
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Parahydrogen-induced polarization study of imine hydrogenations mediated by a metal-free catalyst†
Danila O.
Zakharov
 a,
Konstantin
Chernichenko‡
b,
Kristina
Sorochkina
ab,
Timo
Repo
b and
Vladimir V.
Zhivonitko
*a
a,
Konstantin
Chernichenko‡
b,
Kristina
Sorochkina
ab,
Timo
Repo
b and
Vladimir V.
Zhivonitko
*a
aNMR Research Unit, Faculty of Science, University of Oulu, P.O. Box 3000, Oulu, 90014, Finland. E-mail: vladimir.zhivonitko@oulu.fi
bDepartment of Chemistry, University of Helsinki, A. I. Virtasen Aukio 1, 00014 Helsinki, Finland
First published on 29th August 2022
Abstract
Parahydrogen-induced polarization is a nuclear spin hyperpolarization technique that can provide strongly enhanced NMR signals for catalytic hydrogenation reaction products and intermediates. Among other matters, this can be employed to study the mechanisms of the corresponding chemical transformations. Commonly, noble metal complexes are used for reactions with parahydrogen. Herein, we present a PHIP study of metal-free imine hydrogenations catalyzed by the ansa-aminoborane catalyst QCAT. We discuss the reaction mechanism by showing the pairwise nature of the initial hydrogen activation step that leads to the formation of the negative net nuclear spin polarization of N–H hydrogen in the QCAT-H2 intermediate, enabling the further transfer of parahydrogen-originating protons to the imine substrate with the accumulation of hyperpolarized amine products. Parahydrogen-induced polarization also demonstrates the reversibility of the catalytic cycle.
Introduction
Amines are an important class of compounds widely used in various crucial areas such as the food, agrochemical and pharmaceutical industries.1,2 There are several synthetic methods employed to produce amines, among which metal-catalyzed reductions of imines, enamines, and nitriles are the most important ones.3 These approaches, however, use costly noble metals and can produce toxic chemical wastes.4 As an alternative, metal-free reductions of imines are known, such as those with hydroborate reagents.5 Other reported methods include Brønsted acid-catalyzed reduction with Hantzsch esters as the hydrogen source,6,7 Lewis base-catalyzed hydrosilylation with trichlorosilane8,9 and intermolecular hydroamination.10 In addition, hydrogenations of imines and enamines using frustrated Lewis pairs (FLPs) — pairs of sterically hindered Lewis acids and bases — were reported.11–13 In these reactions, derivatives of B(C6F5)3 are typically involved as Lewis acids forming intermolecular FLPs with the nitrogen-containing reagents. FLPs and molecular hydrogen generate hydroborates in situ as active catalytic species, thus bridging the gap between traditional metal-catalyzed hydrogenations and hydroborate reducing agents. In 2008, it was shown that the intramolecular FLP 1-{2-[bis(pentafluorophenyl)boryl]benzyl}-2,2,6,6-tetramethylpiperidine, CAT (Scheme 1), can catalyze the hydrogenation of imines and enamines by molecular hydrogen, demonstrating high activities.14 Thereafter, several alike ansa-aminoboranes (AABs) were studied in hydrogenation reactions. Among them, QCAT (Scheme 1) showed the best activity towards the hydrogenations of imines.15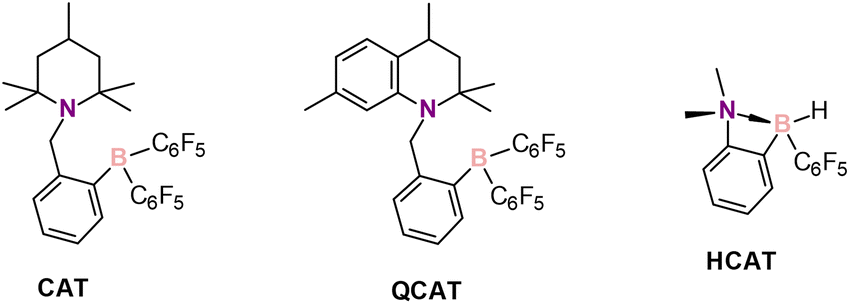 | ||
| Scheme 1 Selected AABs used in hydrogenations with molecular hydrogen. | ||
Understanding the mechanistic aspects of catalytic transformation can play a crucial role in improving catalyst performance. In this respect, NMR spectroscopy offers a versatile toolbox to study reaction mechanisms. However, the detection of possible catalytic intermediates leading to product formation is usually not a trivial task, since highly active species are typically present in low concentrations that are hard to observe using conventional NMR methodology. This can become feasible if the sensitivity of the NMR methods is increased by means of nuclear spin hyperpolarization to boost signal intensities.16 Parahydrogen-induced polarization (PHIP) is a specifically useful hyperpolarization technique in catalytic studies as hyperpolarization occurs naturally upon the chemical activation of H2 molecules enriched in the para nuclear spin isomer.17,18 When parahydrogen is activated by a catalyst, strongly enhanced NMR signals of the reaction intermediates or subsequent products can be observed, providing mechanistic information. To observe the hyperpolarization, H2 activation must be pairwise, i.e., the pair of H atoms must not be separated completely at least at the initial step.19
PHIP has demonstrated its power by proving the mechanisms of hydrogenation of various metal complex catalysts, especially in the detection of dihydride intermediates.19,20 For example, it was used to study the kinetics of the formation of [IrCO(H)2I(PPh3)2] after the light-induced reductive elimination of hydrogen from that species;21 to detect cationic RhI intermediates with coordinated arene rings;22 to determine the pathways of H2 exchange in (μ-H)2Os3(CO)10 clusters;23 to understand ligand exchange in [IrCO(H)2Cl(PPh3)2] and [IrH2Cl(PPh3)3];24 and in many other interesting mechanistic investigations19,20,25 including heterogeneous catalytic systems.26
Although PHIP was mostly used to study reactions mediated by metal-containing catalysts,19,20,27,28 it was reported that metal-free catalysts can activate parahydrogen, leading to PHIP effects.29,30 Specifically, AABs were the first FLPs to show spin hyperpolarization effects in reactions with parahydrogen.29,31,32 Later, several pnictogen biradicaloids were also documented as efficient metal-free parahydrogen activators.33,34 Interactions of the discussed CAT and QCAT AABs with parahydrogen were shown to provide enhanced signals of NH and BH hydrogens of the corresponding CAT-H2 and QCAT-H2 adducts, respectively.29 In general, until very recently, PHIP effects with metal-free compounds were observed exclusively for the catalyst–parahydrogen adducts formed at the initial H2 activation step. In 2022, we reported the nuclear spin hyperpolarization of various alkenes achieved by alkyne hydrogenations with parahydrogen over a metal-free hydroborane catalyst HCAT (Scheme 1).35
Herein, we show that the metal-free AAB catalyst QCAT can hyperpolarize various amines in imine hydrogenations with parahydrogen, providing among other matters information about the hydrogenation catalytic process. We observed unusual one-hydrogen hyperpolarization of product amine NH protons in the 1H NMR spectra. We demonstrate the reversibility of the catalytic cycle of imine hydrogenation catalyzed by QCAT via the observation of hyperpolarized amine when it was used as an individual initial reagent, shedding light on the mechanism of the involved catalytic transformations.
Results and discussion
Several aromatic imines were used to study hydrogenation with parahydrogen (Scheme 2). The reaction was performed at room temperature under 6 bar of 92% parahydrogen-enriched H2, referred to as para-H2 in the following text.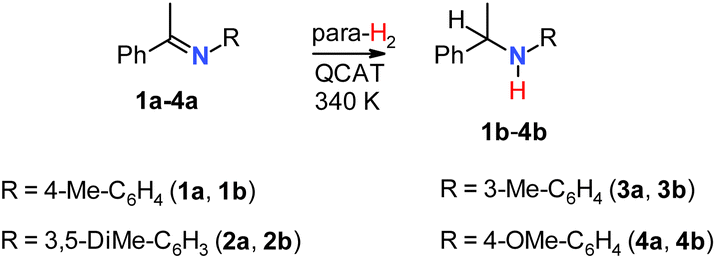 | ||
| Scheme 2 The studied imine hydrogenations. | ||
In the first experiments, the NMR spectra were recorded after introducing para-H2 into solutions of QCAT (0.04 M) and imines (0.13 M) in toluene-d8, examining the reaction in a high magnetic field (9.4 T). Although imine structures have some influence on the observed hyperpolarization effects, the resulting 1H NMR spectra generally demonstrated similar features. For instance, Fig. 1a illustrates the results of the hydrogenation of 1a into 1b with para-H2 at 340 K. There are several signals that reveal the hyperpolarization effects via antiphase and negative in-phase multiplets. The four antiphase doublets centred at ca. 4.0 ppm correspond to the B–H group hydrogen of the QCAT-H2 adduct; the splitting is induced by coupling to the spin-3/2 11B nucleus. The observation of antiphase signals is typical for high-field experiments according to the PASADENA experiment theory for weakly coupled spin-systems.36 In contrast, the N–H group signal of QCAT-H2 at ca. 8.8 ppm shows a mixture of broad antiphase and in-phase negative signal contributions. The formation of the net negative polarization in the high-field experiment is unusual as coherent spin dynamics cannot lead to this effect.35 However, it is crucial for the observation of the hyperpolarized amine 1b as discussed below. As seen in Fig. 1a, the N–H group signal of the hydrogenation product 1b appears as a negative in-phase signal at ca. 3.4 ppm (Fig. 1a), most likely inheriting the net negative polarization from the N–H hydrogen of QCAT-H2.
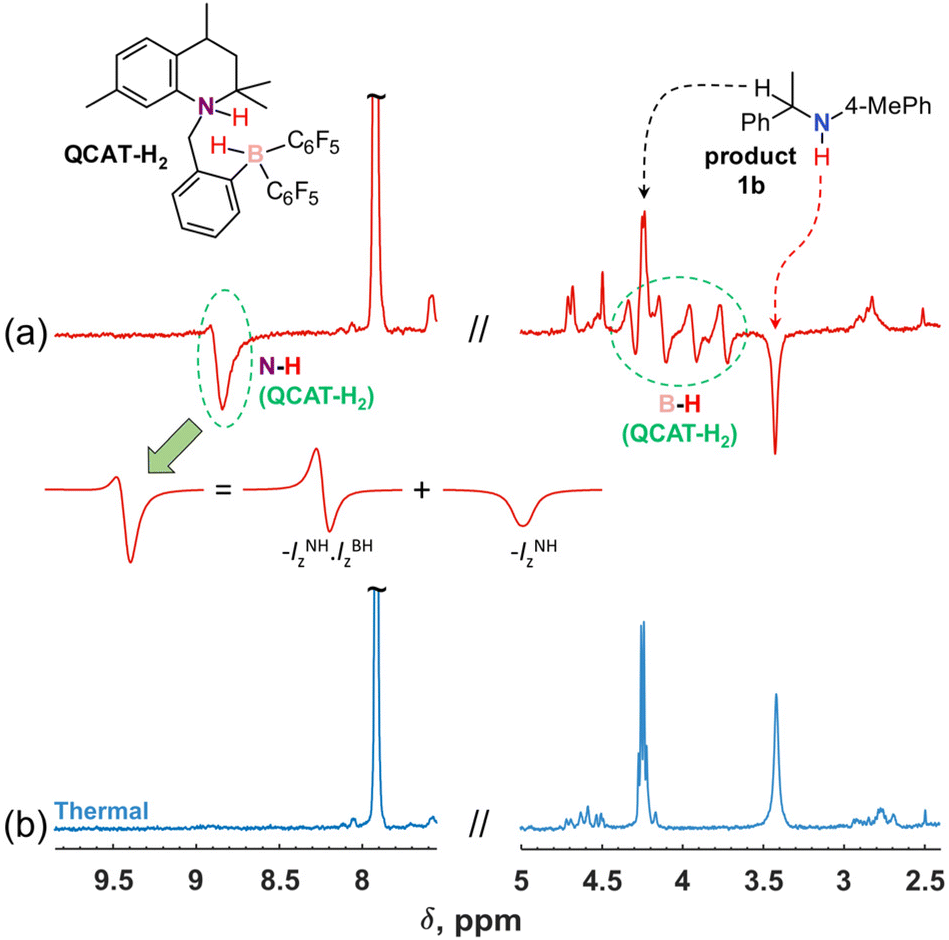 | ||
| Fig. 1 1H NMR spectra of the QCAT-catalyzed hydrogenation of (1E)-N-(4-methylphenyl)-1-phenylethan-1-imine (1a) with para-H2. The red spectrum (a) was acquired using a π/4 pulse just after introducing para-H2 into a solution containing 0.04 M QCAT and 0.13 M of 1a in toluene-d8 at 340 K. The enhanced antiphase signals correspond to the B–H group hydrogen (centred at 4.0 ppm) of the QCAT-H2 adduct. The N–H group hydrogen of QCAT-H2 (8.8 ppm) revealed a mixed antiphase/in-phase signal shape (see the decomposition). The N–H group hydrogen of amine product 1b (3.4 ppm) revealed an enhanced in-phase negative signal. The blue spectrum (b) was recorded after the hyperpolarization relaxed at 340 K in 1 min, approaching the thermal equilibrium. | ||
Generally, the formation of a hyperpolarized QCAT-H2 adduct is in accord with the proposed four-step mechanism of imine hydrogenations over QCAT (Scheme 3 and Scheme S1 in the ESI†).15 According to this process, a hydrogen molecule is activated by QCAT, yielding the QCAT-H2 adduct (Step I). The following proton transfer from the N–H group of QCAT-H2 to the imine nitrogen atom results in an unstable ionic pair intermediate (Step II). The consequent hydride transfer from the anion to the cation leads to the dative QCAT-amine adduct (Step III). The last stage of the hydrogenation cycle releases the product amine and recovers the catalyst (Step IV). Either Step II or Step IV is the rate-determining step, depending on the substrate and catalyst structures.15
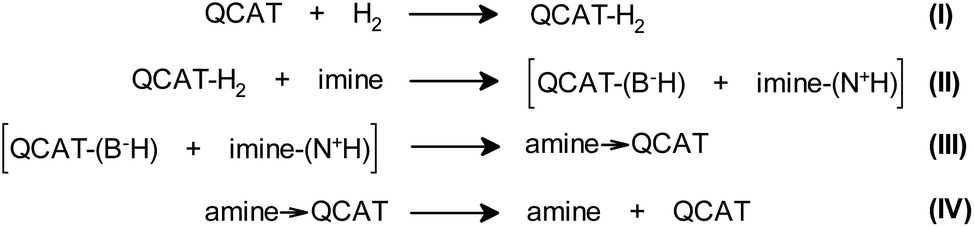 | ||
| Scheme 3 Proposed mechanism of imine hydrogenation over the QCAT catalyst. | ||
Importantly, Step I is pairwise and should lead to hyperpolarization after para-H2 activation.29 The formation of the final amine product 1bvia Steps II–IV either may or may not be pairwise, since the formation of the unlinked [QCAT-(B−H) + imine-(N+H)] ion pair at Step II can mix hydrogens coming from different para-H2 molecules, preventing the pairwise H2 transfer to form 1b and observation of the antiphase contribution. In terms of magnetisation modes, this means that the two-spin order, –IzNH·IzBH, inherited from para-H2, must be destroyed by the non-pairwise process at Step II. In contrast, the single-spin net polarization, –IzNH, created by the QCAT-H2 intermediate at Step I, can survive the non-pairwise transfer and result in hyperpolarized amine 1b.
The non-pairwise nature of Step II can be confirmed to an extent indirectly. For instance, the hydrogenation of 1a using borane catalyst B(C6F5)3 does not lead to any hyperpolarization effects (see the ESI, Fig. S11†). In that case, the activation of para-H2 is performed by an intermolecular borane–imine FLP, meaning that the intermolecular nature of FLPs prevents the observation of PHIP. A similar conclusion was drawn while comparing alkyne and alkene hydrogenations with intra- and intermolecular FLPs, respectively,35 where the latter did not show any hyperpolarization effects but the former did (see the ESI, Fig. S13 and S14†). At the same time, we cannot rule out completely the possibility that the two-spin orders survived the chemical transformation because the J-coupling constant between the vicinal N–H and C–H protons in 1a is small, which can lead to the mutual cancellation of the antiphase components.
As we can be sure that the source of the negative net polarization of the N–H group proton in 1b is the QCAT-H2 intermediate, it is worth briefly discussing the reasons for its formation in Step I. There are examples of one-hydrogen polarization reported for metal complex catalysts, e.g., in hydroformylation reactions (oneH-PHIP effect)37 and the exchange of water ligands.38 The observations were rationalized as a result of the para-H2-derived protons exhibiting strong coupling in an intermediate, thus, accumulating net polarization via coherent dynamics before the transfer to the target substrate. However, in the case of QCAT-H2, those protons are in the weak coupling regime (Δω = 2 kHz at 9.4 T; JHH = 3 Hz). In principle, the theory of the PASADENA effect in the case of weakly coupled systems predicts a negligible contribution to single-spin polarization, as kinetic averaging of the density operator leaves almost exclusively only –IzNH·IzBH, when para-H2 is added to highly magnetically inequivalent sites.36 Moreover, knowing that the sign of the J-coupling constant is positive, the theory predicts positive possible single-spin polarization created by the coherent mixing on the N–H proton site in QCAT-H2,35,36 whereas our experiments show negative single-spin polarization. Most likely, the two-spin → single-spin-order transformation (–IzNH·IzBH → –IzNH) is induced by a cross-correlated relaxation process, e.g., via correlated fluctuations of the dipole–dipole coupling of the N–H⋯H–B proton pair and the chemical shift anisotropy of the N–H proton in the QCAT-H2 adduct.39,40 This seems to be a common feature of the H2 adducts of AABs, as similar effects have been observed and discussed in para-H2 activations for a variety of such systems.29,32,35 Indeed, a comparison of the hydrogenation of imine 1a with para-H2 in two different magnetic fields (9.4 and 14.1 T) revealed an increase in the net negative polarization level by a factor of 1.3–1.4 at the higher magnetic field for both QCAT-H2 and the amine product 1b (see Fig. S6 in ESI†). This observation indicates that the chemical shift anisotropy should indeed play an important role as its effect increases with the increase in the magnetic field strength. However, a more detailed discussion of this transformation requires extensive dedicated experiments that are beyond the scope of the current publication and will be done in the future. Experimental results on the hydrogenations of other imines (2a–4a) at 340 K are summarized in Fig. 2.
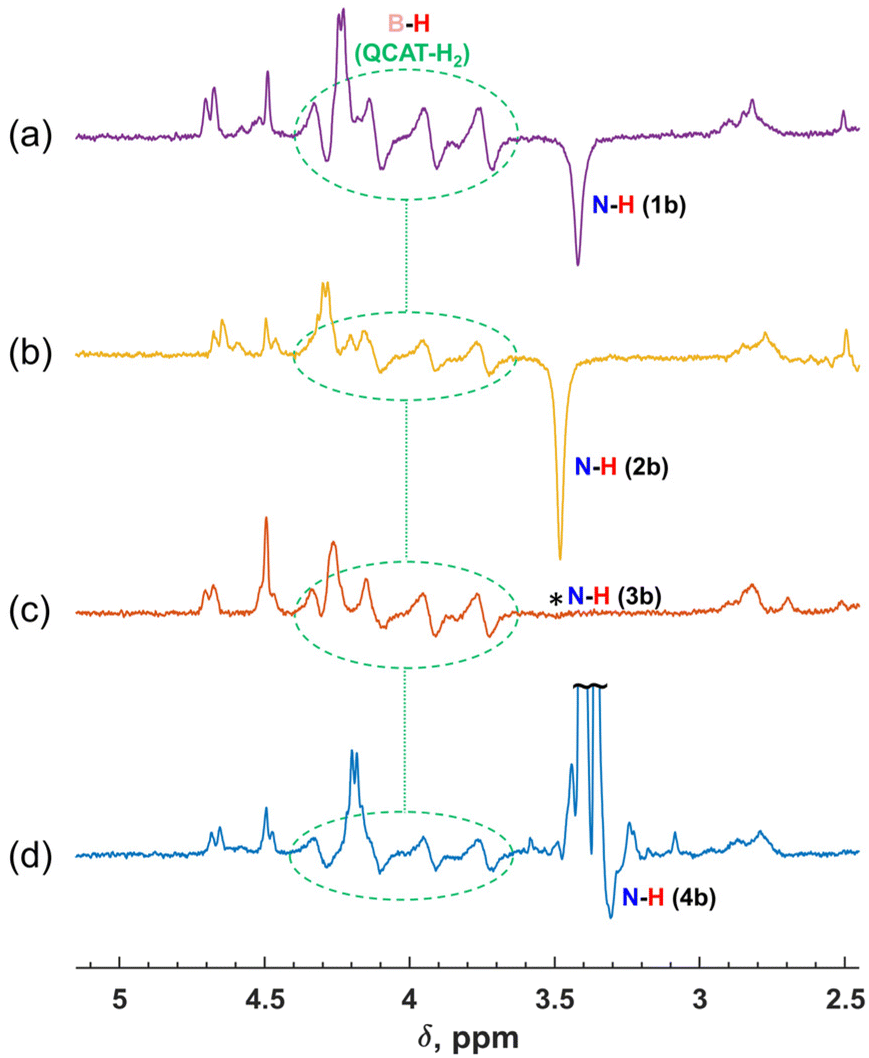 | ||
| Fig. 2 (a)–(d) 1H NMR spectra showing enhanced resonances observed in the hydrogenations of imines 1a–4a with para-H2 over the QCAT catalyst at 340 K in toluene-d8, respectively. The spectra were measured at the first moments after introducing para-H2 into the corresponding imine–QCAT solutions. Spectral regions with the B–H group signals of the QCAT-H2 intermediate and the N–H signals of the amine products 1b–4b are displayed. Dotted ovals highlight the B–H group signals of QCAT-H2. The positions of the amine products are shown with text labels. The position of the signal of the N–H group hydrogen of 3b disappeared after introducing para-H2, which is marked with an asterisk in (c). | ||
In all cases, the hyperpolarization effects were visible for para-H2 originating hydrogens in the QCAT-H2 intermediate and the N–H proton in amine products. The strengths of amine hyperpolarization, however, vary from one product to another. The in-phase negative signal of the N–H group hydrogen of 2b observed via the hydrogenation of 2a was slightly stronger as compared with that of 1b. In principle, the use of more basic and more sterically hindered imines should lead to higher conversion rates,15 and we could expect a somewhat higher intensity of the enhanced signal for 2b. In the case of 3b, the negative signal was not visible. Instead, the thermal signal of this amine disappeared after adding a fresh portion of para-H2, implying the cancellation of the enhanced negative and thermal positive signals. The negative net polarization of the N–H group hydrogen was also observed in the case of 4b, similarly to 1b and 2b. However, it was not that clearly visible due to the overlapping of the N–H and OCH3 signals in the 1H NMR spectra. Overall, the estimated signal enhancement factors due to hyperpolarization are 10 (1b), 15 (2b), 7 (3b) and 10 (4b) (see Table S1 in the ESI†), which are not record high values for PHIP. Theoretically, these factors can be more than four orders of magnitude for the 1H nuclei in a 9.4 T magnetic field,36 while the experimentally observed values are typically smaller but may be more than three orders of magnitude, e.g., in hydrogenations of alkynes with para-H2 using cationic Rh complexes as catalysts. Nevertheless, the rather moderate enhancements seen in our case still provided valuable qualitative information about the mechanistic features and consistency in the observed effects for different imine structures.
Further experiments showed that raising the temperature to 360 K can lead to the observation of a negative net polarization also for orthohydrogen (ortho-H2; see the results for 1b in Fig. S1 in the ESI†). Moreover, the C–H group hydrogen of the amine product revealed the hyperpolarization as well as the N–H group hydrogen. It was also visible that negative components of the B–H antiphase doublets of QCAT-H2 become stronger than the positive ones. It seems that under these experimental conditions, the QCAT + H2 ↔ QCAT-H2 exchange is fast enough to release hyperpolarized free ortho-H2 into the reaction “cocktail”, which in turn is transferred into the amine product via Steps I–IV, contributing to the polarization of both C–H and N–H hydrogens. This observation pushed us to conclude that reversible exchange processes can play a significant role in the whole catalytic cycle at higher temperatures, allowing observation of the hyperpolarization effects when we substitute imine for amine as a substrate in the reaction mixture. In other words, we could expect to go through Steps I–IV in the reverse order and return, polarizing the amine substrate without alteration of its chemical structure. Indeed, when the experiments were repeated with QCAT, para-H2 and commercial amine 1b in the reaction mixture, the negative signal of the N–H group hydrogen of 1b was observed at 360 K (Fig. 3a). The concentrations of the QCAT catalyst and amine were approximately on the same level as in the discussed imine hydrogenation reactions. We believe that these experiments show that basically all the mechanistic reaction steps shown in Scheme 3 must be reversible to a significant extent to enable the observation of the hyperpolarized amine without the formal hydrogenation of an unsaturated substrate.
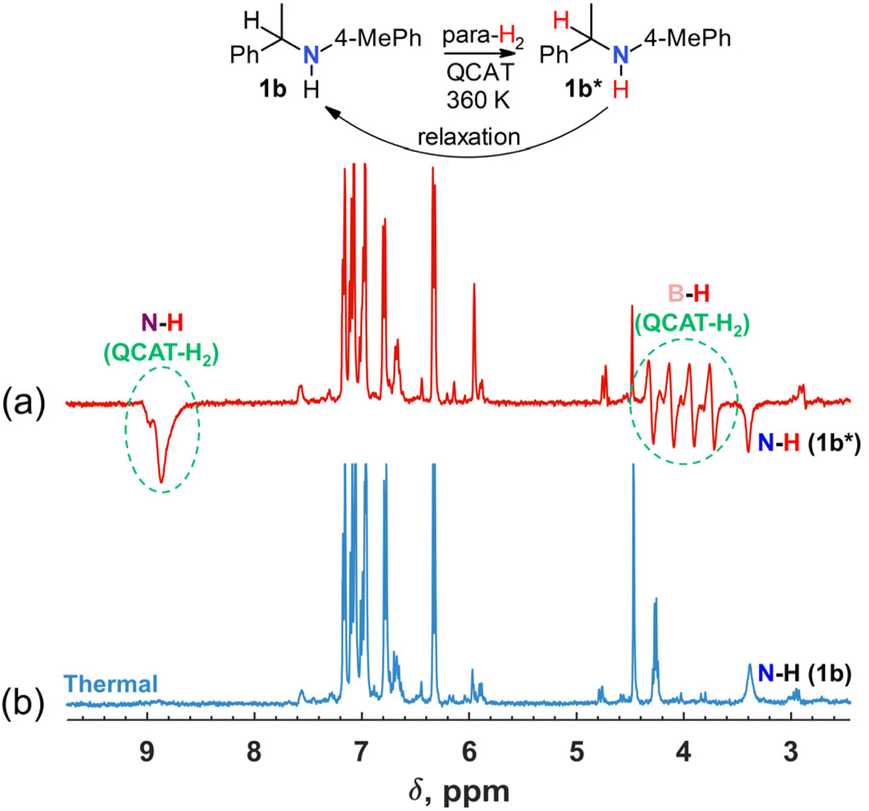 | ||
| Fig. 3 1H NMR spectra acquired during the reaction of para-H2, QCAT and 1b. The red spectrum (a) was acquired using a π/4 pulse just after introducing para-H2 into a solution containing 0.04 M QCAT and 0.04 M of 1b in toluene-d8 at 360 K. The negative signal of the N–H proton of amine 1b is clearly visible. The signal of the α-C–H hydrogen of 1b completely vanished, indicating a weak hyperpolarization of this group. The blue spectrum (b) was recorded after relaxation to the thermal equilibrium. | ||
The reverse Step III requires the abstraction of hydride from activated C(sp3)–H bonds in primary or secondary amines. The feasibility of the abstraction was reported for strong Lewis acids, such as B(C6F5)3.41 For example, hydride abstraction from the activated α-position in NEt2Ph proceeds in a facile manner to yield the corresponding iminium hydroborate. It can interact with the initial amine, deprotonating iminium with the formation of enamine and ammonium hydroborate salt.41 A similar reversibility was also documented for the abstraction of α-C–H hydrogens in amines by QCAT. For instance, QCAT demonstrated a good racemization activity of amine 4b,15 which requires the elimination of α-hydrogen,42 proving the ability of QCAT to dehydrogenate amines. The reversibility of Step I was known from previous studies.29 In addition to this, our experiments show that Steps II and IV can be also reversible. The observed hyperpolarization effect of the reversible process was weaker as compared with that of the imine hydrogenation (see the N–H signals in Fig. 1a and 3a), but provided important mechanistic information. We also found in a test experiment an even weaker but still detectable hyperpolarization of the amino group in 1,2,3,4-tetrahydro-2,2,4,7-tetramethylquinoline, which does not have α-C–H hydrogen in the structure, in the exchange experiment at 360 K (Fig. S9 in the ESI†). This implies that the direct N–H⋯H–N exchange between QCAT-H2 and the target amine may contribute to hyperpolarization. We should note that a similar proton transfer from the hyperpolarized QCAT-H2 may serve as a route to sensitize other materials under exchange. This is the foundation of SABRE RELAY43 and PHIP-X,44 which obviously should be explored in future works with metal-free para-H2 activators.
Finally, we tested the influence of QCAT concentration on the nuclear spin hyperpolarization effects. In addition to the described experiments with 33 mol% QCAT (Fig. 1 and 2), we conducted several reactions using lower catalyst loadings (see the ESI, Fig. S10†). The hydrogenation of imine 4a with para-H2 using 1–3 mol% of QCAT at 330 K did not lead to the observation of the enhanced antiphase and/or negative in-phase signals in the 1H NMR spectra. Since the signals of QCAT-H2 and the amine product 4b were extensively hampered by the intense signals of the substrate, we were unable to see any hyperpolarization effects. The reaction rate also was significantly lower as compared with the high catalyst loading. In the experiment using 10 mol% QCAT, the rate significantly increased, and we observed hyperpolarization of the NH and BH hydrogens of QCAT-H2 similarly to those with 33 mol% loadings. We noticed that the strength of these signals increased with the increase in the reaction temperature. The negative signal of the NH hydrogen of the amine product quite significantly overlapped with other signals in the spectrum in the case of 4b.
Conclusions
Ansa-aminoboranes can provide interesting capabilities for hydrogenation catalysis without metals. Specifically, QCAT shows a relatively high activity in imine hydrogenations which can be tailored further to get even better results. Knowledge of the mechanistic details can extremely be helpful in this regard. In this study, we showed that parahydrogen-induced polarization can provide useful insights into the activation of H2 by QCAT, proving the pairwise nature of this step. Moreover, using the single-spin negative net polarization created in QCAT-H2 intermediates upon parahydrogen activation, it was possible to track the transfer of hydrogens to several imine substrates leading to the formation of hyperpolarized amine products. This is the first demonstration of the production of PHIP hyperpolarized amines from imines using metal-free catalysts. We also showed that when amines are used as initial substrates in the reaction with QCAT and parahydrogen their N–H group hydrogens can become hyperpolarized without any formal chemical structure changes, proving the reversibility of all the steps of the catalytic cycle. The variation in the catalyst concentration revealed the reaction rate alterations and the decrease in the amplitudes of the hyperpolarization effects at low catalyst loadings. An order of magnitude signal enhancements were observed for N–H hydrogen in amine products. We believe that further development of metal-free imine hydrogenation can lead to much higher enhancements, ideally reaching the levels of metal-containing catalysts. Generally, this study shows that PHIP-assisted mechanistic studies provide important mechanistic information about metal-free hydrogenations, as proved many times similarly for metal-containing catalysts19,20,45,46 and, in addition to this study, only once for a metal-free catalyst.35Author contributions
DOZ: investigation, validation, and writing – original draft; KC: conceptualisation, validation, and writing – review and editing; KS: validation and writing – review and editing; TR: conceptualisation, funding acquisition, resources, supervision, validation, and writing – review and editing; VVZ: conceptualisation, funding acquisition, resources, supervision, validation, visualisation, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Academy of Finland (grant no. 323480) and the University of Oulu (Kvantum Institute). T. R. acknowledges the Academy of Finland for the financial support (grant no. 276586). The authors thank Dr Victor Sumerin (the University of Helsinki, former member) for the synthesis of the QCAT catalyst.References
- A. Kleemann and J. R. Engel, Pharmaceutical Substances: Syntheses, Patents, Applications, Thieme, Stuttgart, New York, 3rd edn, 1999 Search PubMed.
- A. Albert, Selective Toxicity: The Physico-chemical Basis of Therapy, Chapman and Hall, London, New York, 7th edn, 1985 Search PubMed.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, Wiley-VCH, Weinheim, 2nd rev. and enl. edn, 2004 Search PubMed.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS.
- E. W. Baxter and A. B. Reitz, in Organic Reactions, 2002, vol. 59, pp. 1–714 Search PubMed.
- S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- M. Rueping, E. Sugiono and F. R. Schoepke, Synlett, 2010, 852–865, DOI:10.1055/s-0029-1219528.
- A. V. Malkov, K. Vrankova, S. Stoncius and P. Kocovsky, J. Org. Chem., 2009, 74, 5839–5849 CrossRef CAS.
- F. M. Gautier, S. Jones and S. J. Martin, Org. Biomol. Chem., 2009, 7, 229–231 RSC.
- J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795–813 CrossRef CAS.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- D. J. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131 RSC.
- P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Frohlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS.
- V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskelä, T. Repo, P. Pyykko and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117–14119 CrossRef CAS PubMed.
- V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef CAS.
- J.-H. Ardenkjaer-Larsen, G. S. Boebinger, A. Comment, S. Duckett, A. S. Edison, F. Engelke, C. Griesinger, R. G. Griffin, C. Hilty, H. Maeda, G. Parigi, T. Prisner, E. Ravera, J. van Bentum, S. Vega, A. Webb, C. Luchinat, H. Schwalbe and L. Frydman, Angew. Chem., Int. Ed., 2015, 54, 9162–9185 CrossRef CAS PubMed.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- B. J. Tickner and V. V. Zhivonitko, Chem. Sci., 2022, 13, 4670–4696 RSC.
- S. B. Duckett and N. J. Wood, Coord. Chem. Rev., 2008, 252, 2278–2291 CrossRef CAS.
- B. Procacci, P. M. Aguiar, M. E. Halse, R. N. Perutz and S. B. Duckett, Chem. Sci., 2016, 7, 7087–7093 RSC.
- R. Giernoth, P. Huebler and J. Bargon, Angew. Chem., Int. Ed., 1998, 37, 2473–2475 CrossRef CAS PubMed.
- S. Aime, W. Dastru, R. Gobetto, F. Reineri, A. Russo and A. Viale, Organometallics, 2001, 20, 2924–2927 CrossRef CAS.
- C. J. Sleigh, S. B. Duckett and B. A. Messerle, Chem. Commun., 1996, 2395–2396 RSC.
- S. B. Duckett, in Encyclopedia of Spectroscopy and Spectrometry, ed. J. C. Lindon, G. E. Tranter and D. W. Koppenaal, Elsevier, Academic Press, 3rd edn, 2016, pp. 527–534 Search PubMed.
- K. V. Kovtunov, O. G. Salnikov, V. V. Zhivonitko, I. V. Skovpin, V. I. Bukhtiyarov and I. V. Koptyug, Top. Catal., 2016, 59, 1686–1699 CrossRef CAS.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- S. B. Duckett and C. J. Sleigh, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 34, 71–92 CrossRef CAS.
- V. V. Zhivonitko, V.-V. Telkki, K. Chernichenko, T. Repo, M. Leskelä, V. Sumerin and I. V. Koptyug, J. Am. Chem. Soc., 2014, 136, 598–601 CrossRef CAS PubMed.
- L. E. Longobardi, C. A. Russell, M. Green, N. S. Townsend, K. Wang, A. J. Holmes, S. B. Duckett, J. E. McGrady and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 13453–13457 CrossRef CAS PubMed.
- V. V. Zhivonitko, K. Sorochkina, K. Chernichenko, B. Kotai, T. Foldes, I. Papai, V.-V. Telkki, T. Repo and I. Koptyug, Phys. Chem. Chem. Phys., 2016, 18, 27784–27795 RSC.
- K. Sorochkina, V. V. Zhivonitko, K. Chernichenko, V. V. Telkki, T. Repo and I. V. Koptyug, J. Phys. Chem. Lett., 2018, 9, 903–907 CrossRef CAS.
- V. V. Zhivonitko, J. Bresien, A. Schulz and I. V. Koptyug, Phys. Chem. Chem. Phys., 2019, 21, 5890–5893 RSC.
- V. V. Zhivonitko, H. Beer, D. O. Zakharov, J. Bresien and A. Schulz, ChemPhysChem, 2021, 22, 813–817 CrossRef CAS PubMed.
- D. O. Zakharov, K. Chernichenko, K. Sorochkina, S. Yang, V. V. Telkki, T. Repo and V. V. Zhivonitko, Chem. – Eur. J., 2022, 28, e202103501 CrossRef CAS PubMed.
- C. R. Bowers, in Encyclopedia of Nuclear Magnetic Resonance, ed. D. M. Grant and R. K. Harris, Wiley, Chichester, 2002, vol. 9, pp. 750–769 Search PubMed.
- A. B. Permin and R. Eisenberg, J. Am. Chem. Soc., 2002, 124, 12406–12407 CrossRef CAS PubMed.
- M. Emondts, D. Schikowski, J. Klankermayer and P. P. M. Schleker, ChemPhysChem, 2018, 19, 2614–2620 CrossRef CAS.
- A. Kumar, R. C. R. Grace and P. K. Madhu, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 37, 191–319 CrossRef CAS.
- S. Aime, W. Dastru, R. Gobetto, A. Russo, A. Viale and D. Canet, J. Phys. Chem. A, 1999, 103, 9702–9705 CrossRef CAS.
- J. Paradies, Eur. J. Org. Chem., 2019, 283–294 CrossRef CAS.
- E. J. Ebbers, G. J. A. Ariaans, J. P. M. Houbiers, A. Bruggink and B. Zwanenburg, Tetrahedron, 1997, 53, 9417–9476 CrossRef CAS.
- W. Iali, P. J. Rayner and S. B. Duckett, Sci. Adv., 2018, 4, eaao6250 CrossRef PubMed.
- K. Them, F. Ellermann, A. N. Pravdivtsev, O. G. Salnikov, I. V. Skovpin, I. V. Koptyug, R. Herges and J.-B. Hövener, J. Am. Chem. Soc., 2021, 143, 13694–13700 CrossRef CAS PubMed.
- V. V. Zhivonitko, I. V. Skovpin, K. C. Szeto, M. Taoufik and I. V. Koptyug, J. Phys. Chem. C, 2018, 122, 4891–4900 CrossRef CAS PubMed.
- V. V. Zhivonitko, I. V. Skovpin, M. Crespo-Quesada, L. Kiwi-Minsker and I. V. Koptyug, J. Phys. Chem. C, 2016, 120, 4945–4953 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, results of supplementary experiments. See DOI: https://doi.org/10.1039/d2dt02178a |
| ‡ Present address: Discovery, Product Development & Supply (DPDS), Janssen Pharmaceutical Companies of Johnson & Johnson, Turnhoutseweg 30, 2340 Beerse, Belgium. |
| This journal is © The Royal Society of Chemistry 2022 |