
Ruthenium(II) complexes with phosphonate-substituted phenanthroline ligands: synthesis, characterization and use in organic photocatalysis†
Gleb V.
Morozkov
 a,
Anton S.
Abel
*a,
Mikhail A.
Filatov
b,
Sergei E.
Nefedov
c,
Vitaly A.
Roznyatovsky
a,
Andrey V.
Cheprakov
a,
Alexander Yu.
Mitrofanov
ad,
Ilia S.
Ziankou
ad,
Alexei D.
Averin
a,
Irina P.
Beletskaya
a,
Julien
Michalak
d,
Christophe
Bucher
*e,
Laurent
Bonneviot
e and
Alla
Bessmertnykh-Lemeune
*de
a,
Anton S.
Abel
*a,
Mikhail A.
Filatov
b,
Sergei E.
Nefedov
c,
Vitaly A.
Roznyatovsky
a,
Andrey V.
Cheprakov
a,
Alexander Yu.
Mitrofanov
ad,
Ilia S.
Ziankou
ad,
Alexei D.
Averin
a,
Irina P.
Beletskaya
a,
Julien
Michalak
d,
Christophe
Bucher
*e,
Laurent
Bonneviot
e and
Alla
Bessmertnykh-Lemeune
*de
aLomonosov Moscow State University, Department of Chemistry, 1-3, Leninskie Gory, Moscow, 119991, Russia. E-mail: antonabel@list.ru
bSchool of Chemical and Pharmaceutical Sciences, Technological University Dublin, City Campus, Grangegorman, Dublin 7, Ireland
cN.S. Kurnakov Institute of General and Inorganic Chemistry RAS, 119991, Leninsky pr., 31, Moscow, Russian Federation
dInstitut de Chimie Moléculaire de l'Université de Bourgogne, UMR CNRS 6302, Université Bourgogne Franche-Comté, 9 Avenue Alain Savary, 21078 Dijon, France
eENS de Lyon, UMR 5182, CNRS, Laboratoire de Chimie, 69342 Lyon, France. E-mail: alla.lemeune@ens-lyon.fr; christophe.bucher@ens-lyon.fr
First published on 27th June 2022
Abstract
Ru(II) complexes with polypyridyl ligands play a central role in the development of photocatalytic organic reactions. This work is aimed at the structural modification of such complexes to increase their photocatalytic efficiency and adapt them for the preparation of reusable photocatalytic systems. Nine [Ru(phen)(bpy)2]2+-type complexes (bpy = 2,2′-bipyridine, phen = 1,10-phenanthroline) (Ru-Pcat) bearing the P(O)(OEt)2 substituent attached to the phen core directly or through a 1,4-phenylene linker were synthesized and characterized by spectroscopic and electrochemical techniques. The coordination mode of phen ligands was confirmed by single crystal X-ray analysis. The (spectro)electrochemical data show that the first electron transfer in Ru-Pcat takes place on the phen ligand. The emission maxima and quantum yields are strongly affected by the substitution pattern, reaching the far-red region (697 nm) for Ru-3,8P2. The singlet oxygen quantum yields of Ru-Pcat were evaluated using the chemical trapping method. Finally, the photocatalytic performance of Ru-Pcat in the oxidation of sulfides with molecular oxygen was investigated. Both dialkyl and alkyl aryl sulfides were quantitatively transformed into sulfoxides under irradiation with a blue LED in the acetonitrile–water mixture (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) using a low loading of 0.005–0.05 mol% Ru(II) photocatalysts. To rationalize the effect of phosphonate substituents on the photocatalytic efficiency, comparative kinetic studies of (1) 4-nitrothioanisole oxidation proceeding predominantly via the electron transfer pathway and (2) oxidation of dibutyl sulfide wherein singlet oxygen serves as an oxidant have been performed. It was demonstrated that complexes with the P(O)(OEt)2 substituent at positions 4 and 7 outperform the benchmark photocatalyst Ru-(bpy)3 and the parent complex Ru-phen in the reactions proceeding through electron transfer (reductive quenching photocatalytic cycle). The TON in the oxidation of 4-methoxythioanisole was found to be as high as 1000000 that is, to our knowledge, the highest among previously reported photocatalysts. In contrast, upon separating the P(O)(OEt)2 group and the phen core with the 1,4-phenylene linker, singlet oxygen quantum yields significantly increase that favors reactions proceeding through energy transfer (the oxidation of dibutyl sulfide in our case). Thus, both series of Ru(II) complexes prepared in this work are promising for the improvement of known photocatalytic reactions and the development of new transformations.
:1) using a low loading of 0.005–0.05 mol% Ru(II) photocatalysts. To rationalize the effect of phosphonate substituents on the photocatalytic efficiency, comparative kinetic studies of (1) 4-nitrothioanisole oxidation proceeding predominantly via the electron transfer pathway and (2) oxidation of dibutyl sulfide wherein singlet oxygen serves as an oxidant have been performed. It was demonstrated that complexes with the P(O)(OEt)2 substituent at positions 4 and 7 outperform the benchmark photocatalyst Ru-(bpy)3 and the parent complex Ru-phen in the reactions proceeding through electron transfer (reductive quenching photocatalytic cycle). The TON in the oxidation of 4-methoxythioanisole was found to be as high as 1000000 that is, to our knowledge, the highest among previously reported photocatalysts. In contrast, upon separating the P(O)(OEt)2 group and the phen core with the 1,4-phenylene linker, singlet oxygen quantum yields significantly increase that favors reactions proceeding through energy transfer (the oxidation of dibutyl sulfide in our case). Thus, both series of Ru(II) complexes prepared in this work are promising for the improvement of known photocatalytic reactions and the development of new transformations.
Introduction
Visible light-active Ru(II) and Ir(III) complexes with chelating ligands containing pyridyl residues (VATMPY complexes) such as 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen) and 2,2′,6′,2′′-terpyridine have been intensively studied for more than a half of century due to their relevance to molecular electronics, host–guest modeling, functional supramolecular systems and labeling of biomolecules, and electro- and photocatalysis.1–6 They also found application in organic synthesis as photosensitizers for the oxidation of unsaturated compounds, phosphines and sulfides using molecular oxygen.7 During the last decade, impressive progress has been achieved in their application as catalysts in visible light-driven photoredox catalytic reactions.8–10 The excited states of Ru(II) or Ir(III) complexes with bpy ligands have both the oxidizing (Ru(III) or Ir(IV)) and reducing (bpy˙−) sites11 and can mediate a wide range of organic reactions proceeding through both reductive or oxidative quenching photocatalytic cycles.Among many light-active organic compounds and metal complexes, which were successfully used as photoredox catalysts, Ru(II) and Ir(III) complexes have attracted particular interest due to their high light absorptivity, high reduction and oxidation potentials and exceptional photostability. Moreover, the redox and spectroscopic properties of these complexes can be fine-tuned by the introduction of substituents at the periphery of polypyridyl ligands.12 The catalyst screening has been recognized as an efficient tool in photoreaction optimization, but the number of Ru(II) and Ir(III) complexes which have been investigated is rather limited and [Ru(bpy)3]2+ is still a benchmark photocatalyst among Ru(II) complexes.10 This is in part due to the laborious synthesis of these complexes and the rather obscure understanding of structure–activity relationships in photocatalytic reactions.
Some relevant information on the substituent effect in photocatalytic reactions can be gathered from the data obtained in more mature fields where Ru(II) and Ir(III) complexes were applied, e.g., in electron transfer processes, light harvesting, binding to DNA and water splitting.13–18 For instance, an investigation of electron transfer reactions between ruthenium(II) complexes with bpy ligands and triethylamine (NEt3) showed that the back electron transfer can be retarded by introducing hydrophobic substituents in the pyridine ring that favors the chemical transformation of intermediates arising after the initial electron transfer step.19 This observation led to one of the early examples of photoredox catalyzed reactions, i.e., the oxidative hydrolysis of NEt3 to acetaldehyde,19 which led to a conclusion that the efficiency of photoredox catalysts is governed not only by the electronic properties of substituents, but also by their hydrophobicity and steric effects.19,20 In recent years, the development of structure–activity relationships for VATMPY complexes has attracted some attention.21–23 However, despite the increasing amount of data on the photophysical and redox properties of such compounds, the development of reliable guidelines for designing complexes, which meet specific performance criteria remains a challenge.24
Our work is focused on the optimization of the Ru(II) photocatalyst efficiency. Although only a single polypyridyl ligand is required to acquire MLCT transitions in these complexes,14 the replacement of one bpy ligand in the coordination shell of the ruthenium atom by another specific chelator can have a detrimental effect on the photophysical properties and reactivity.25 Taking this into account, we explored the functionalization of polypyridyl ligands as a way to increase the catalytic efficiency of Ru(II) photocatalysts. In this work, we synthesized two series of mixed ligand [Ru(phen)(bpy)2]2+-type complexes (Ru-Pcat), in which the phen ligand is functionalized with the diethoxyphosphoryl group (P(O)(OEt)2) (Fig. 1) and investigated their photocatalytic properties in the photooxidation of sulfides to sulfoxides by molecular oxygen. In the first series (Ru-P), the phosphorus substituent is directly bonded to the phen core, while in the second series (Ru-PPh), these fragments are separated by a 1,4-phenylene spacer, which allows for a systematic tuning of the photophysical and redox properties of the studied complexes. 1,10-Phenanthrolines bearing a phosphonate substituent at position 2 were excluded from these investigations because the bleaching of Ru(II) complexes which involves the photoinduced ligand dissociation may be accelerated in these complexes due to steric strain around the Ru(II) center.
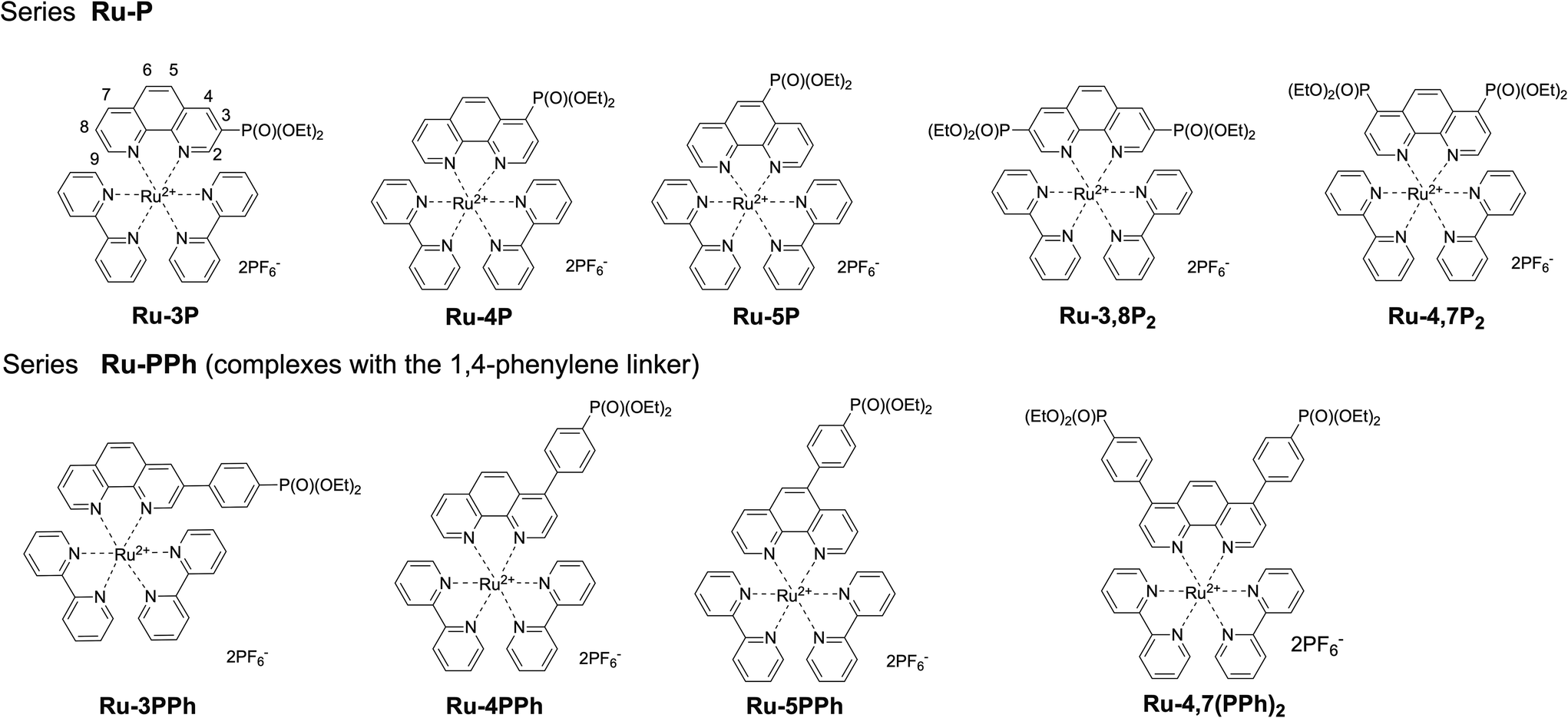 | ||
| Fig. 1 Structures of Ru(II) complexes Ru-Pcat investigated in this work. | ||
The hydrophilic and strongly electron-withdrawing P(O)(OEt)2 group could offer several benefits relevant to photocatalytic applications such as increasing solubility of photocatalysts in solvents with medium polarity and fine tuning of their redox potentials. Moreover, diesters of phosphonic acids can be easily transformed into phosphonic acids or their mono esters, both useful for further development of water-soluble photocatalysts. Finally, the phosphonate substituent is a well-known anchoring group for grafting organic compounds and metal complexes onto inorganic supports such as titanium or zirconium oxides.26,27 Heterogeneous photocatalysts prepared according to this strategy could be made reusable which is an important goal in sustainable organic synthesis involving precious metal complexes.
Ru(II) complexes bearing phosphonate-substituted bpy ligands were prepared for photovoltaic applications28–35 and recently used in organic photocatalysis.36–38 We were interested in attaching this group to the phen ligand, which is known for inertness in ligand exchange reactions. The coordination lability of ligands is a serious drawback often observed in photocatalytic reactions to account for the non-radiative relaxation of excited photocatalysts and photobleaching.39
Our initial efforts in the studies of phosphonate-substituted Ru(II) complexes involved their synthesis, characterization and application in the oxidation of sulfides by molecular oxygen. By comparing these complexes and common Ru(II) photocatalysts such as tris(2,2′-bipyridine)ruthenium(II) hexafluorophosphate (Ru-(bpy)3), bis(2,2′-bipyridine)(1,10-phenanthroline)ruthenium(II) hexafluorophosphate (Ru-phen) and tris(1,10-phenanthroline)ruthenium(II) hexafluorophosphate (Ru-(phen)3), we gained insight into the influence of the substitution pattern on the photocatalytic properties. The results obtained in this work emphasize the potential utility of 4-substituted and 4,7-disubstituted complexes as photocatalysts in reactions involving either electron transfer or energy transfer. Notably, an unprecedented TON of almost 1000000 was achieved in the photooxidation of 4-methoxythioanisole to sulfoxide using 4,7-di-phosphonate-substituted complex Ru-4,7P2 as a photocatalyst.
When our work was in progress,40 the synthesis of Ru-5P and its use in water splitting were reported.41
Results and discussion
Synthesis
Phen ligands bearing diethyl phosphonate groups directly attached to the phen core 3P, 4P, 5P, 3,8P2 and 4,7P2 were prepared from halogen-substituted 1,10-phenanthrolines using the Hirao reaction as was previously reported by us.42 Ligands bearing the 1,4-phenylene spacer were obtained as shown in Scheme 1 by using the Suzuki coupling reaction of halogen-substituted 1,10-phenanthrolines and 4-(diethoxyphosphoryl)phenylboronic acid (see the ESI†).43 All isomeric mono-substituted derivatives 3PPh, 4PPh, 5PPh and diphosphonate 4,7(PPh)2 were prepared in good yields after the optimization of the reaction conditions. The coupling reaction of 4-chloro-1,10-phenanthrolines and 4,7-dichloro-1,10-phenanthrolines smoothly proceeded in the presence of the Pd(OAc)2/PCy3 catalytic system. In contrast, more reactive bromides and the Pd(PPh3)4 catalyst were required for the preparation of 3- and 5-substituted phen ligands. The 1H, 13C and 31P spectra of ligands thus obtained are presented in Fig. S36–S47.†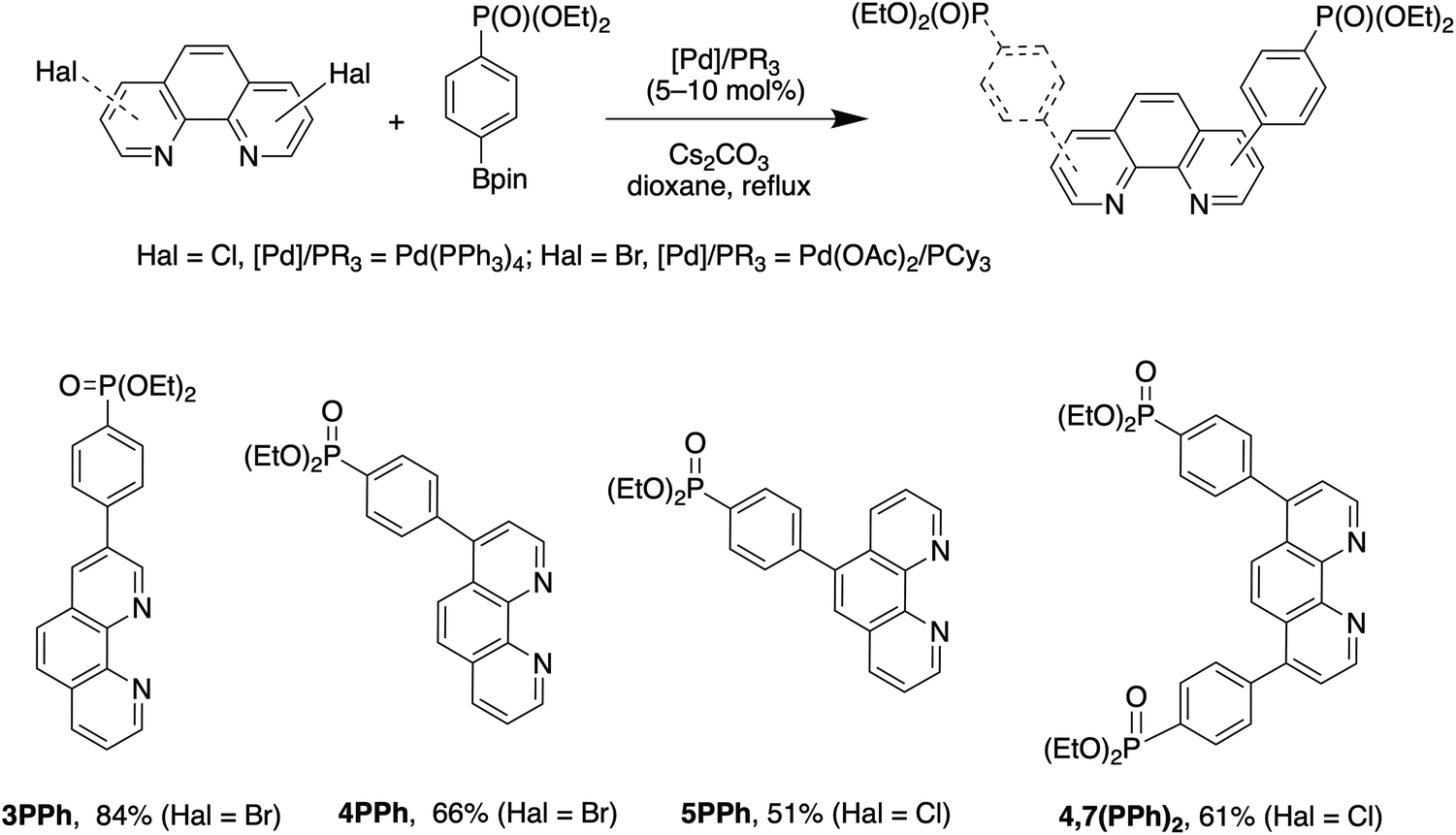 | ||
| Scheme 1 Synthesis of 4-(diethoxyposphoryl)phenyl-substituted 1,10-phenanthrolines 3PPh, 4PPh, 5PPh and 4,7(PPh)2. | ||
The synthesis of all Ru(II) complexes described here follows the well-known synthetic approach for the preparation of mixed ligand complexes. For each of the ligands, Ru(II) complexes Ru-Pcat (Fig. 1) were prepared by reacting the ligand and cis-[Ru(bpy)2Cl2]·2H2O in dry ethanol at 100 °C in a MW reactor. All complexes were isolated as hexafluorophosphate salts by the addition of ammonium hexafluorophosphate to partially evaporated reaction mixtures. The complexes were obtained as orange or crimson solids in 50–90% yields, similarly to that previously reported for mixed ligand Ru(II) complexes with phen ligands.16,44
The purity of Ru-Pcat was confirmed during their structural characterization by mass spectrometry and NMR spectroscopy. The successful coordination of the phen ligand was confirmed by the observation of the [M − PF6]+ ion peak with a matching isotopic pattern as the most prominent peak in the high resolution mass spectra.
Complexes Ru-Pcat were found to be soluble in polar and protic organic solvents such as chloroform, dichloromethane, THF, ethanol, methanol, acetonitrile, DMF and DMSO.
All compounds Ru-Pcat showed high photostability as was proved by irradiating their solutions in acetonitrile or acetonitrile/water (10:1 v/v) solvent mixtures with blue LED for 24 h (Fig. S1†). Such a high photostability is in agreement with the enhanced photostability of the other reported Ru(II) complexes with electron-withdrawing substituents in bpy ligands.14 This substituent effect observed in bpy complexes was explained by a decrease in the probability of dd excited state formation, which are prone to the ligand exchange reactions.
Solid-state structures
Additional structural information on the newly synthesized complexes was obtained by single crystal X-ray analysis of representative compounds. Single crystals of Ru-5P and Ru-4,7P2 suitable for X-ray analysis were grown by a slow diffusion of toluene in CHCl3/MeOH solutions of the complexes.Complexes Ru-5P and Ru-4,7P2 are crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . A summary of the crystallographic data is given in Table S1 (ESI†). The molecular plots of Ru-5P and Ru-4,7P2 are shown in Fig. 2 and 3, respectively. Overall, the geometries of these cationic complexes are similar to that of parent Ru-phen.45 The six-coordinated ruthenium center is chelated by nitrogen atoms from two bpy ligands and one phen ligand bearing phosphonate groups and adopts the expected octahedral geometry. Selected bond lengths and angles are reported in the captions of Fig. 2 and 3 and in Table S2.† All metal nitrogen bond lengths (Ru–N = 2.045–2.067 Å) lay in the usual range18,44–47 and are almost insensitive to the presence of the phosphonate substituent and its position in the phenanthroline ring. The angles comprising the coordination sphere differ by as much as 11° from the ideal right angles due to the narrow bite angles of these heterocyclic ligands. Dihedral angles between the plane of heteroaromatic ligands and the N–Ru–N plane containing nitrogen atoms of this ligand are slightly larger in Ru-4,7P2 (4.14°, 4.93° and 5.37°) as compared to those observed in Ru-5P (1.96°, 2.72° and 5.43°). These deviations were proposed as a criteria of steric hindrance in the coordination environment of the Ru center.44 However, the corresponding parameters of non-substituted complex Ru-phen are rather similar to those of Ru-4,7P2 (Table S2†). This led us to conclude that these angles are determined by molecular packing in crystals rather than by the steric hindrance of the metal center.
. A summary of the crystallographic data is given in Table S1 (ESI†). The molecular plots of Ru-5P and Ru-4,7P2 are shown in Fig. 2 and 3, respectively. Overall, the geometries of these cationic complexes are similar to that of parent Ru-phen.45 The six-coordinated ruthenium center is chelated by nitrogen atoms from two bpy ligands and one phen ligand bearing phosphonate groups and adopts the expected octahedral geometry. Selected bond lengths and angles are reported in the captions of Fig. 2 and 3 and in Table S2.† All metal nitrogen bond lengths (Ru–N = 2.045–2.067 Å) lay in the usual range18,44–47 and are almost insensitive to the presence of the phosphonate substituent and its position in the phenanthroline ring. The angles comprising the coordination sphere differ by as much as 11° from the ideal right angles due to the narrow bite angles of these heterocyclic ligands. Dihedral angles between the plane of heteroaromatic ligands and the N–Ru–N plane containing nitrogen atoms of this ligand are slightly larger in Ru-4,7P2 (4.14°, 4.93° and 5.37°) as compared to those observed in Ru-5P (1.96°, 2.72° and 5.43°). These deviations were proposed as a criteria of steric hindrance in the coordination environment of the Ru center.44 However, the corresponding parameters of non-substituted complex Ru-phen are rather similar to those of Ru-4,7P2 (Table S2†). This led us to conclude that these angles are determined by molecular packing in crystals rather than by the steric hindrance of the metal center.
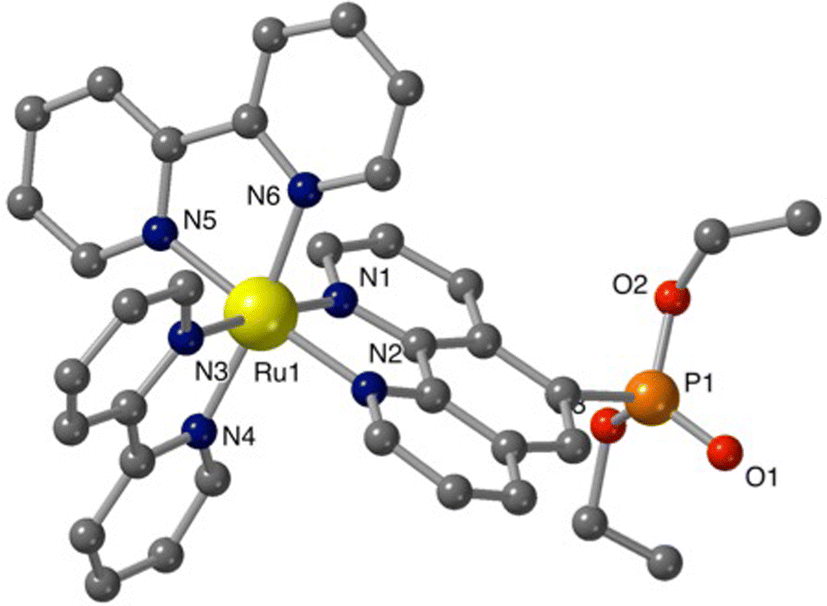 | ||
Fig. 2 Structure of cationic complex Ru-5P. Hexafluorophosphate ions, hydrogen atoms and minor disordered parts are omitted for clarity. Selected bond lengths (Å): Ru–N(1–6) = 2.052(5)–2.067(5); P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(1) = 1.451(7); P–O(2,3) = 1.5666(7), 1.5669(6); P–C(7) = 1.799(7). O(1) = 1.451(7); P–O(2,3) = 1.5666(7), 1.5669(6); P–C(7) = 1.799(7). | ||
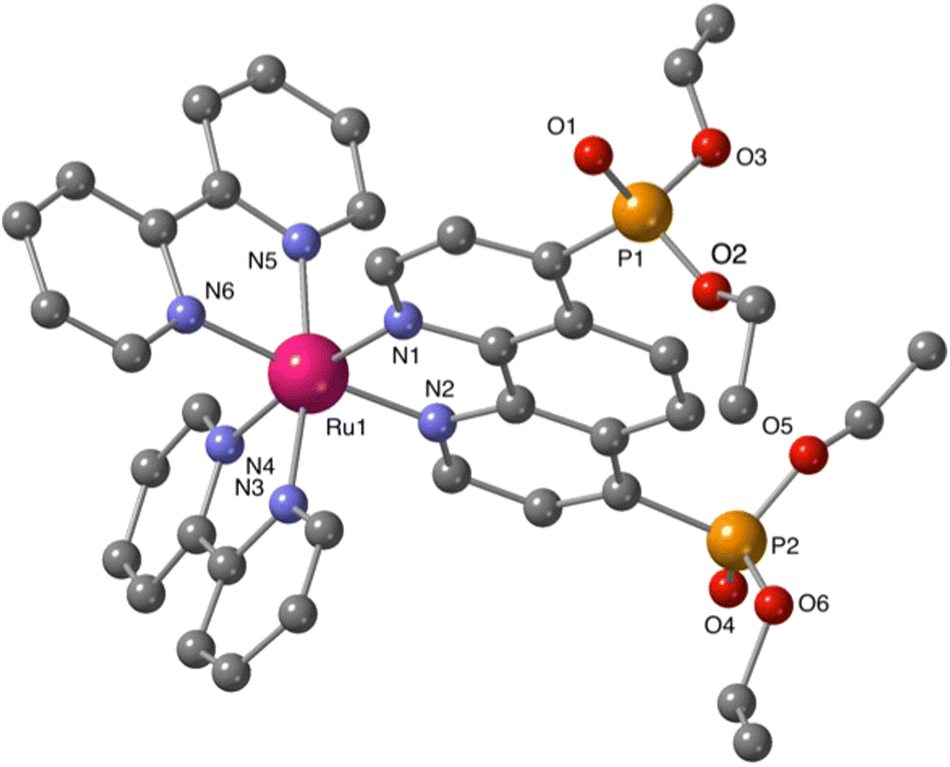 | ||
| Fig. 3 Structure of cationic complex Ru-4,7P2. Hexafluorophosphate ions, hydrogen atoms and minor disordered parts are omitted for clarity. Selected bond lengths (Å): Ru–N(1–6) = 2.045(5)–2.067(5); PO(1) = 1.489(8); P–O(2,3) = 1.54(1), 1.59(1); P–C(7) = 1.803(6). | ||
Thus, the introduction of the P(O)(OEt)2 substituent in 4 and 5 positions of the phen ligand does not significantly change the structural parameters of the coordination sphere of the ruthenium atom.
Characterization in solution
Kinetic stability and solution structures of studied complexes were confirmed by NMR spectroscopy. Complex Ru-4P was chosen as a representative example and investigated in detail in acetonitrile-d3 solution by NMR spectroscopy. In 1H and 13C{1H} NMR spectra (Fig. 4 and S2–S4†), characteristic signals of ethoxy groups of the P(O)(OEt)2 substituent display chemical shifts that are similar to those observed in the free ligand 4P. This is in accordance with the crystal structures of Ru-5P and Ru-4,7P2 in which the phosphonate substituent is non-coordinated.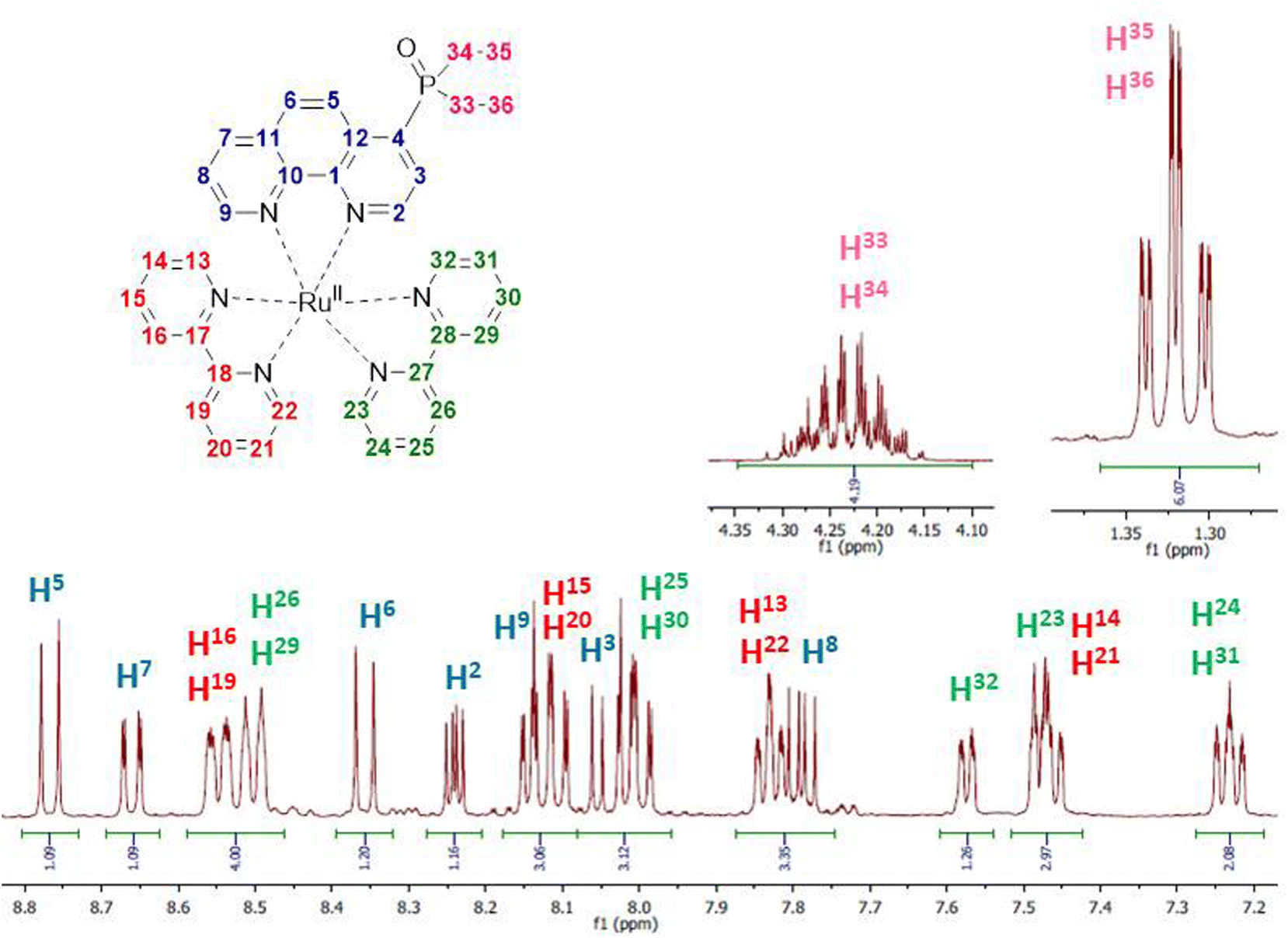 | ||
| Fig. 4 Selected regions of the 1H NMR spectrum of Ru-4P in acetonitrile-d3. | ||
Chemical shifts and coupling constants observed in the 1H NMR spectrum of Ru-4P are summarized in Table S3.† All proton resonances of the phen ligand were easily attributed using the COSY spectrum after the initial assignment of H-3 and H-8 protons (see Fig. 4 for proton labels), which appeared as doublets of doublets at δH 8.03 and 7.79 ppm with typical coupling constants of JH–H = 5.3 Hz, 3JH–P = 14.7 Hz and JH–H = 5.3 Hz, JH–H = 8.3 Hz, respectively. The remaining 12 signals in the aromatic region apparently belong to 16 protons of two bpy ligands. Eight triplets and eight doublets, expected for four pyridine rings, are overlapping but can be distinguished by integration. Four Hα protons of pyridine rings are seen as distinct doublets (δH 7.48, 7.57, 7.82 and 7.84 ppm). Three triplets and one doublet have double intensity. The remaining two triplets and two doublets display very similar chemical shifts (ΔδH < 0.05 in each pair of signals). These paired and strongly overlapped signals are clearly observed in the PSYCHE 1H NMR spectrum (Fig. S3b†). Using the COSY spectrum (Fig. S5†), four sets of pyridine protons were identified. Their protons H-16, H-19, H-26 and H-29 appeared as two doublets with double intensity and more strongly deshielded in comparison with Hα protons at δH 8.50 and 8.54 ppm. The absence of cross peaks through three bands for these protons in the gHMBCAD spectrum (Fig. S9†), enabled their assignment to the protons of two different bpy ligands. This finally allowed for the tentative assignment of two proton sets of bpy ligands.
Surprisingly, all resonances of one bpy molecule are shifted upfield as compared to the corresponding signals of the second bpy ligand. At the same time, the corresponding protons of two pyridine rings in each bpy chelator exhibit identical or very similar chemical shifts and appear as signals with double intensity except those of Hα protons. The only remaining uncertainty at this point is the decision on which of the two bpy ligands is located at the trans position to the phen ligand.
It is also worth noting that our proton assignment of bpy ligands disagrees with the signal attribution in a series of mixed ligand complexes [Ru(phen)(bpy)2]2+ bearing 5-substituted phenanthrolines, which was reported by Ye et al.44 and Rau et al.41 and were based on only COSY measurements and common sense arguments.
All carbon signals observed in the 13C NMR spectrum conventionally associated with heterocyclic carbon signals were unambiguously assigned using gHSQCAD (CH carbon atoms) and gHBMCAD (quaternary carbon atoms) spectra (Fig. S7–S9†). Their chemical shifts and 13C–31P coupling constants are summarized in Table S3.† The 31P NMR spectrum also proves the non-coordinated character of the phosphonate group.
1H and 31P{1H} NMR spectra of other complexes Ru-Pcat are in good agreement with the proposed structures and are shown in the ESI (Fig. S48–S65†).
Electrochemistry
Ru-Pcat complexes (Fig. 1) were studied by electrochemical methods to assess the effect of the substitution pattern on the redox potentials and the stability of electrogenerated species. The half-wave or peak potential values were obtained from cyclic voltammetry (CV) measurements, which were carried out under N2 in acetonitrile (MeCN) containing tetra-n-butylammonium hexafluorophosphate (TBAPF6, 0.1 M solution) are summarized in Table 1, which also includes the data for reference compounds Ru-(bpy)3, Ru-phen and Ru-(phen)3.| Oxidation | Reduction | ||||||
|---|---|---|---|---|---|---|---|
| E 1a | E 1c (L10/˙−) | E 2c (L20/˙−) | E 3c (L30/˙−) | E 4c | E 5c | E 6c | |
| a Half wave potential value measured at 0.1 V s−1. b Peak potential value measured at 0.1 V s−1. In our experimental conditions, E1/2 [Fc+/Fc] = 83 mV. Conversion to SCE-referenced potential values can be obtained by the addition of +298 mV.49 c Previously reported data for this compound (in DMF, 0.1 M Bu4NBF4, vs. Fc+/Fc): E1a = 0.75 V, E1c (L10/˙−) = −1.70 V, E2c (L20/˙−) = −2.00 V, E3c (L30/˙−) = −2.20.41 d Broad peak with shoulder. | |||||||
| Ru-phen | 0.975 (73)a | −1.645 (66)a | −1.83 (62) | −2.09 (71)a | −2.77b | −3.02b,c | |
| Ru-(phen)3 | 0.975 (71) | −1.68b | −1.805b | ||||
| Ru-(bpy)3 | 0.97 (75)a | −1.64 (62)a | −1.83 (57)a | −2.075 (61)a | −2.76b | ||
| Ru-3P | 1.03 (80)a | −1.45b | −1.81 (83)a | −2.09 (144)a | |||
| Ru-4P | 1.025 (73)a | −1.415 (63)a | −1.78 (63)a | −2.01 (61)a | −2.40b | −2.775b | |
| Ru-5P | 1.005 (97)a | −1.55b | −1.79 (91)a | −2.05 (95)a | −2.76b | −2.89b | |
| Ru-3,8P2 | 1.08 (94)a | −1.23 (73)a | −1.71 (77)a | −1.92 (68)a | −2.2 (75)a | −2.83b | |
| Ru-4,7P2 | 1.08 (99)a | −1.335 (71)a | −1.765 (99)a | −1.96 (123)a | −2.31b | −2.86b,c | |
| Ru-3PPh | 0.995 (76)a | −1.525 (86)a | −1.80 (68)a | −2.04 (73)a | −2.27 (83)a | −2.49 (96)a | −2.835b,d |
| Ru-5PPh | 0.985 (82)a | −1.60 (74)a | −1.815 (89)a | −2.08 (92)a | −2.47b | −2.765b | |
| Ru-4,7(PPh)2 | 0.98 (89)a | −1.55 (68)a | −1.78 (65)a | −2.00 (67)a | −2.205 (65)a | −2.43 (73)a | −2.69 (116)a |
All investigated complexes undergo multiple electron transfers typical of Ru(II) polypyridyl complexes,48 giving CV curves with up to 6 consecutive ligand-centered reduction and single oxidation waves observed in the accessible potential window. The CV curves recorded for the Ru-PPh series and the reference compound Ru-phen are shown in Fig. 5.
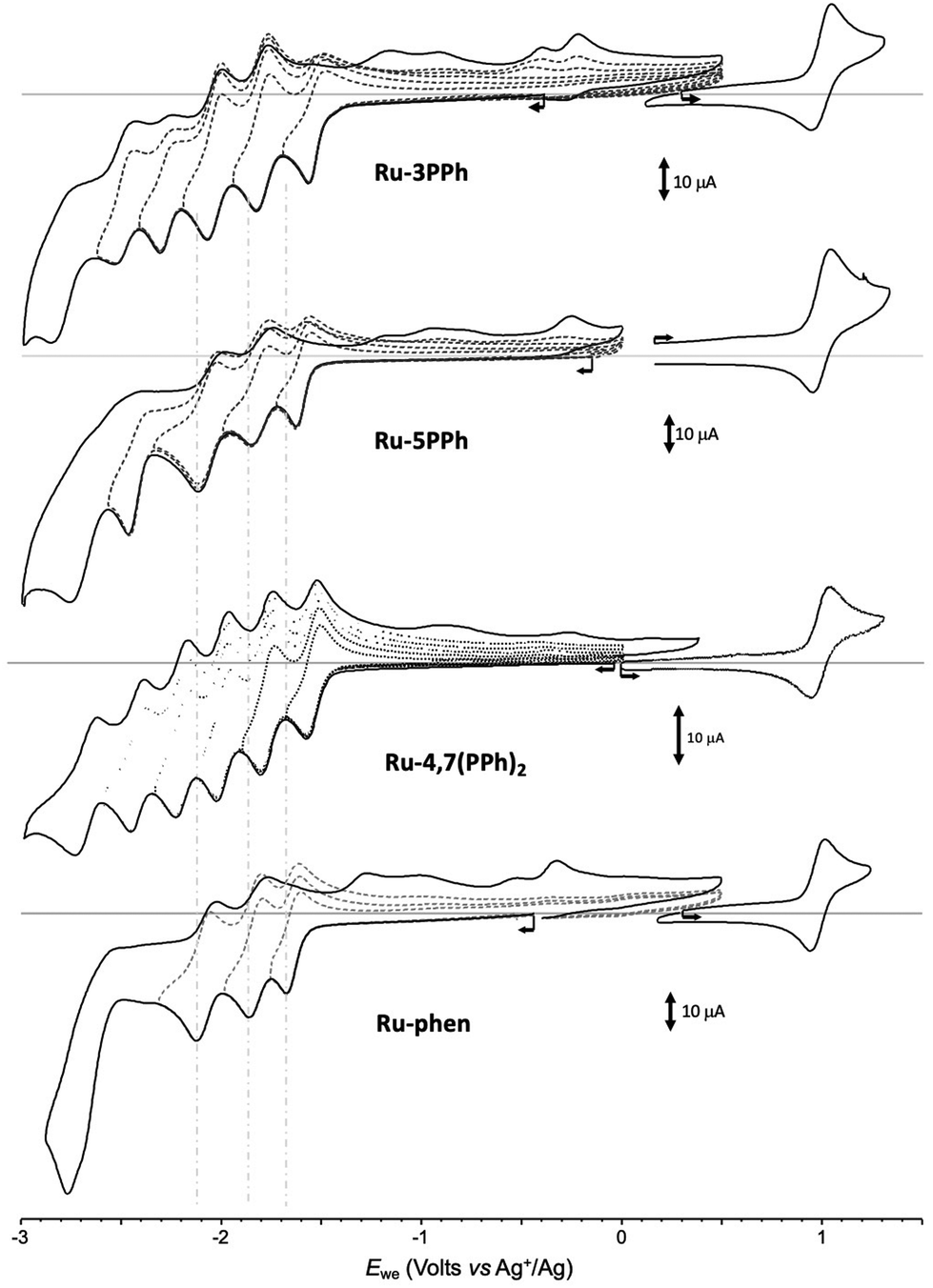 | ||
| Fig. 5 Voltamperometric curves of MeCN solutions of Ru-3PPh, Ru-5PPh, Ru-4,7(PPh)2 and Ru-Phen (1 × 10−3 M + 0.1 M in TBAPF6) recorded at a vitreous carbon working electrode (∅ = 3 mm, 0.1 V s−1). | ||
The effect of the electron-withdrawing P(O)(OEt)2 substituent is first revealed through a shift of the one-electron reversible oxidation wave attributed50 to the oxidation of the Ru center (Ru2+/Ru3+). As can be seen by comparing the values presented in Table 1, the introduction of P(O)(OEt)2 substituent(s) in one or more of the available positions of the phen core of Ru-phen makes the oxidation of the corresponding complexes more difficult due to a decrease in the effective charge density at the metal center. The negative inductive effect of P(O)(OEt)2 leads to a 55 and 50 mV shift in E1/2 values for Ru-3P and Ru-4P, respectively, even though in these compounds the substituent remains fairly distant from the metal center (4 and 5 bonds, respectively). Attachment of the second P(O)(OEt)2 group to the opposite pyridine ring (Ru-3,8P2 and Ru-4,7P2) has a similar effect (+50–55 mV per substituent), to reach an overall positive shift of about +100–105 mV regardless its position. The introduction of the P(O)(OEt)2 in position 5 (complex Ru-5P) also influences the oxidation process but leads to a much weaker positive shift in potential (30 mV). Such positive shifts of the first oxidation potentials were already observed in Ru(II) complexes with polypyridyl ligands substituted by electron-acceptors including phosphonate groups.51 Our results are also in agreement with previous reports demonstrating that there is a strong correlation between E1/2 (Ru2+/Ru3+) values and Hammett constants of substituents introduced in position 4 of the pyridine ring.17
As expected, the negative inductive effect of P(O)(OEt)2 is significantly lowered when this substituent is separated by a 1,4-phenylene linker in the Ru-PPh series. Positive potential shifts of only 10 and 20 mV are observed for Ru-5PPh and Ru-3PPh. Here again, the amplitude of the shift correlates with the number of bonds introduced between the phosphorous and Ru centers (5 and 4 bonds respectively). Surprisingly, the oxidation potential of the di-substituted complex Ru-4,7(PPh)2 is found to be shifted by only 5 mV despite the presence of two phosphorous substituents.
As can be seen from Table 1 and Fig. 5, S10–S21,† numerous reduction waves are observed in the CV curves of the investigated compounds Ru-P and Ru-PPh, which agrees with the known ability of bpy and phen ligands to undergo two consecutive one-electron reductions in the accessible potential range.50 We found that the shape and number of these waves depend on the substitution pattern. Six consecutive reversible reduction waves appear well separated in the CV curve of Ru-3PPh and Ru-4,7(PPh)2 while only 5 or 4 waves of different intensities are observed for Ru-5PPh and Ru-Phen, respectively (Fig. 5). Those features are well interpreted as a stepwise reduction of each ligand, with a sequence depending on the relative position of their π* orbitals.47 The three first waves can be attributed to a stepwise one-electron reduction of the bpy (bpy/bpy˙−) and phen ligands (phen/phen˙−). The relative order, and most importantly, the identification the phen-centered reduction among these signals, was determined by comparing the potential values collected with the Ru-P series. We found the first reduction wave to be reversible at 100 mV s−1 for all complexes in this series except for Ru-3P and Ru-5P, for which partial reversibility could only be observed for scan rates above 2 V s−1. These two compounds were therefore excluded from the comparison discussed below focusing on the half-wave potential values measured at 100 mV s−1 for Ru-4P, Ru-3,8P2 and Ru-4,7P2. The first three ligand-based reductions noted E1c (L10˙−), E2c (L20/˙−) and E3c (L30/˙−) are observed in the range of −1.23 to −1.415 V (ΔE = 185 mV), −1.71 to −1.81 (ΔE = 100 mV), and −1.92 to −2.01 (ΔE = 90 mV), respectively. A rapid overview of those data reveals that the introduction of one or two EWGs on the phen ligand is mainly reflected in the shifts of the first reduction wave, with a maximum value of about 185 mV observed between Ru-4P and Ru-3,8P2. It should also be noted that there is a rather large shift of about 100 mV between the E1c values calculated for the isomeric complexes Ru-4,7P2 and Ru-3,8P2, which is consistent with the above conclusion that the inductive effect is stronger when the substituent is introduced in position 3. We also found that the first reduction waves systematically appear at significantly lower potentials than those of the reference complexes Ru-(bpy)3 and Ru-phen reduction. All these data led us to attribute the first wave observed at E1c (L10/˙−) to the one-electron reduction of the phen ligand and the two following ones E2c (L20/˙−) and E3c (L30/˙−) to the reduction of the bipyridines.
This hypothesis is indirectly supported by the irreversible character of the first reduction waves in the CV curves of Ru-3P and Ru-5P complexes which is attributed to the existence of a chemical step coupled to the formation of phen˙− species. This instability resulting from the presence of a single EWG at positions 3 or 5 is currently not understood and all the more surprising since it is not observed for Ru-4P and the disubstituted derivatives under the same conditions.
It should be noted that the proposed attribution strongly differs from the experimental data for Ru-phen and theoretical studies so far reported for mixed Ru(II) complexes, containing bpy and phen ligands, suggesting that the bpy ligand is easier to reduce than the phen.52,53 These reported observations are consistent with our experimental data collected with the reference Ru-(bpy)3, Ru-(phen)3 and Ru-phen complexes (Table 1) showing that the reduction of the phen ligand is only slightly negatively shifted (by about −40 mV) compared to that of bpy and that the largest difference between the Ru-(bpy)3 and Ru-phen complexes is observed in the third reduction process E3c (L30/˙−). Our attribution of the first wave in Ru-P to the phen-centered reduction is easily understood taking into account the strong EW character of the P(O)(OEt)2 group which makes the reduction of the phen ligand much easier than that of the unsubstituted bipyridines.
In the Ru-PPh series, the first reduction potentials E1c (L10/˙−) are also positively shifted and more sensitive to substitution than E3c (L30/˙−) with the exception of Ru-4,7(PPh)2. Thus, the attribution of the reduction peaks in this series using this criterion is ambiguous. A comparison of potential values obtained for Ru-3PPh, Ru-5PPh and Ru-phen provides however strong support of our hypothesis that the electron is also localized on the phen ligand for these reduced complexes.
Spectroelectrochemistry (SEC) measurements can provide useful information for deciphering the reduction mechanisms involved in ruthenium(II) mixed-ligand complexes.52 Attribution of the first reduction wave to the reduction of the phen ligands in the Ru-P series was thus further supported by SEC. These measurements were carried out in thin-layer cells upon collecting time-resolved UV–vis spectra during the first reduction of selected compounds. The curves obtained for Ru-4P, Ru-4,7P2, Ru-(bpy)3 and Ru-(phen)3 are shown in Fig. 6. The one-electron reduction of Ru-4P and Ru-4,7P2 leads to similar changes including the development of a broad and intense band in the range 460–480 nm with a shoulder at about 600 nm. These features contradict with those collected under the same conditions for the reference compound Ru-(bpy)3, where the first reduction of the bpy ligand leads to the appearance of more red shifted signals above 500 nm along with additional weak signals at about 784 and 867 nm. Appearance of the broad absorption band at a shorter wavelength (600–650 nm) is a characteristic signature of the phen˙− radical anion as reported previously52 and confirmed by us for Ru-(phen)3.
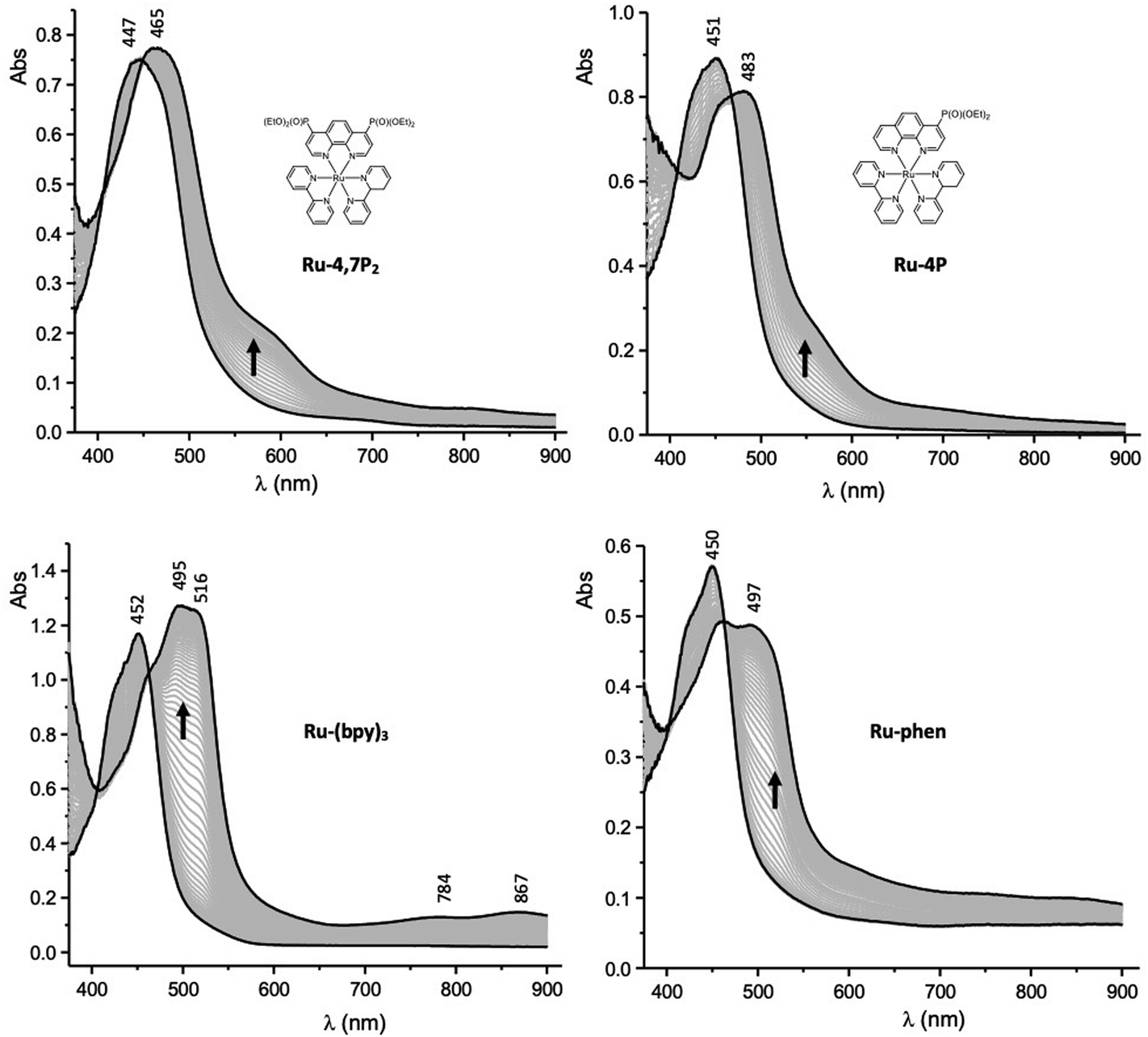 | ||
| Fig. 6 UV–vis spectra recorded during the first one-electron reduction of Ru-4,7P2, Ru-4P, Ru-(bpy)3 and Ru-phen in MeCN containing 0.1 M TBAPF6 (working electrode: Pt, l = 0.5 mm). | ||
Altogether, the electrochemical and spectroelectrochemical data discussed above reveal that the addition of the P(O)(OEt)2 group to Ru-phen leads to a large positive shift of the first reduction wave whose magnitude depends on the substitution pattern. The two following bpy-centered reduction waves as well as the Ru-centered oxidation wave are also affected but to a much lesser extent in most of the complexes. The greater sensitivity of the ligand-based reductions as compared to the metal-based oxidation can be explained by the localization of the P(O)(OEt)2 substituents in the ligand. We also found that the addition of a phenylene linker between the phen core and the P(O)(OEt)2 group lowers the substituent effect on the reduction potentials and can be used for the fine tuning of redox potentials. Most importantly all collected data demonstrate that the first electron transfer is centered on the phen ligand in Ru-P and most probably in Ru-PPh series in contrast to the parent complex Ru-phen. We can expect that the localization of the first electron transfer on the phen ligand in Ru-Pcat could have a great influence on both the photophysical and photocatalytic properties of these complexes.
Photophysical properties
The electronic absorption and emission spectra of the newly synthesized Ru-Pcat complexes and reference compounds in MeCN are presented in Fig. S22† and summarized in Table 2. All complexes exhibit intense absorption bands in the 250–350 nm region, which can be assigned to the ligand-centered π–π* electronic transitions.54,55 The spectra also show characteristic broad absorption bands in the longer wavelength region (400–500 nm) due to overlapping spin-allowed metal–to–ligand charge transfer (MLCT) and interligand bpy/phen-based charge transfer (LLCT) transitions. The observed absorption bands are broader compared to those of the reference Ru-phen complex (Fig. 7) probably due to an increased number of MLCT and LLCT transitions between bpy and phosphorylated phen ligands.15,56 Positions of the absorption maxima for Ru-Pcat are relatively insensitive to the substitution pattern in the phen ligand, except for (3-phosphonate)-substituted derivatives. For complexes Ru-3P and Ru-3,8P2, the lower energy transition is shifted to shorter wavelengths (443 and 435 nm, respectively) compared to Ru-phen (450 nm). An additional low intensity shoulder at 481 nm was observed for Ru-3,8P2 in line with previous reports on Ru(II) complexes with phen ligands.57–60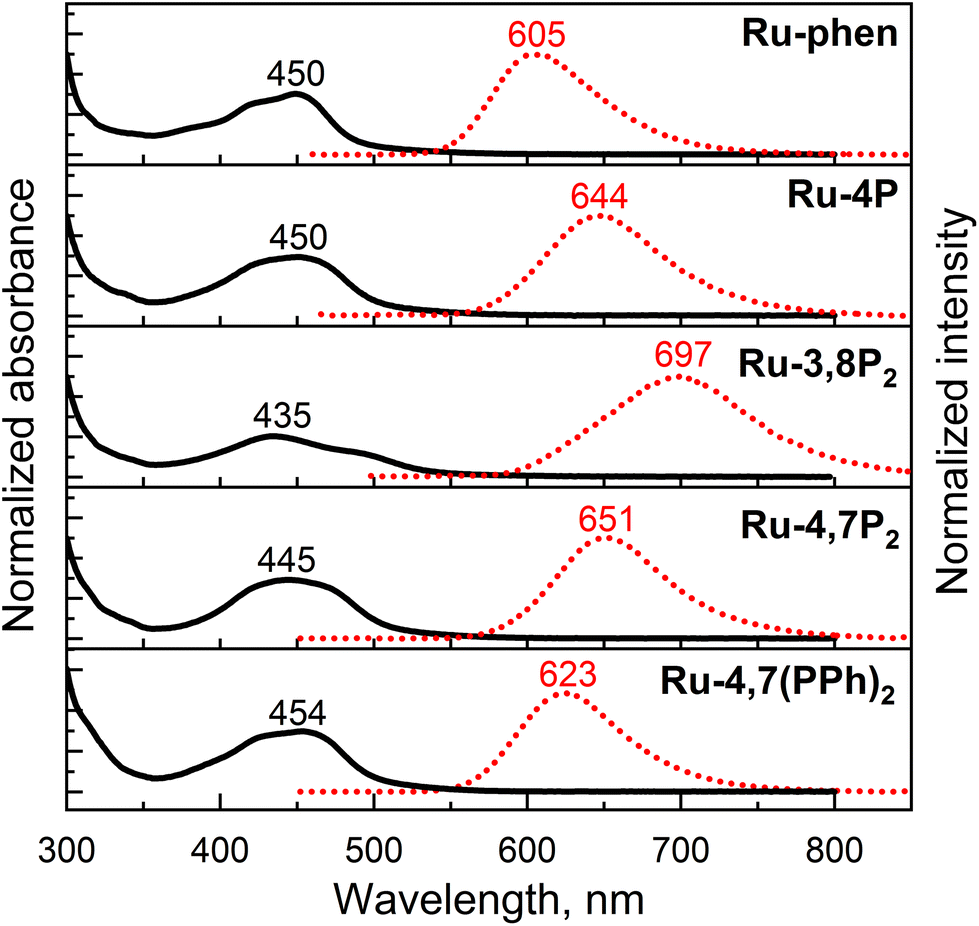 | ||
| Fig. 7 Absorption (black solid line) and emission (red dotted line) spectra of representative Ru-Pcat complexes and reference compound Ru-phen in deaerated MeCN. Emission was excited at 450 nm. | ||
| Compound | λ abs (nm) (ε × 10−3 (M−1 cm−1)) |
λ
ema (nm) |
Φ
emb |
Φ
Δc |
|---|---|---|---|---|
| a Emission was excited at 450 nm. b Measured in deaerated acetonitrile at ambient temperatures relative to a solution of Ru-(bpy)3 as a standard.64 c Measured in air-saturated acetonitrile using Ru-(bpy)3 as a reference photosensitizer.65 d Previously reported data for this compound: λabs (ε × 10−3) 450 (15.08), 287 (56), 265 (51) nm (M−1 cm−1); λem = 601 nm.41 e Ru-4,7Ph2 = bis(2,2′-bipyridine)(4,7-diphenyl-1,10-phenanthroline)ruthenium(II) hexafluorophosphate. Taken from ref. 53. | ||||
| Ru-(bpy)3 | 451 (14.0), 286 (85.0) | 609 | 0.095 | 0.57 |
| Ru-phen | 450 (12.1), 283 (49), 272 (42) | 605 | 0.096 | 0.54 |
| Ru-3P | 443 (13.5), 285 (63.9), 268 (65.4) | 641 | 0.078 | 0.65 |
| Ru-4P | 450 (14.1), 283 (58.0), 272 (59.0) | 644 | 0.085 | 0.53 |
| Ru-5P | 449 (16.2), 285 (62.2), 265 (58.0) | 603 | 0.126 | 0.55 |
| Ru-3,8P2 | 481 (6.8), 435 (10.2), 284 (52.0), 272 (62.0) | 697 | 0.021 | 0.28 |
| Ru-4,7P2 | 445 (17.4), 261 (85.0) | 651 | 0.101 | 0.74 |
| Ru-3PPh | 453 (13.0), 283 (80.0) | 614 | 0.121 | 0.68 |
| Ru-4PPh | 453 (17.5) | 613 | 0.181 | 0.75 |
| Ru-5PPh | 450 (14.6), 285 (59.0) | 606 | 0.121 | 0.61 |
| Ru-4,7Ph2 | 454 (19.9), 284 (90.7) | 614 | 0.155 | - |
| Ru-4,7(PPh)2 | 454 (18.0), 278 (90.0) | 623 | 0.253 | 0.73 |
All studied complexes exhibit rather intense luminescence emission at room temperature which is strongly quenched in the presence of oxygen (Fig. S22†). A broad structureless emission band with maximum at 605 nm was observed for 5-substituted compounds Ru-5P and Ru-5PPh similarly to Ru-phen. Introduction of a single phosphonate group in positions 3 and 4 resulted in a strong red shift (256410–285714 cm−1 (35–39 nm)) of the emission maxima. Such a red shift of the emission is commonly observed for Ru(II) complexes with ligands bearing electron-withdrawing substituents.14 Surprisingly, the second phosphonate substituent strongly further red shifts the emission band (285714 cm−1 (53 nm)) only if this substituent is introduced in position 8. The luminescence band of Ru-4,7P2 (651 nm) is red-shifted by only 1000000 cm−1 (10 nm) relative to that of the Ru-4P complex. Emission quantum yield values (Φem) of all complexes bearing the phosphonate group in the phen ring (Series Ru-P, Fig. 1) are comparable with those of Ru-bpy (0.095) and Ru-phen (0.096), except for Ru-3,8P2 (0.021).
For complexes with a phenylene linker (Ru-PPh series, Fig. 1), the observed red shifts are substantially smaller compared to the correspondingly substituted complexes in Ru-P series. At the same time, the emission quantum yields fall in the range of 0.121 to 0.253 reaching the maximum for Ru-4,7(PPh)2 that is about 2.5-fold higher than the Φem of Ru-phen. Such high quantum yields values were previously reported for di-aryl-substituted phen complexes represented here by Ru-4,7Ph2 (Table 2).59 Notably, the introduction of the phosphonate substituent in the phenyl ring allowed for a significant increase (from 0.155 to 0.253) of the luminescence quantum yield and hydrophilicity of complex Ru-4,7Ph2, which was actively investigated as a valuable DNA marker.61,62
The apparent Stokes shifts for the complexes of both series are comparable to that of the parent Ru-phen (64516 cm−1), except for compounds bearing a phosphonate group in position 4 and Ru-3,8P2, for which they are increased in the following order: Ru-4,7(PPh)2 (59172 cm−1) < Ru-4P (51546 cm−1) < Ru-4,7P2 (47847 cm−1) < Ru-3,8P2 (46296 cm−1).
Thus, absorption and particularly emission of [Ru(phen)(bpy)2]2+-type complexes can be fine-tuned by varying the number and position of P(O)(OEt)2 groups in the phen ring. Complex Ru-3,8P2 is interesting as it is the only example in Ru-Pcat series that exhibits an emission band in the first near IR region. In contrast to previously reported29,34 complexes bearing the phosphonate group in the bpy ring, the introduction of this substituent in the phen core decreases the emissivity of the resulting Ru(II) complexes only in the case of Ru-3,8P2. Emissive properties are generally related to the efficiency of photooxidation reactions mediated by molecular oxygen.63 Thus, Ru-3,8P2 seems to be a less promising candidate for photocatalytic oxidation reactions studied in this work.
This conclusion was proved by measuring the singlet oxygen quantum yields (ΦΔ) for complexes Ru-Pcat using the chemical trapping method with 1,9-dimethylanthracene (DMA) in acetonitrile. Upon irradiation of air-saturated solutions containing each complex at 488 nm, DMA selectively reacts with singlet oxygen forming the corresponding endoperoxide. Ru-Pcat complexes showed no absorption change in the course of irradiation (Fig. S23†). The change of DMA absorbance with time is linear (Fig. 8, inset), allowing to obtain ΦΔ value from comparison with the reference Ru-(bpy)3. All complexes displayed excellent ability to generate singlet oxygen with the ΦΔ value comparable to or higher than those of referenced photosensitizers Ru-phen and Ru-(bpy)3 (0.54 and 0.57, respectively). For 4 and 7-substituted complexes, ΦΔ values reach up to 0.75 with the exception for Ru-4P. It is worth to note that these values are comparable with those of conventional organic photosensitizers such as Methylene Blue (0.5) and Rose Bengal (0.8).66 Thus, Ru-Pcat complexes are promising photocatalysts for the oxidation of organic compounds by molecular oxygen.
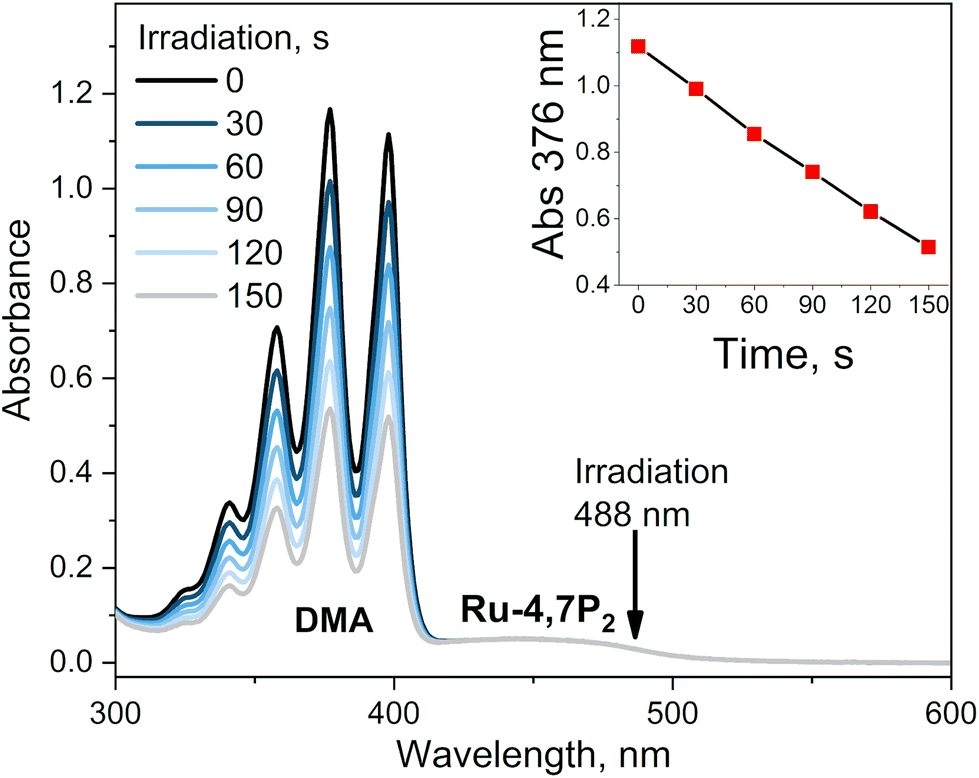 | ||
| Fig. 8 Example of the photosensitized oxidation of 1,9-dimethylanthracene in the presence of Ru-4,7P2 in air saturated acetonitrile solution irradiated with 488 nm laser (10 mW cm−2). Inset: Change of absorbance at 376 nm with time. | ||
DFT studies
To get insight into the electronic structure of complexes Ru-Pcat, DFT calculations were carried out on all complexes with the Firefly quantum chemistry package,67 which is partially based on the GAMESS (US)68 source code. The calculations were performed using the B3LYP functional with the STO 6-31G(d,p) basis set for all elements except Ru, for which we used the Stuttgart valence basis set and pseudopotential.69 The optimized geometries of complexes Ru-Pcat are depicted in Fig. S24–S33† and the calculated structural parameters are summarized in Table S4 (see the ESI†). A satisfactory agreement of calculated and experimental data was observed for complexes Ru-5P and Ru-4,7P2 (Table S4†).Isodensity plots of HOMO and LUMO orbitals of representative complexes Ru-P and Ru-PPh along with a diagram showing HOMO and LUMO energy levels are presented in Fig. 9. A set of π* orbitals associated with phen and bpy ligands are close in energy and the highest-energy occupied orbitals also give a set with very similar energies due to small contributions of ligands in these orbitals wherein the electron density is found primarily on the metal center. This is consistent with the broad light absorption manifold observed in the experimental spectra.
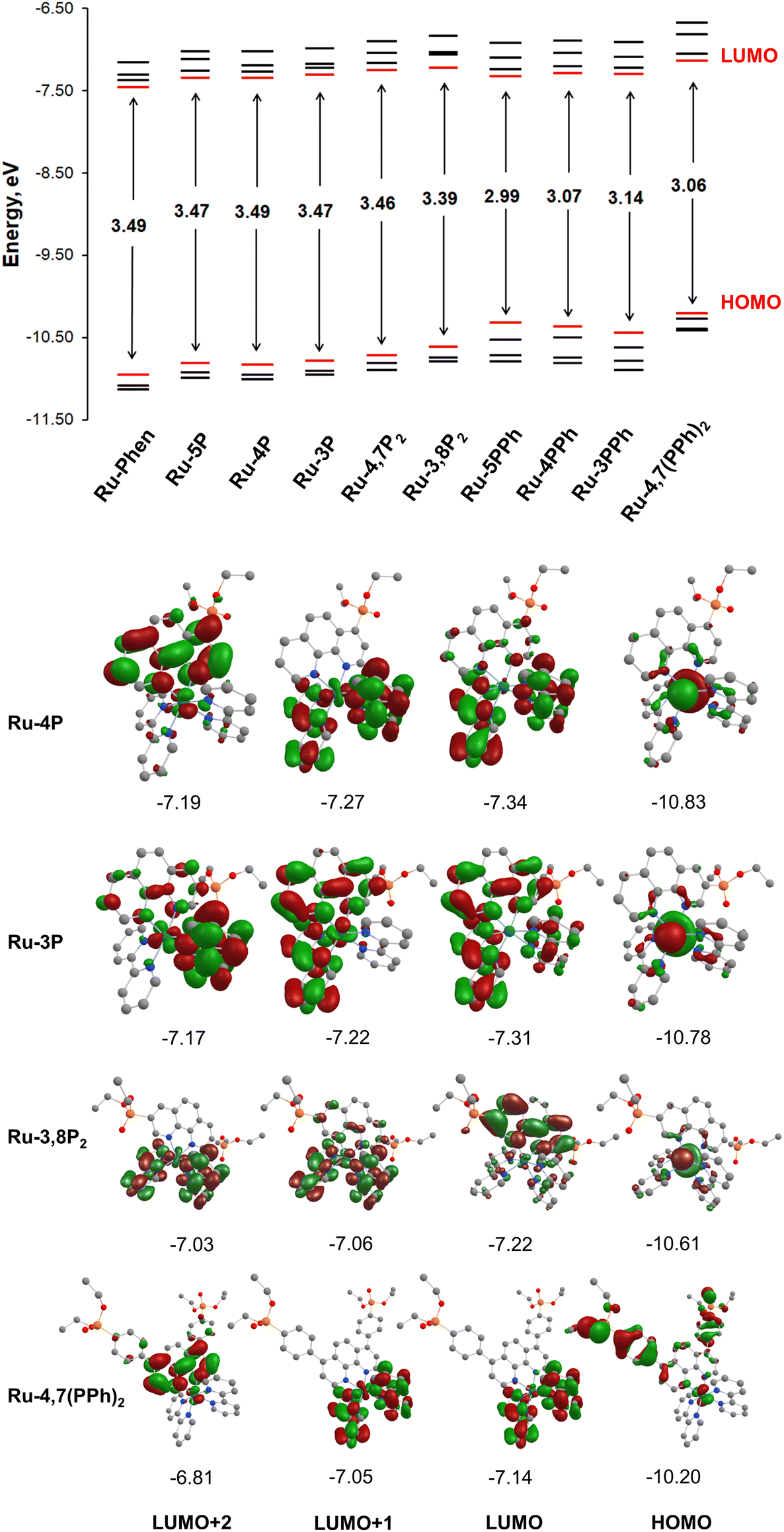 | ||
| Fig. 9 (Top) Frontier orbital energies obtained from DFT calculations for Ru-cat. (Bottom) The isodensity plot of HOMO and LUMO orbitals for the representative set of chromophores Ru-cat obtained from DFT calculations illustrating effects of ligand substituents. | ||
There is no orbital mixing for the phenanthroline scaffold and the P(O)(OEt)2 substituent in frontier orbitals for complexes of Ru-P series, and the energies of HOMO and LUMO orbitals vary to only a slight extent depending on the substitution pattern (Tables S5–S10†). By comparing the frontier orbitals of phosphonate-substituted complexes to those of the parent Ru-phen complex a close similarity can be noticed when one or two phosphonate substituents are attached to position 4 and 5 of the phen ring (Ru-5P, Ru-4P and Ru-4,7P2) (Fig. 9 and Tables S5–S8†). As shown in Fig. 9 for complex Ru-4P, the main contribution to HOMO, HOMO−1 and HOMO−2 comes from d orbitals but there is also a partial ligand character in particular for HOMO−1 (phen ligand) and HOMO−2 (bpy ligand). The LUMO and LUMO+1 levels are situated on both bpy ligands, while the π* orbitals of the phen ligand give the main contribution to LUMO+2 and LUMO+3.
The HOMOs of Ru-3P and Ru-3,8P2 are very similar to those of other Ru-P complexes that is expected because the substituent is still quite distant from the metal d orbitals which are the main contributors to HOMOs (Tables S8 and S10†). In contrast, the electron density of LUMO of Ru-3P is found on π* orbitals of all three heteroaromatic ligands. A significant phen and bpy character is also observed in LUMO+1 and LUMO+2 despite the electron density being delocalized on only two ligands in both orbitals. The electron density distribution in the LUMOs of Ru-3,8P2 resembles that of Ru-phen with only one exception that the order of LUMOs situated on the π* orbitals of bpy and phen ligands is changed. In Ru-3,8P2, the LUMO and LUMO+3 are localized mainly on the π* orbitals of the phen ligand while the electron density of other two unoccupied frontier orbitals is found on bpy ligands. Accordingly, the energies of MLCT and LLCT transitions in 3-substituted derivatives can be significantly different from those observed in other Ru-P complexes as it is experimentally observed in electronic absorption and luminescence spectra.
Ru(II) complexes with the phenylene linker (Ru-PPh) differ from the parent complex Ru-phen in the character of HOMO orbitals. The main contribution to the HOMOs comes from π orbitals of the phenylene linker and the phosphonate substituent. The electron density is found mainly on the d orbitals only at the HOMO−3 level for Ru-5PPh and HOMO−2 and HOMO−3 for Ru-3PPh and Ru-4PPh (Tables S11–S13†). In the complex, Ru-4,7(PPh)2, all HOMOs are constructed from the d orbitals and π orbitals of the phenylene linker and the phosphonate substituent with a small amount of electron density observed on the d orbitals (Table S14†). In contrast, their LUMOs are rather similar to those of Ru-phen.
It is known that the presence of low-lying dd states can strongly decrease the excited-state lifetimes and photostability of Ru(II) complexes.14 The absence of such states in all studied complexes is likely to account for their high photostability. In contrast, our attempt to use the calculated MO energies for deeper understanding of the oxidation and reduction potentials showed a limited value of such simple DFT calculations. Complex Ru-3,8P2 was the only compound, for which DFT calculations agree with the assignment of the first reduction potentials discussed above. We tried to overcome the inconsistence of experimental and theoretical data by changing the STO 6-31G(d,p) basis to CRENBL, LANL2DZ or Stuttgart 1997 RSC ECP for ruthenium atoms and to Jorge-TZP for other atoms and also compared our results with data reported by other groups.23,32,53,70 All our calculations gave similar electron density distributions in frontier orbitals for Ru-phen and were comparable to the previously reported data. Similar inconsistences of experimental and calculated data were already discussed by others.70 Also we have not attempted to perform computations of excited states by the TD-DFT method. Our previous experience as well as common knowledge say that this method, though quite reasonable in the computations of organic chromophores and coordination compounds involving the transitions of purely ligand-to-ligand nature, tends to give inconsistent if not merely ridiculous predictions when it comes to states including heavy transition metals of 2-nd and 3rd rows. At the same time going beyond TD-DFT to more sophisticated levels of theory is far beyond our current computational facilities.71–74
Photocatalytic properties
Visible light-promoted reactions are started with the absorption of a photon by a photocatalyst (PC), leading to the generation of an excited state *PC. Thus formed, the high energy species *PC can take part in chemical transformations, which happens due to either energy transfer (the EnT photocatalysis) or electron transfer (the redox photocatalysis or photoredox catalysis) to substrates or oxidative or reductive quenchers. Rapid insight into the efficiency of a photocatalyst in both types of processes is possible by using specific experimental and calculated parameters.The EnT photocatalytic reactions are less common and most of them involve oxidation reactions by molecular oxygen.7,75 Energy transfer from an excited ruthenium(II) complex to oxygen molecule can generate singlet oxygen (Scheme 2, pathway A, eq. 2) and this step is usually rate-limiting in the oxygenation reaction. Thus, the singlet oxygen quantum yield (ΦΔ) (Table 2) is a prerequisite for the preliminary evaluation of photocatalyst's efficiency in oxidation reactions.
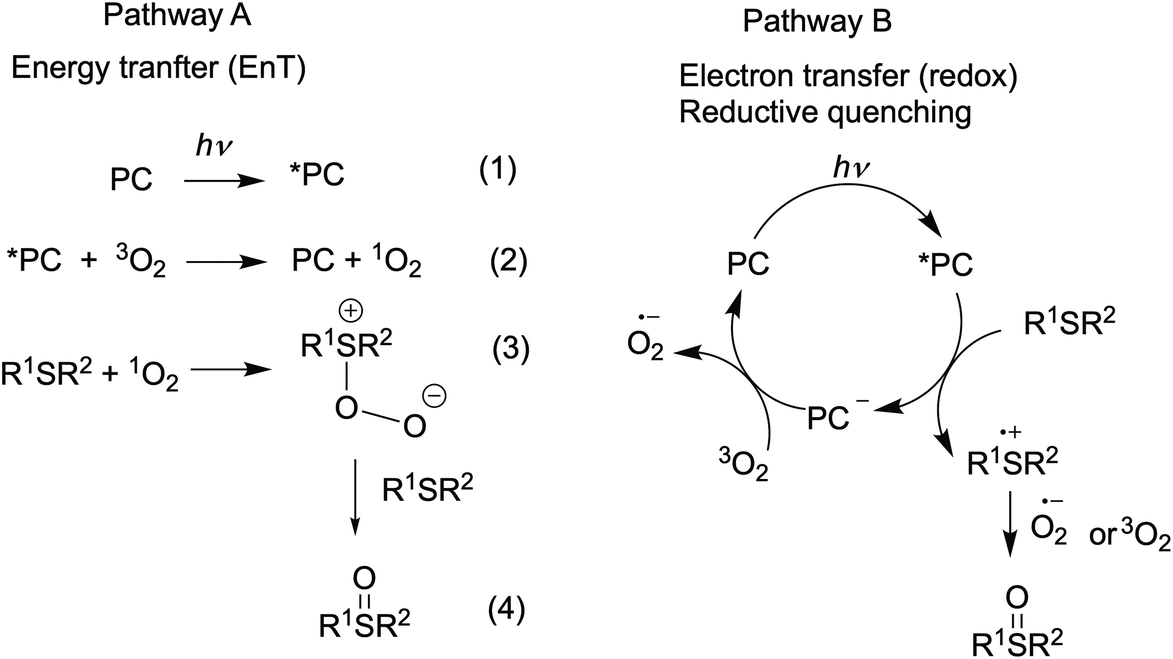 | ||
| Scheme 2 Two possible pathways for the photooxidation of sulfides by molecular oxygen. | ||
The ability of Ru(II) complexes to initiate electron transfer after absorption of a photon depends on redox potentials of excited species, while further evolution of the catalytic cycle may involve redox-reactions of the ground states.10,76 Since excited Ru(II) complexes can be involved in both reductive and oxidative quenching photocatalytic cycles, both first oxidation and reduction potentials of the excited and ground states influence the performance in photoredox catalysis. The redox potentials of the ground state are available from electrochemical studies. Formal redox potentials of the excited state can be estimated by different methods70,77 and simplified calculations76,77 based on CV and spectroscopic data are commonly used in organic photocatalysis. The data obtained for the Ru-Pcat complexes are summarized in Table S15.† On comparing the redox potentials of phosphorous-substituted complexes Ru-P, it can be concluded that the substitution pattern does influence all redox potentials and that the introduction of EWG can be expected to favor the reductive quenching photocatalytic cycle.
It is also worth mentioning that most phosphonate-substituted complexes Ru-Pcat exhibit similar absorption manifolds in the region covered by blue LED, allowing to safely ignore probable effects of the difference in absorptivity on their photocatalytic activity thus simplifying their comparison in photocatalytic reactions.
To investigate the catalytic properties of Ru-Pcat, the oxidation of sulfides into sulfoxides by molecular oxygen was chosen as a model reaction. This reaction attracts considerable interest due to its relevance to biochemistry, warfare agent disposal, environmental consequences of fuel desulfurization and in organic synthesis.78–84 The overoxidation of sulfides to sulfones and the cleavage of S–C and (S)C–H bonds are commonly observed as side reactions85–87 and thus selective methods for the transformation of different sulfides are in high demand in particular in asymmetric synthesis and in the development of pharmaceuticals involving selective late-stage oxygenation.83,88 Explosive peracids and peroxides are commonly used in industry to prepare sulfoxides and sulfones on a large scale.89 Their replacement by less dangerous reagents in the production of fine chemicals is highly desirable. Photocatalysis allows to perform these oxidation reactions using molecular oxygen as a terminal oxidant. We have chosen this reaction as a model reaction to study the photocatalytic properties of Ru-Pcat not only due to its practical importance, but also because the reaction mechanism was thoroughly investigated at least in the case of organic photocatalysts75,90–92 that simplifies the understanding of substituent effects on the reaction course.
It was shown that the photooxidation of sulfides by oxygen proceeds through either of the two mechanisms.75,90–92 The reaction can be initiated by energy transfer from the excited-state photocatalyst (*PC) to oxygen molecule (Scheme 2, pathway A) or by electron transfer from sulfide molecule to the excited-state PC (Scheme 2, pathway B). Alkyl sulfides are commonly oxidized by the generated singlet oxygen (1O2), while aryl sulfides tend to react through both mechanisms with most of the organic photocatalysts.91,93–95 We decided to evaluate the performance of our complexes in redox and EnT photocatalytic reactions by studying sulfoxidation of aryl sulfides and alkyl sulfides, respectively.
First, the photocatalytic efficiency of Ru(II) complexes in this reaction was investigated (Scheme 3). Dialkyl sulfides and methyl aryl sulfides can be quantitively oxidized by slowly bubbling molecular oxygen through their solution in the MeCN/H2O (10:1 v/v) mixture containing Ru-phen or Ru-(bpy)3. In contrast to the previously reported oxidation in chloroform saturated by water,96 under these conditions the reaction proceeded selectively even for the most problematic methyl benzyl sulfide,86,97,98 for which the side reactions of C–S and (S)C–H cleavage are commonly observed.86,97,98 The oxidation was not observed in the absence of water and without irradiation of the reaction mixtures. Dialkyl sulfides were found to be more reactive than aryl sulfides and gave the products quantitively in less than 4 h with the amount of catalyst as low as 0.005 mol%. As in the case of organic photocatalysts, the rate of oxidation of aryl methyl sulfides depended on the substitution in the aromatic ring, and electron acceptor groups at the phenyl ring decreased the reaction rate. Nevertheless, increasing the amount of the photocatalyst up to 0.05 mol% allowed for the oxidation of aryl methyl sulfides to obtain target sulfoxides in quantitative yield within 6 h of irradiation. However, diphenyl sulfide did not react under these conditions even after increasing the loading Ru-phen and Ru-(bpy)3 up to 2 mol%. Sulfoxides thus obtained can be easily isolated in pure form (>98%) without additional purification by column chromatography. All complexes Ru-Pcat were found to be efficient in these reactions and gave the target sulfoxides in quantitative yields.
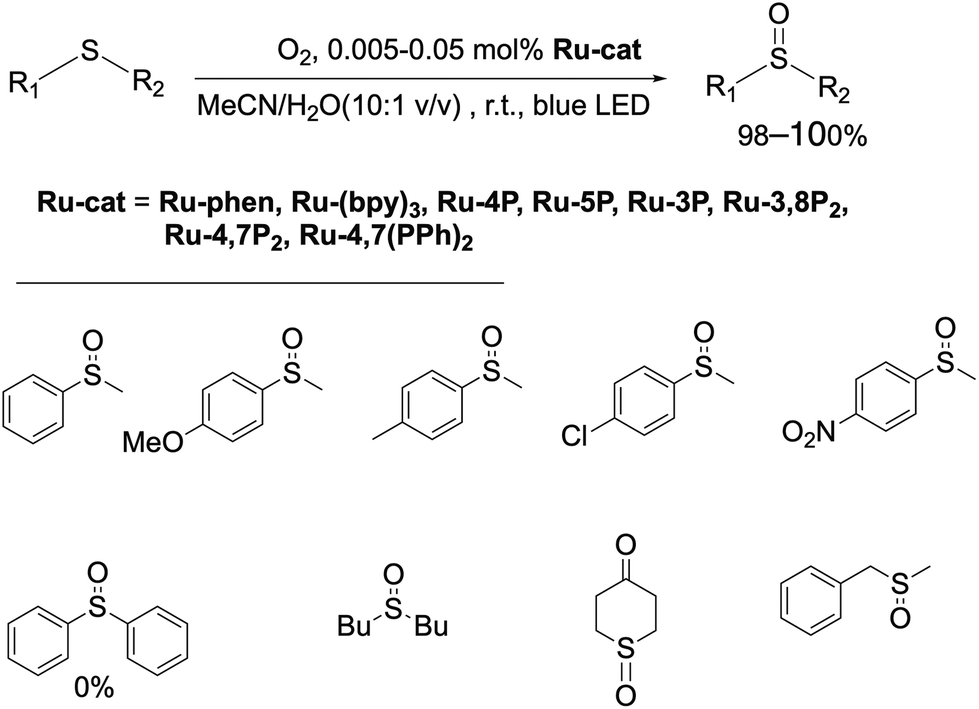 | ||
| Scheme 3 Photocatalytic synthesis of sulfoxides using molecular oxygen as an oxidant. | ||
Encouraged by these results, we undertook kinetic studies to understand the substituent effects on the efficiency of Ru-Pcat complexes. All starting sulfides, complexes and the reaction products were soluble in the MeCN/H2O (10:1 v/v) solvent mixture that allowed for measuring the reaction rates by NMR spectroscopy after withdrawing aliquot samples. The change of conversion over time for the oxidation 4-nitrothioanisole in the presence of 0.05 mol% Ru-Pcat is shown in Fig. 10. The substitution pattern does influence the reaction rate. In the series of isomeric mono-substituted derivatives Ru-P, acceleration of the oxidation was observed in the following range: Ru-5P ≤ Ru-phen < Ru-3P < Ru-4P and the most efficient Ru-4P complex provided the complete conversion by about 2.5-fold more rapidly than Ru-phen and commonly used Ru-(bpy)3. The positive effect of the second phosphonate substituent was less pronounced but still observable when the Ru-4P photocatalyst was replaced by Ru-4,7P2. In contrast, Ru-3,8P2 was less efficient than Ru-3P. The introduction of the phenylene linker between the aromatic ring and the P(O)(OEt)2 group had a detrimental effect on the reaction rate as was evident from comparing Ru-4,7P2 and Ru-4,7(PPh)2 photocatalysts.
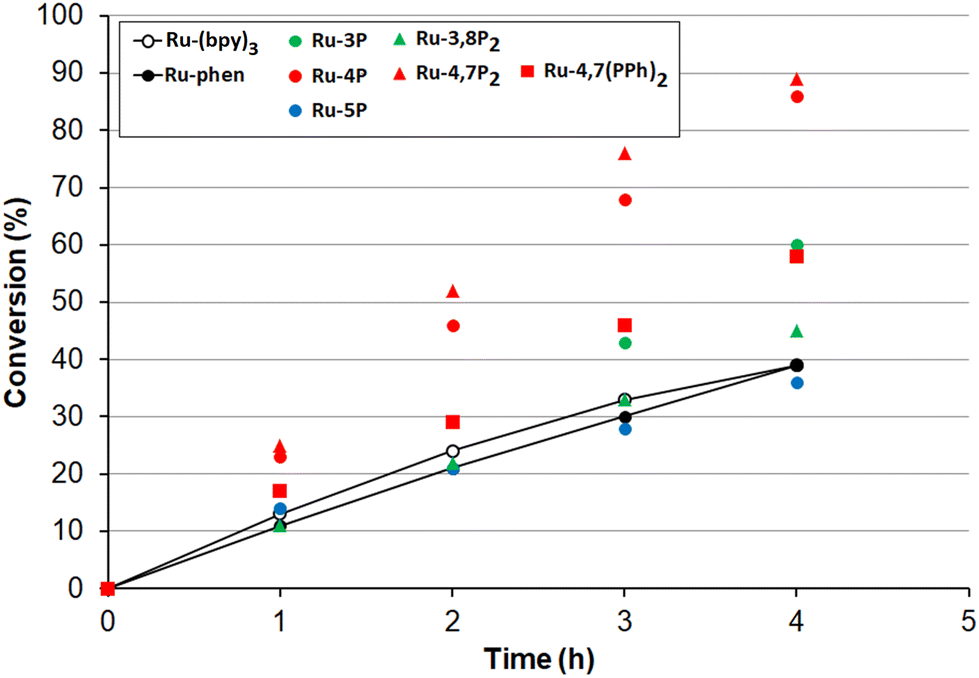 | ||
| Fig. 10 Photooxidation of 4-nitrothioanisole by molecular oxygen in the presence of 0.05 mol% Ru-Pcat and referenced photocatalysts Ru-(bpy)3 and Ru-phen in MeCN/H2O (10:1 v/v) under irradiation of blue LED. | ||
The high efficiency of Ru-4P and Ru-4,7P2 derivatives as compared to Ru-(bpy)3 and Ru-phen complexes in this oxidation reaction (which predominantly proceeds through the reductive quenching catalytic cycle (pathway B, Scheme 2)) can be explained by higher values of their E1/2 (*PC/PC−) potentials. However, correlation between the oxidation rate and this parameter is not as clear for mono-substituted derivatives Ru-3P and Ru-4P, exhibiting the markedly different photocatalytic efficiency while rather similar E1/2 (*PC/PC−) values. This is also understandable because the reduction potential of the excited complex represents an approximative criterion for the evaluation of reaction thermodynamics while kinetics often determines the processes involving short-lived intermediates. On the other hand, we cannot exclude that non-linearity between the reaction rate and redox potentials observed for the Ru-P photocatalysts results from small but noticeable contribution of singlet oxygen in the reaction outcome (Pathway A, Scheme 2).
Next, the oxidation of dibutyl sulfide was investigated because the singlet oxygen mechanism is considered to be operative for this substrate. This sulfide was more reactive than 4-nitroanizole, and the photocatalyst loading was decreased by a factor of 10 to obtain comparable oxidation rates for both sulfides. This indicates that singlet oxygen is an oxidant in this reaction because the bimolecular quenching rate constant of excited-state Ru(II) complexes by dialkyl sulfides and 4-nitrothioanisol is rather similar.99 As shown in Fig. 11, only photocatalysts with P(O)(OEt)2 substituents in positions 4 and 7 gave the sulfoxide more rapidly than Ru-phen and Ru-(bpy)3. Complex Ru-4,7(PPh)2 with the phenylene spacer gave the best results though Ru-4,7P2 and Ru-4P belonging to the series Ru-P were almost as active as this compound. The high efficiency of Ru-4,7(PPh)2 is in good agreement with its increased singlet oxygen quantum yield, though this correlation was not observed for all complexes studied. For instance, complexes Ru-4,7P2 and Ru-4P having rather different capabilities in the generation of 1O2 gave very similar oxidation rates.
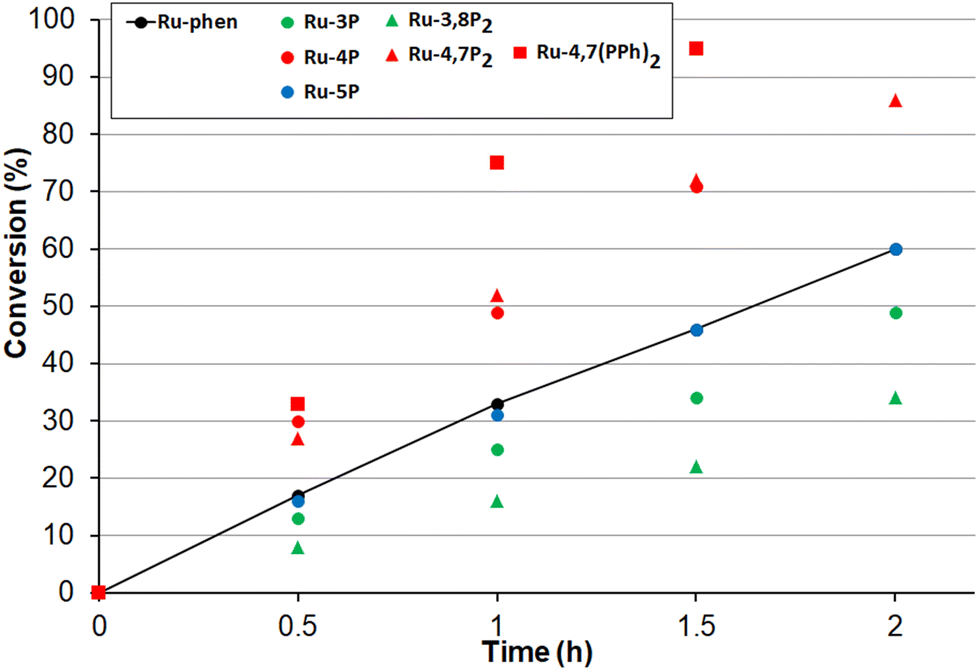 | ||
| Fig. 11 Photooxidation of dibutyl sulfide by molecular oxygen in the presence of 0.005 mol% Ru-Pcat and referenced photocatalysts Ru-(bpy)3 and Ru-phen in MeCN/H2O (10:1 v/v) under irradiation of blue LED. | ||
Next, we tested the performance of the most efficient photocatalysts for the oxidation of other sulfides. In the oxidation of benzyl methyl sulfide, Ru-4,7(PPh)2 and Ru-4,7P2 were as efficient as Ru-4P and all were superior to the Ru-phen complex (Fig. S34†). The oxidation of 4-chlorothioanisol in the presence of Ru-4,7(PPh)2, Ru-4,7P2 or Ru-4P proceeded also faster than with Ru-phen (Fig. S35†).
More theoretical and experimental investigations are certainly required to gain a better understanding of Ru-Pcat photocatalytic properties. Nevertheless, at this point we can safely conclude that complexes Ru-4,7(PPh)2 and Ru-4,7P2 are among the best (in terms of selectivity and the oxidation rate) known photocatalysts so far reported for the oxidation of organic sulfides to sulfoxides.95 Although the direct comparison of reaction TONs for our photocatalysts and those reported earlier is not possible since these values are dependent on the irradiation intensity and other experimental conditions, we evaluated the TON for the oxidation of 4-chlorophenyl methyl sulfide in our conditions. When the most active Ru-4,7P2 photocatalyst was used, the quantitative synthesis of sulfoxide was achieved with a TON of almost 100000, i.e., 100 times higher than reported for the oxidation of more reactive methyl phenyl sulfide in the recent work focused on the photocatalyst optimisation.95 4-Methoxyphenyl methyl sulfoxide was prepared in 99% yield with a TON of about 1000000 and a TOF of about 16000 h−1.
It is worth noting that photocatalytic procedures for the selective formation of sulfoxide from sulfide are still scarce and, in most of them, specific photocatalysts immobilized on solid supports are employed.100–105 Recently the selective photooxidation of sulfides was performed in the presence of Rose Bengal,106 riboflavin95 or BODIPY107 derivatives employing 0.2–2 mol% of photocatalysts. It was also demonstrated that Ir(III) complexes are efficient in this reaction but the reaction scope was limited to alkyl aryl sulfides and 2-hydroxy-4-methylphenyl phenyl sulfide.23
Thus, we can conclude that the complexes Ru-Pcat are promising photocatalysts for performing both redox and EnT photocatalytic reactions. Comparative studies of reactivities of Ru-phen, Ru-4,7(PPh)2 and Ru-4,7-P2 can also be useful in mechanistic investigations of photoreactions.
Conclusions
We report here the study of a series of Ru(II) polypyridyl complexes (Ru-Pcat) of [Ru(phen)(bpy)2]2+-type, which are kinetically stable and have valuable optical and redox properties for application in organic photocatalysis. In this series, the phen ligand is modified with electron-withdrawing substituents P(O)(OEt)2 and 4-(EtO)2(O)PC6H4 in order to tune the redox potentials and excited-state energies and also to gain access to grafting capabilities and developing novel heterogenized photocatalysts.Synthetic approaches were developed to prepare these complexes in good yields. All newly synthesized compounds exhibited high photostability and good solubility in many polar solvents including chlorinated hydrocarbons. The coordination mode of ditopic phen ligands was confirmed by single crystal X-ray analysis of the representative complexes (Ru-5P and Ru-4,7P2). Their kinetic inertness in acetonitrile solutions was shown by NMR studies. Optical, electrochemical and spectroelectrochemical properties were systematically investigated to gain insight into the influence of P(O)(OEt)2 groups on the electronic structure of these complexes. Electrochemical measurements showed that the first reduction ground state potentials vary from −1.60 to −1.23 V (vs. Ag+/Ag) because the incoming electron occupies the π* orbital on the phen ligand in contrast to the parent Ru-phen complex where the bpy ligand lends this MO. The modifications of the phen ligand cause small shifts in the metal-to-ligand charge-transfer absorption energies; the brightness of most complexes is comparable to that of non-substituted Ru-phen and the commonly used Ru-(bpy)3 complex. In contrast, the maximum of the emission band is sensitive to substituents reaching the far-red region for Ru-3,8P2. The electronic structure of complexes Ru-Pcat was obtained by DFT calculations using the B3LYP functional providing a comparison of the newly synthesized compounds and their previously reported analogues.
It was also demonstrated in the studies on the sulfide oxidation by molecular oxygen that the photoredox-catalyzed reactions can be benefited by tuning the structure of the catalyst. An acceleration of aryl sulfide oxidation was observed when the Ru-(bpy)3 photocatalyst was replaced by complexes Ru-4P and Ru-4,7P2.
The introduction of the phenylene linker between the heteroaromatic ring and the phosphonate substituent significantly influences the redox properties of complexes Ru-Pcat, making ligands weaker as electron acceptors. The advantage of photocatalysts with the phenylene linker is observed in the singlet oxygen generation. This makes the complexes Ru-PPh good photocatalysts for the oxidation of organic substrates by molecular oxygen as it was demonstrated in this work by investigating the oxidation of dialkyl sulfides.
It is worth noting that the complexes with a directly attached phosphonate group (Ru-P) and those containing phenylene linkers (Ru-PPh) exhibit different reactivities in the reactions proceeding through energy transfer and photoinduced electron transfer, which could be useful in that they can be used in the mechanistic studies of photoreactions.
Complexes Ru-Pcat are attractive candidates for studies of homogeneous photocatalytic reactions owing to their high solubility in many solvents, high brightness and variable redox potentials that might impact the rate and selectivity of photocatalytic reactions. They are also promising for the development of reusable photocatalytic systems, which is a final goal of our on-going studies. Such systems can be obtained by immobilization of Ru-Pcat on a solid support (metal oxides) or into liquid-phases (liquid–liquid phase separation in which water-soluble Ru(II) complexes are immobilized in the aqueous phase). The wide use of Ru(II) complexes in photocatalysis inevitably poses the questions on their cost, toxicity and future accessibility, which can be solved only by the preparation of reusable photocatalytic systems. The knowledge gathered in this work is an indispensable background in the design of such reusable photocatalytic systems.
Experimental section
Synthesis
General information on materials, methods and synthesis of ligands and complexes Ru-Pcat is present in the ESI.†X-ray crystallography of Ru-5P and Ru-4,7P2
Single crystals of Ru-5P and Ru-4,7P2 were obtained by slow diffusion of toluene in solutions of complexes in the CHCl3/MeOH (1:1) mixture at 4 °C. The measurements were made on a Bruker SMART APEX II diffractometer with a CCD area detector (graphite monochromator, Mo-Kα radiation, λ = 0.71073 Å, ω-scanning, 2θmax = 56°). The semi-empirical method SADABS was applied for the absorption correction.108 The structure was solved by direct methods and refined by the full-matrix least-squares technique against F2 with the anisotropic displacement parameters for all non-hydrogen atoms. All the hydrogen atoms in the complexes were placed geometrically and included in the structure factor calculation in the riding motion approximation. All the data reduction and further calculations were performed using the SAINT and SHELXTL-97.109
Crystallographic data and refinement details are presented in Table S2;† the main geometrical parameters of the studied complexes are listed in Table S3† and Fig. 2 and 3. Full tables of atomic coordinates, bond lengths, and valence angles are deposited in the Cambridge Structural Database (CCDC 2151157 (Ru-5P) and 2159570 (Ru-4,7P2)†).
Structural investigation in solution
All NMR spectra were registered with a Bruker Avance-400 spectrometer in CDCl3 or CD3CN. 2D NMR spectra of Ru-4P were recorded on the Agilent 400-MR instrument. Chemical shifts are expressed in parts per million (ppm), referenced on the δ scale by using residual non-deuterated solvent signals as internal standard for 1H and 13C NMR spectroscopy and external phosphonic acid (H3PO4) for 31P NMR spectroscopy. The coupling constants are expressed in units of frequency (Hz).Electrochemical and spectroelectrochemical characterization
Acetonitrile (Acros Organics, extra-dry with molecular sieves, water <0.005%) was degassed using a freeze–pump–thaw procedure and used as is in a glovebox under N2. Tetra-n-butylammonium hexafluorophosphate was prepared, purified and dried using standard procedures.Cyclic voltammetry and voltammetry with rotating disc electrodes (RDE) were recorded using a SP300 Bilogic potentiostat. Analytical studies were conducted under N2 (glove box) in a standard three-electrode electrochemical cell. Tetra-n-butylammonium hexafluorophoshate was used as supporting electrolytes (0.1 M). An automatic ohmic drop compensation procedure (biologic ZIR) was systematically performed when using cyclic voltammetry. Vitreous carbon (∅ = 3 mm) working electrodes (CH Instruments) were polished with 1 mm diamond paste before each recording. Voltamperometry with a rotating disk electrode (RDE) was carried out with a radiometer (CTV101 radiometer analytical) equipment at a rotation rate of 500 rad min−1 using a glassy carbon RDE tip (∅ = 3 mm).
Spectroelectrochemical measurements were carried out at room temperature under N2 (glove box) in dedicated batch “thin layer” type of cells (0.5 or 1 mm optical path lengths, Pt mesh electrodes, ALS Co. Ltd) using a biologic SP300 potentiostat coupled to a MCS 601 UV-NIR Zeiss spectrophotometer. The counter-electrode was a platinum wire isolated from the electrolytic solution through an ionic bridge. Ag/AgNO3 (CH Instruments, 10−2 M + TBAP 10−1 M in CH3CN) was used as a reference electrode. Ferrocene was ultimately used as an internal reference. Measurements were carried out upon scanning the working electrode potential at 20 mV s−1 between the open circuit potential and a chosen final potential followed by microelectrolysis (1–5 min) at this potential.
Photophysical measurements
UV–vis spectra were recorded in solutions using a PerkinElmer Lambda 900 UV/VIS/NIR Spectrometer (1 cm path length quartz cell). Emission spectra were measured using a PerkinElmer LS 55 Luminescence Spectrometer. Emission quantum yields of all compounds were measured relative to the Ru-(bpy)3 in acetonitrile (Φem = 0.095)64 and calculated using a standard procedure.110Singlet oxygen generation quantum yield measurements were performed according to the literature.111 To a solution of the 1O2 trap, 1,9-dimethylanthracene (DMA), with an optical density of around 1.4 in the air-saturated MeCN corresponding Ru(II) complex was added in a quartz cuvette, and its absorbance was adjusted to around 0.1 at the wavelength of irradiation. The solution in the cuvette was irradiated with 488 nm laser (Melles Griot 43 series i laser, 543R-AP-AO1) at a constant power density of 10 mW cm−2. The absorption spectra of the solutions were measured every 30 s. The slope of plots of absorbance of DMA at 376 nm vs. irradiation time for each photosensitizer was calculated.
Singlet oxygen quantum yields were calculated based on the equation:
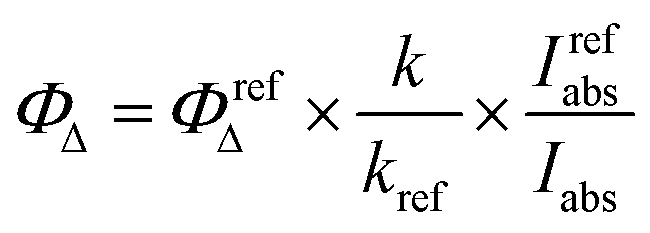
Photocatalytic oxidation of sulfides
Standard 0.01 M solutions of Ru-(bpy)3, Ru-phen and Ru-Pcat were prepared dissolving 0.05 mmol Ru(II) complexes in an acetonitrile/water (10:1 v/v) solvent mixture in 5 ml volumetric flasks.
:1 v/v) was added to obtain a solution of reagents in 2 mL of the solvent mixture. The reaction was irradiated with blue LED (30 W) for 4–6 h while gently bubbling oxygen. When the reaction was complete, the mixture was diluted with 7 mL of water and extracted with methylene chloride (3 × 5 mL). The combined extracts were dried over sodium sulfate and evaporated under reduced pressure at room temperature. The yield and purity of the products were determined by 1H NMR using toluene as an internal standard.
Dialkyl sulfides were oxidized using 0.005 mol% photocatalysts and the oxidation of aryl sulfides was performed in the presence of 0.05 mol% photocatalysts as shown in Scheme 2. This catalyst loading was also used in kinetic studies (Fig. 10, 11, S34 and S35†) which were performed by using the same procedure. The reactions were periodically monitored by NMR spectroscopy after withdrawing aliquot samples.
Author contributions
J. M., A. Yu. M. and A. S. A. performed the synthesis of P ligands and Ru-P complexes. I. S. Z. prepared PPh ligands. G. V. M. optimized the synthesis of PPh ligands and Ru-P complexes and developed synthetic approaches to Ru-PPh complexes. M. A. F. performed the photophysical characterization of all complexes and writing of the original draft of this article section. A. D. A. participated in the characterization (NMR and mass spectroscopy) of newly synthesized compounds. V. A. R. performed specific NMR experiments allowing for the signal assignment in the spectra of Ru-4P. S. E. N. was responsible for single crystal X-ray characterization. C. B. performed the electrochemical and spectroelectrochemical characterization of Ru complexes and writing of the original draft of this article section. A. V. C. performed DFT calculations. A. B.-L. studied the photocatalytic properties of Ru-Pcat complexes. I. P. B. was responsible for data analysis, manuscript editing and funding acquisition. L. B. participated in manuscript editing. A. B.-L. and A. S. A. were responsible for the project conceptualization, supervision, data analysis, writing of the article and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. K. A. Lyssenko, Dr Y. Bretonnière and Dr S. Denis-Quanquin for help and useful discussions. This work was supported by the Russian Science Foundation (grant no. 20-73-00103). It was accomplished in the frame of the collaborative agreement between ENS de Lyon and Lomonosov Moscow State University (2012-2021). The 2D NMR spectra were recorded on the Agilent 400-MR instrument purchased by the MSU Development Program.References
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- F. Bella, C. Gerbaldi, C. Barolo and M. Grätzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC.
- D. L. Ashford, M. K. Gish, A. K. Vannucci, M. K. Brennaman, J. L. Templeton, J. M. Papanikolas and T. J. Meyer, Chem. Rev., 2015, 115, 13006–13049 CrossRef CAS PubMed.
- P. Alreja and N. Kaur, RSC Adv., 2016, 6, 23169–23217 RSC.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148–6169 RSC.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- C. P. Anderson, D. J. Salmon, T. J. Meyer and R. C. Young, J. Am. Chem. Soc., 1977, 99, 1980–1982 CrossRef CAS.
- T.-J. J. Kinnunen, M. Haukka, M. Nousiainen, A. Patrikka and T. A. Pakkanen, J. Chem. Soc., Dalton Trans., 2001, 2649–2654 RSC.
- P. A. Mabrouk and M. S. Wrighton, Inorg. Chem., 1986, 25, 526–531 CrossRef CAS.
- K. R. Bargawi, A. Llobet and T. J. Meyer, J. Am. Chem. Soc., 1988, 110, 7751–7759 CrossRef CAS.
- A. E. Curtright and J. K. McCusker, J. Phys. Chem. A, 1999, 103, 7032–7041 CrossRef CAS.
- X. Xiao, J. Sakamoto, M. Tanabe, S. Yamazaki, S. Yamabe and T. Matsumura-Inoue, J. Electroanal. Chem., 2002, 527, 33–40 CrossRef CAS.
- N. A. F. Al-Rawashdeh, S. Chatterjee, J. A. Krause and W. B. Connick, Inorg. Chem., 2014, 53, 294–307 CrossRef CAS PubMed.
- S. S. Bhat, A. S. Kumbhar, P. Lönnecke and E. Hey-Hawkins, Inorg. Chem., 2010, 49, 4843–4853 CrossRef CAS PubMed.
- P. J. DeLaive, J. T. Lee, H. W. Sprintschnik, H. Abruna, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1977, 99, 7094–7097 CrossRef CAS.
- T. Rajendran, P. Thanasekaran, S. Rajagopal, G. A. Gnanaraj, C. Srinivasan, P. Ramamurthy, B. Venkatachalapathy, B. Manimaran and K.-L. Lu, Phys. Chem. Chem. Phys., 2001, 3, 2063–2069 RSC.
- E. Deponti and M. Natali, Dalton Trans., 2016, 45, 9136–9147 RSC.
- H. Shalan, A. Colbert, T. T. Nguyen, M. Kato and L. Cheruzel, Inorg. Chem., 2017, 56, 6558–6564 CrossRef CAS PubMed.
- L.-P. Li and B.-H. Ye, Inorg. Chem., 2019, 58, 7775–7784 CrossRef PubMed.
- S. DiLuzio, T. U. Connell, V. Mdluli, J. F. Kowalewski and S. Bernhard, J. Am. Chem. Soc., 2022, 144, 1431–1444 CrossRef CAS PubMed.
- M. Liras, M. Pintado-Sierra, M. Iglesias and F. Sánchez, J. Mater. Chem. A, 2016, 4, 17274–17278 RSC.
- C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed.
- G. Guerrero, J. G. Alauzun, M. Granier, D. Laurencin and P. H. Mutin, Dalton Trans., 2013, 42, 12569–12585 RSC.
- H. Zabri, I. Gillaizeau, C. A. Bignozzi, S. Caramori, M.-F. Charlot, J. Cano-Boquera and F. Odobel, Inorg. Chem., 2003, 42, 6655–6666 CrossRef CAS PubMed.
- P. Wang, C. Klein, J.-E. Moser, R. Humphry-Baker, N.-L. Cevey-Ha, R. Charvet, P. Comte, S. M. Zakeeruddin and M. Grätzel, J. Phys. Chem. B, 2004, 108, 17553–17559 CrossRef CAS.
- M. R. Norris, J. J. Concepcion, C. R. K. Glasson, Z. Fang, A. M. Lapides, D. L. Ashford, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2013, 52, 12492–12501 CrossRef CAS PubMed.
- K. Neuthe, F. Bittner, F. Stiemke, B. Ziem, J. Du, M. Zellner, M. Wark, T. Schubert and R. Haag, Dyes Pigm., 2014, 104, 24–33 CrossRef CAS.
- D. L. Ashford, M. K. Brennaman, R. J. Brown, S. Keinan, J. J. Concepcion, J. M. Papanikolas, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2015, 54, 460–469 CrossRef CAS PubMed.
- M. M. Raber, M. D. Brady, L. Troian-Gautier, J. C. Dickenson, S. L. Marquard, J. T. Hyde, S. J. Lopez, G. J. Meyer, T. J. Meyer and D. P. Harrison, ACS Appl. Mater. Interfaces, 2018, 10, 22821–22833 CrossRef CAS PubMed.
- M. D. Brady, L. Troian-Gautier, R. N. Sampaio, T. C. Motley and G. J. Meyer, ACS Appl. Mater. Interfaces, 2018, 10, 31312–31323 CrossRef CAS PubMed.
- S. L. Esarey and B. M. Bartlett, Langmuir, 2018, 34, 4535–4547 CrossRef CAS PubMed.
- P. Farràs, C. Di Giovanni, J. N. Clifford, E. Palomares and A. Llobet, Coord. Chem. Rev., 2015, 304–305, 202–208 CrossRef.
- S. Reischauer, V. Strauss and B. Pieber, ACS Catal., 2020, 10, 13269–13274 CrossRef CAS.
- S. Reischauer and B. Pieber, ChemPhotoChem, 2021, 5, 716–720 CrossRef CAS.
- A.-C. Laemmel, J.-P. Collin and J.-P. Sauvage, Eur. J. Inorg. Chem., 1999, 383–386 CrossRef CAS.
- A. Yu Mitrofanov, PhD thesis, Université de Bourgogne and Lomonosov Moscow state university, Dijon, France and Moscow, Russia, 2015.
- S. Amthor, P. Keil, D. Nauroozi, D. Perleth and S. Rau, Eur. J. Inorg. Chem., 2021, 4790–4798 CrossRef CAS.
- A. Mitrofanov, A. B. Lemeune, C. Stern, R. Guilard, N. Gulyukina and I. Beletskaya, Synthesis, 2012, 3805–3810 CAS.
- A. Mitrofanov, S. Brandès, F. Herbst, S. Rigolet, A. Bessmertnykh-Lemeune and I. Beletskaya, J. Mat. Chem. A, 2017, 5, 12216–12235 RSC.
- B.-H. Ye, L.-N. Ji, F. Xue and T. C. W. Mak, Transition Met. Chem., 1999, 24, 8–12 CrossRef CAS.
- B.-H. Ye, X.-M. Chen, T.-X. Zeng and L.-N. Ji, Inorg. Chim. Acta, 1995, 240, 5–11 CrossRef CAS.
- D. S. Eggleston, K. A. Goldsby, D. J. Hodgson and T. J. Meyer, Inorg. Chem., 1985, 24, 4573–4580 CrossRef CAS.
- D. P. Rillema, D. S. Jones, C. Woods and H. A. Levy, Inorg. Chem., 1992, 31, 2935–2938 CrossRef CAS.
- C. M. Elliott and E. Hershenhart, J. Am. Chem. Soc., 1982, 104, 7519–7526 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- M. Montalti, S. Wadhwa, W. Y. Kim, R. A. Kipp and R. H. Schmehl, Inorg. Chem., 2000, 39, 76–84 CrossRef CAS PubMed.
- P. S. Braterman, J.-I. Song and R. D. Peacock, Spectrochim. Acta, Part A, 1992, 48, 899–903 CrossRef.
- N. Yoshikawa, S. Yamabe, S. Sakaki, N. Kanehisa, T. Inoue and H. Takashima, J. Mol. Struct., 2015, 1094, 98–108 CrossRef CAS.
- T. Véry, D. Ambrosek, M. Otsuka, C. Gourlaouen, X. Assfeld, A. Monari and C. Daniel, Chem. – Eur. J., 2014, 20, 12901–12909 CrossRef PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–834 CrossRef CAS PubMed.
- A. Ghosh, B. Ganguly and A. Das, Inorg. Chem., 2007, 46, 9912–9918 CrossRef CAS PubMed.
- C. T. Lin, W. Boettcher, M. Chou, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1976, 98, 6536–6544 CrossRef CAS.
- P. C. Alford, M. J. Cook, A. P. Lewis, G. S. G. McAuliffe, V. Skarda, A. J. Thomson, J. L. Glasper and D. J. Robbins, J. Chem. Soc., Perkin Trans. 2, 1985, 705–709 RSC.
- H. Suzuki, T. Kanbara and T. Yamamoto, Inorg. Chim. Acta, 2004, 357, 4335–4340 CrossRef CAS.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- J. K. Barton, US Pat, 5157032, 1992 Search PubMed.
- K. Mori, M. Kawashima, K. Kagohara and H. Yamashita, J. Phys. Chem. C, 2008, 112, 19449–19455 CrossRef CAS.
- H. Ishida, S. Tobita, Y. Hasegawa, R. Katoh and K. Nozaki, Coord. Chem. Rev., 2010, 254, 2449–2458 CrossRef CAS.
- A. A. Abdel-Shafi, P. D. Beer, R. J. Mortimer and F. Wilkinson, J. Phys. Chem. A, 2000, 104, 192–202 CrossRef CAS.
- J. Al-Nu'airat, I. Oluwoye, N. Zeinali, M. Altarawneh and B. Z. Dlugogorski, Chem. Rec., 2021, 21, 315–342 CrossRef PubMed.
- A. A. Granovsky, Firefly version 8, https://classic.chem.msu.su/gran/firefly/index.html Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. Dolg, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
- D. F. Zigler, Z. A. Morseth, L. Wang, D. L. Ashford, M. K. Brennaman, E. M. Grumstrup, E. C. Brigham, M. K. Gish, R. J. Dillon, L. Alibabaei, G. J. Meyer, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2016, 138, 4426–4438 CrossRef CAS PubMed.
- D. F. Zigler, Z. A. Morseth, L. Wang, D. L. Ashford, M. K. Brennaman, E. M. Grumstrup, E. C. Brigham, M. K. Gish, R. J. Dillon, L. Alibabaei, G. J. Meyer, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2016, 138, 4426–4438 CrossRef CAS PubMed.
- F. Alary, J. L. Heully, L. Bijeire and P. Vicendo, Inorg. Chem., 2007, 46, 3154–3165 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- S. Grimme and M. Parac, ChemPhysChem, 2003, 4, 292–295 CrossRef CAS PubMed.
- A. Dreuw and M. Wormit, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CAS.
- E. L. Clennan, Acc. Chem. Res., 2001, 34, 875–884 CrossRef CAS PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- V. Balzani, F. Bolletta, M. T. Gandolfi and M. Maestri, in Organic chemistry and theory. Topics in current chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 1978, pp. 1–64 Search PubMed.
- S. Miller, Environ. Sci. Technol., 1990, 24, 1286–1289 CrossRef.
- K. A. Stingl and S. B. Tsogoeva, Tetrahedron: Asymmetry, 2010, 21, 1055–1074 CrossRef CAS.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2015, 54, 9001–9005 CrossRef CAS PubMed.
- X. Lang, W. Hao, W. R. Leow, S. Li, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 5000–5005 RSC.
- G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834–3860 RSC.
- G. Laudadio, N. J. W. Straathof, M. D. Lanting, B. Knoops, V. Hessel and T. Noël, Green Chem., 2017, 19, 4061–4066 RSC.
- Sulfur Chemistry, ed. X. Jiang, Springer, Berlin, 2019 Search PubMed.
- E. Baciocchi, C. Rol, E. Scamosci and G. V. Sebastiani, J. Org. Chem., 1991, 56, 5498–5502 CrossRef CAS.
- E. Baciocchi, C. Crescenzi and O. Lanzalunga, Tetrahedron, 1997, 53, 4469–4478 CrossRef CAS.
- E. Baciocchi, T. Del Giacco, M. F. Gerini and O. Lanzalunga, Org. Lett., 2006, 8, 641–644 CrossRef CAS PubMed.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- E. Baciocchi, T. D. Giacco, F. Elisei, M. F. Gerini, M. Guerra, A. Lapi and P. Liberali, J. Am. Chem. Soc., 2003, 125, 16444–16454 CrossRef CAS PubMed.
- S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem. – Eur. J., 2006, 12, 4844–4857 CrossRef CAS PubMed.
- S. M. Bonesi, M. Fagnoni and A. Albini, Eur. J. Org. Chem., 2008, 2612–2620 CrossRef CAS.
- H. Yokoi, A. Hatta, K. Ishiguro and Y. Sawaki, J. Am. Chem. Soc., 1998, 120, 12728–12733 CrossRef CAS.
- E. Baciocchi, T. Del Giacco, F. Elisei and A. Lapi, J. Org. Chem., 2006, 71, 853–860 CrossRef CAS PubMed.
- T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef.
- G. Bianchini, A. Scarso, G. L. Sorella and G. Strukul, Chem. Commun., 2012, 48, 12082–12084 RSC.
- S. M. Bonesi, M. Mella, N. d'Alessandro, G. G. Aloisi, M. Vanossi and A. Albini, J. Org. Chem., 1998, 63, 9946–9955 CrossRef CAS.
- E. L. Clennan, W. Zhou and J. Chan, J. Org. Chem., 2002, 67, 9368–9378 CrossRef CAS PubMed.
- E. Rajkumar and S. Rajagopal, Photochem. Photobiol. Sci., 2008, 7, 1407–1414 CrossRef CAS PubMed.
- J.-M. Zen, S.-L. Liou, A. S. Kumar and M.-S. Hsia, Angew. Chem., Int. Ed., 2003, 42, 577–579 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- H. Yang and H. Fei, Dalton Trans., 2017, 46, 2751–2755 RSC.
- X. Zhang, X. Zhang, J. A. Johnson, Y.-S. Chen and J. Zhang, J. Am. Chem. Soc., 2016, 138, 8380–8383 CrossRef CAS PubMed.
- Z.-Y. Xu, Y. Luo, D.-W. Zhang, H. Wang, X.-W. Sun and Z.-T. Li, Green Chem., 2020, 22, 136–143 RSC.
- H.-P. Liang, Q. Chen and B.-H. Han, ACS Catal., 2018, 8, 5313–5322 CrossRef CAS.
- X. Gu, X. Li, Y. Chai, Q. Yang, P. Li and Y. Yao, Green Chem., 2013, 15, 357–361 RSC.
- W. Li, Z. Xie and X. Jing, Catal. Commun., 2011, 16, 94–97 CrossRef CAS.
- G. Sheldrick, Bruker AXS Inc., Madison, USA, 1997.
- Bruker SAINT: Area-Detector Integration Sofware, 2012, Madison, Wisconsin, USA, 2012 Search PubMed.
- M. Levitus, Methods Appl. Fluoresc., 2020, 8, 033001 CrossRef CAS PubMed.
- L. Gou, C. N. Coretsopoulos and A. B. Scranton, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1285–1292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HR-ESI mass spectra, UV–vis spectra, and NMR spectra for the synthesized compounds; additional data on X-ray structures, electrochemistry, photocatalysis and DFT calculations. CCDC 2151157 and 2159570. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01364a |
| This journal is © The Royal Society of Chemistry 2022 |