin A (6) showed weak cytotoxic effects on HepG2 and SKMel2 cell lines with IC50 values of 7.53 ± 0.70 and 9.93 ± 0.95 μM, respectively.Author contributions
PV Kiem, NX Nhiem, BH Tai contributed to research idea and writing; DT Trang, DTT Hang, DT Dung contributed to isolation; NT Cuc, PH Yen, PTT Huong, NT Mai, LT Huyen contributed to structure elucidation and cytotoxic evaluation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Vietnam Academy of Science and Technology under grant number TĐDLB0.01/20-22.Notes and references
- M. F. Mehbub, J. Lei, C. Franco and W. Zhang, Mar. Drugs, 2014, 12, 4539 CrossRef PubMed.
- C. Calcabrini, E. Catanzaro, A. Bishayee, E. Turrini and C. Fimognari, Mar. Drugs, 2017, 15, 310 CrossRef PubMed.
- V. A. Stonik and S. A. Kolesnikova, Mar. Drugs, 2021, 19, 327 CrossRef CAS PubMed.
- J. L. McCormick, T. C. McKee, J. H. Cardellina II, M. Leid and M. R. Boyd, J. Nat. Prod., 1996, 59, 1047–1050 CrossRef CAS PubMed.
- G. Ryu, S. Matsunaga and N. Fusetani, J. Nat. Prod., 1996, 59, 512–514 CrossRef CAS PubMed.
- D. Tasdemir, G. C. Mangalindan, G. P. Concepcion, S. M. Verbitski, S. Rabindran, M. Miranda, M. Greenstein, J. N. A. Hooper, M. K. Harper and C. M. Ireland, J. Nat. Prod., 2002, 65, 210–214 CrossRef CAS PubMed.
- M. Fouad, R. A. Edrada, R. Ebel, V. Wray, W. E. G. Mueller, W. H. Lin and P. Proksch, J. Nat. Prod., 2006, 69, 211–218 CrossRef CAS PubMed.
- Y. D. Boyko, C. J. Huck and D. Sarlah, J. Am. Chem. Soc., 2019, 141, 14131–14135 CrossRef CAS PubMed.
- C. J. Huck, Y. D. Boyko and D. Sarlah, Acc. Chem. Res., 2021, 54, 1597–1609 CrossRef CAS PubMed.
- Z. Rao, S. Deng, H. Wu and S. Jiang, J. Nat. Prod., 1997, 60, 1163–1164 CrossRef CAS.
- M. L. Bourguet-Kondracki, A. Longeon, C. Debitus and M. Guyot, Tetrahedron Lett., 2000, 41, 3087–3090 CrossRef CAS.
- J. A. Clement, M. Li, S. M. Hecht and D. G. I. Kingston, J. Nat. Prod., 2006, 69, 373–376 CrossRef CAS PubMed.
- S. Aoki, M. Sanagawa, Y. Watanabe, A. Setiawan, M. Arai and M. Kobayashi, Bioorg. Med. Chem., 2007, 15, 4818–4828 CrossRef CAS PubMed.
- M. Hirashima, K. Tsuda, T. Hamada, H. Okamura, T. Furukawa, S. I. Akiyama, Y. Tajitsu, R. Ikeda, M. Komatsu, M. Doe, Y. Morimoto, M. Shiro, R. W. M. van Soest, K. Takemura and T. Iwagawa, J. Nat. Prod., 2010, 73, 1512–1518 CrossRef CAS PubMed.
- J. Li, B. Xu, J. Cui, Z. Deng, N. J. de Voogd, P. Proksch and W. Lin, Bioorg. Med. Chem., 2010, 18, 4639–4647 CrossRef CAS PubMed.
- N. Tanaka, R. Momose, A. Shibazaki, T. Gonoi, J. Fromont and J. I. Kobayashi, Tetrahedron, 2011, 67, 6689–6696 CrossRef CAS.
- J. Li, H. Zhu, J. Ren, Z. Deng, N. J. de Voogd, P. Proksch and W. Lin, Tetrahedron, 2012, 68, 559–565 CrossRef CAS.
- D. T. Trang, D. T. Dung, N. X. Nhiem, N. T. Cuc, P. H. Yen, D. T. T. Hang, T. M. Linh, N. C. Mai, P. T. T. Huong, B. H. Tai and P. V. Kiem, Tetrahedron Lett., 2022, 89, 153607 CrossRef CAS.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
- W. H. Zhang and C. T. Che, J. Nat. Prod., 2001, 64, 1489–1492 CrossRef CAS PubMed.
- F. Lv, Z. Deng, J. Li, H. Z. Fu, R. W. M. van Soest, P. Proksch and W. Lin, J. Nat. Prod., 2004, 67, 2033–2036 CrossRef PubMed.
- A. C. Herrera Braga, S. Zacchino, H. Badano, M. G. Sierra and E. A. Rúveda, Phytochemistry, 1984, 23, 2025–2028 CrossRef.
- L. Di Bari, G. Pescitelli, C. Pratelli, D. Pini and P. Salvadori, J. Org. Chem., 2001, 66, 4819–4825 CrossRef CAS PubMed.
- P. V. Kiem, D. T. Dung, P. H. Yen, N. X. Nhiem, T. H. Quang, B. H. Tai and C. V. Minh, Phytochem. Lett., 2018, 26, 199–204 CrossRef CAS.
- M. Tsuda, M. Ishibashi, K. Agemi, T. Sasaki and J. i. Kobayashi, Tetrahedron, 1991, 47, 2181 CrossRef CAS.
- A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2022 10.1039/D1NP00076D.
Footnote |
| † Electronic supplementary information (ESI) available: HR-ESI-MS, NMR, and ECD spectra of new compounds would be found. See DOI: 10.1039/d2ra01674e |
| This journal is © The Royal Society of Chemistry 2022 |