
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of homogeneous (U,Am)O2 and (U,Pu,Am)O2 nanopowders†
Jean-François
Vigier
 *a,
Daniel
Freis
a,
Olaf
Walter
a,
Oliver
Dieste Blanco
a,
Daniel
Bouëxière
a,
Evelyn
Zuleger
a,
Natalia
Palina
b,
Tonya
Vitova
b,
Rudy J. M.
Konings
a and
Karin
Popa
*a
*a,
Daniel
Freis
a,
Olaf
Walter
a,
Oliver
Dieste Blanco
a,
Daniel
Bouëxière
a,
Evelyn
Zuleger
a,
Natalia
Palina
b,
Tonya
Vitova
b,
Rudy J. M.
Konings
a and
Karin
Popa
*a
aEuropean Commission, Joint Research Centre (JRC), Karlsruhe, Germany. E-mail: jean-francois.vigier@ec.europa.eu; karin.popa@ec.europa.eu
bInstitute for Nuclear Waste Disposal (INE), Karlsruhe Institute of Technology, P.O. 3640, D-76021 Karlsruhe, Germany
First published on 31st August 2022
Abstract
This paper details the first dedicated production of homogeneous nanocrystalline particles of mixed actinide oxide solid solutions containing americium. The target compositions were U0.75Pu0.20Am0.05O2, U0.90Am0.10O2 and U0.80Am0.20O2. After successful hydrothermal synthesis and chemical characterisation, the nanocrystals were sintered and their structure and behaviour under self-irradiation were studied by powder XRD. Cationic charge distribution of the as-prepared nanocrystalline and sintered U0.80Am0.20O2 materials was investigated applying U M4 and Am M5 edge high energy resolution XANES (HR-XANES). Typical oxidation states detected for the cations are U(IV)/U(V) and Am(III)/Am(IV). The measured crystallographic swelling was systematically smaller for the as-synthesised nanoparticles than the sintered products. For sintered pellets, the maximal volumetric swelling was about 0.8% at saturation, in line with literature data for PuO2, AmO2, (U,Pu)O2 or (U,Am)O2.
1. Introduction
One of the sustainability goals for nuclear reactors of the fourth generation (Gen-IV), as defined by the Generation IV International Forum (GIF), is the minimisation of the nuclear waste and the reduction of the long-term stewardship burden.1 Since plutonium and the minor actinides (MA) are largely responsible for the long-term radiotoxicity of the spent nuclear fuel,2,3 their separation and transmutation into short-lived isotopes in fast reactor systems can significantly contribute to this goal.4 The reuse of fissile isotopes from nuclear waste in the form of mixed oxide fuel is an integral part of the nuclear energy strategy in some EU member states and contributes to the sustainable usage of nuclear material resources. Similarly, the recycling of MA could lead to a further reduction of the radiotoxic inventory to be placed in the final deposits.5The GIF sodium fast reactor advanced fuel project (SFR AF) targets at the development of minor actinide bearing fuels for irradiation in future SFR.6 Within the frame of this project, the Joint Research Centre investigates the performance of such fuels but also safe and reliable preparation routes. The preparation of MOX fuels containing MA requires energy and manpower, the workers then being exposed to radiation. Therefore, there is a need for development of processes that improve the quality of powders, decrease the reaction temperature and the total time of the process.
Oxalate thermal decomposition is a method largely applied for AnO2 production, being appropriate for quantitative separation and recycling of actinides, as well as for fuel production or reprocessing of spent fuel.7,8 Such powders are difficult to sinter by conventional routes9 because of their platelet shape.
The hydrothermal decomposition (called also “decomposition under hot compressed water”) of actinide oxalates has been recently proposed by our group as an innovative approach for the safe and secure synthesis of oxide fuel.10 AnO2 (An = Th, U, Np, Pu) end-members have been prepared by this method.10–14 Associated U1−xAnxO2 mixed oxides (An = Th, Pu) were obtained as well by using this method.15,16 Such powders are typically composed of crystallites of 10 nm or smaller, softly agglomerated at a submicrometric scale due to the low temperature of the decomposition.11
During our extensive studies on such compounds, we have noticed the divergent decomposition behaviour of the tetravalent plutonium and cerium oxalates.11 Previous results indicate that the hydrothermal decomposition of the plutonium oxalate hexahydrate leads to formation of PuO2 nano crystals. On the other hand, decomposition of cerium oxalate gives inconclusive results in the 60–350 °C temperature range. The product of the cerium oxalate decomposition at 400 °C/250 bar is the hexagonal Ce(CO3)(OH), in which cerium has a trivalent oxidation state. Keeping in mind the analogy between Ce(III) and Am(III), similar behaviour was expected for the decomposition of americium oxalate. On the other hand, work on (U,Pu)-mixed oxides show that the americium contained in the PuO2 source is fully incorporated in the oxide nanoparticles (up to 1% out of the total Pu-content).16
The present report addresses this matter, namely the feasibility of the methodology for the production of nanocrystalline (U,Am)O2 and (U,Pu,Am)O2 solid solutions. Such systems are particularly complex due to the high oxygen potential, chemical disorder in the cationic sublattice, and significant self-irradiation effects. Thus, the stability of the solid solutions of different chemical composition and forms (powder or pellets, under- or fully stoichiometric in oxygen) under α self-irradiation is reported. Mechanical and structural characterization of the sintered products is also presented in this paper.
2. Experimental
Caution! Americium-241 is a highly radioactive isotope (t1/2 = 432.8 years, specific activity of 126.8 GBq g−1). Moreover, the use of plutonium-239 (as the main component of the Pu-source) imply additional radiological hazard (t1/2 = 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) 110 years, specific activity of 2.8 GBq g−1). Natural uranium is only weakly radioactive. All work presented in this paper has been carried out in radiological laboratories licensed for handling actinides, equipped with radiation shielding and remote handling tools.
110 years, specific activity of 2.8 GBq g−1). Natural uranium is only weakly radioactive. All work presented in this paper has been carried out in radiological laboratories licensed for handling actinides, equipped with radiation shielding and remote handling tools.
2.1 Sample preparation and mechanical processing
For the synthesis of (U,Am)O2 and (U,Pu,Am)O2 nanopowders we have used the method of hydrothermal decomposition of mixed oxalates, as originally described in Walter et al.10 in addition, cerium was used as surrogate in order to check whether it can properly simulate the Am-behaviour in (U,Am)O2 nanopowders over the all compositional range.U(IV) aqueous solution was obtained by electroreduction of UO2(NO3)2 solution in HNO3 (4 mol L−1) containing 0.5 mol L−1 of hydrazine. The Pu(IV) solution was produced by dissolution of PuO2 in HNO3 (8 mol L−1). The Am(III) solution was obtained by dissolution of AmO2 in HNO3 (6 mol L−1), while the Ce(III) solution by direct dissolution of Ce(NO3)3·6H2O (Sigma, 99.99%) in milli-Q water. The metal concentration in all these solutions was of about 0.5 mol L−1.
Then, the nitrate actinide solutions were mixed in the desired molar ratio. The mixed solution was precipitated with excess (10–20%) of oxalic acid (aqueous solution of 0.5 mol L−1). The readily formed precipitate was separated from the acidic solution and washed repeatedly with milli-Q water until pH = 7. The oxalate precipitate (about 1.5–2.0 g), together with 6.5 mL of milli-Q water, was treated hydrothermally for 3.5 h at 220 °C and autogenic pressure (estimated to 25–35 bar). The process was conducted in an autoclave made out of stainless steel and containing a Teflon inset of 20 mL (Fig. S1†). The final product was washed with water, ethanol and acetone.
The first experimental trial was performed in order to produce (U,Pu,Am)O2 nanopowders and sintered pellets (Fig. 1a). Thus, the nc-powder was pressed uniaxially at 500 MPa to green discs. Some discs were placed into a molybdenum crucible and sintered for 6 h at 1650 °C under an atmosphere of Ar/H2 (4%) and about 2000 ppm of moisturizing water (heating and cooling ramps of 200 °C h−1). Other disks were sintered for 6 h at 1650 °C under an atmosphere of dry Ar/H2 (4%).
 | ||
| Fig. 1 Flowcharts indicating the steps used for the production and conditioning of (U,Pu,Am)O2 (a) and (U,Am)O2 (b) solid solutions. | ||
Further experiments were achieved in order to obtain (U,Am)O2 solid solutions. Since the process used for the production of the (U,Pu,Am)O2 pellets did not result in very high density disks (please see the Results and discussions section), an additional heating stage (1 h at 400 °C under Ar) was introduced in the workflow (Fig. 1b). In this case, the sintering has been performed only under one condition (6 h at 1600 °C under an atmosphere of Ar/H2 (4%) and about 100 ppm of moisturizing water, heating and cooling ramps of 200 °C h−1).
In order to check the limit of the incorporation of trivalent Am-cations in the fluorite structure, additional experiments were performed using cerium as americium substitute. U(IV) and Ce(III) solutions were mixed in molar ratios of 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, 80:20, 70:30, 60:40, 50:50, 40:60, 25:75 and 0:100. All the other procedural steps were the same as described for the synthesis of (U,Am)O2.
:10, 80:20, 70:30, 60:40, 50:50, 40:60, 25:75 and 0:100. All the other procedural steps were the same as described for the synthesis of (U,Am)O2.
2.2 Characterization
In order to avoid any dispersion of radioactive powders into the glovebox, the measurements were performed on about 10 mg of powder immobilized in a bi-component epoxy resin on a sample holder. (Fig. S3†). The measurements were repeated over a two-years timeframe so as to follow the swelling of the oxides as a function of time/radiation dose.
The scanning electron microscope (SEM) used in this work was a Philips XL40 which has the column, chamber and high voltage power supply placed in a glovebox.
UO2 and AmO2 were used as reference to calibrate the respective HR-XANES spectra. The main absorption maximum was set to 3275.5 keV and 3890.8 keV for UO2 and AmO2 respectively.23 The sample cells were placed into a double-containment multi-position cell, where the inner compartment was sealed by 8 μm and the outer compartment by 13 μm Kapton foil, respectively (Fig. S3†). The experimental energy resolution during the U M4 and Am M5 HR-XANES measurements was estimated to be 1 eV and 1.3 eV, respectively. The HR-XANES spectra were measured with step size 0.1 eV from −10 eV to +25 eV from the white line (WL) of the respective edge and 0.5 eV in all other parts of the spectra. At least two spectra were averaged for each sample. The sample, crystals, and detector were enclosed in a box filled with helium to minimize intensity losses due to scattering and absorption of photons in air. A constant helium flow was maintained to keep the oxygen level below 0.1%. No effect of radiation damage in the materials was evident during the measurements.
We estimated the relation between U(IV) and U(V) by (i) assuming that the main absorption maxima of U(IV) and U(V) have the same absorption intensity for 100% U(IV) or U(V) present in a sample; note that the main peak of the spectrum of U(V) will have higher intensity than U(IV) as a result of less electrons in the f states thus the U(V) contribution will be underestimated; (ii) only mixtures of U(IV) and U(V) are present in the two samples studied. Generally, the main peak intensity does not necessarily strictly follow the absorption cross section. But because U(V) and U(IV) occupy the same crystallographic positions and have very similar electronic structure, the approach is justified. The trend in intensity change will follow the change of electron density in the f states, i.e. the uranium oxidation state change. It should be also mentioned that the HR-XANES spectrum is a cut through a resonant inelastic X-ray scattering map and thus the intensity of the U(V) peak is additionally influenced.23 We then calculated the ratio U(IV)/U(V) characteristic peak intensities for the as prepared and sintered samples.
3. Results and discussion
3.1 Chemical characterization
The chemical composition of the synthetized compounds obtained on the basis of the chemical characterization is presented in Table 1. The uranium and plutonium isotopic compositions are reported in the Table S2 as a ESI.†| Target composition | Actual composition | a, Å | Crystal size, nm | Strain |
|---|---|---|---|---|
| U0.75Pu0.20Am0.05O2 | U0.769Pu0.171Am0.056Np0.004O2 | 5.451(1) | 13 ± 2 | 22 × 10−4 |
| U0.90Am0.10O2 | U0.878Am0.114Np0.008O2 | 5.466(1) | 12 ± 2 | 20 × 10−4 |
| U0.80Am0.20O2 | U0.761Am0.223Np0.016O2 | 5.466(1) | 10 ± 2 | 31 × 10−4 |
Due to the ageing process, the americium contains about 7% 237Np, and the plutonium about 2% 241Am. It can be observed that, despite inherent uncertainties in the composition of the reagents (moisture content, presence of the decay products), the final compositions are very close to the targeted ones. For simplification, we will use further the target composition.
3.2 Dimensional measurements
The sintered disks were measured using a micrometer screw gauge (for the diameters) and a dial indicator (for the heights). The weights were measured using a calibrated Sartorius precision balance. The results are summarized in Table S1.† The (U,Am)O2 disks showed geometrical densities between 83% TD (theoretical density) and 96% TD, as function of the composition. However, hydrostatic measurements performed on selected samples result in values that are about 3% higher, explained by the important geometrical deviations due to the axial shrinkage. In a similar manner, the (U,Pu,Am)O2 disks sintered under Ar/H2 + 2000 ppm H2O indicated geometrical densities between 72% TD and 79% TD, while the (U,Pu,Am)O2 disks sintered under dry Ar/H2 atmosphere between 83% TD and 87% TD.3.3 Powder XRD and morphological characterization of the freshly synthesized specimens
 | ||
| Fig. 2 XRD pattern of the nanocrystalline product (nc-U0.75Pu0.20Am0.05O2), U0.75Pu0.20Am0.05O2.00 sintered under moisturized Ar/H2, and U0.75Pu0.20Am0.05O2−x sintered under dry Ar/H2, recorded instantly after synthesis/ conditioning. | ||
Under moisturized Ar/H2 sintering atmosphere, the material was expected to be stoichiometric U0.75Pu0.20Am0.05O2.00. The lattice parameter obtained in this case was 5.4542(5) Å. Under dry Ar/H2 sintering atmosphere, the lattice parameter obtained was equal to 5.4556(5) Å. This higher value compared to the former one is in agreement with the formation of sub-stoichiometric oxide U0.75Pu0.20Am0.05O2−x. However, the lattice parameter difference was very small between the two variations, suggesting a very limited deviation from stoichiometry, with an overall O/M composition of about 1.995 when compared to the U–Pu–O system.25
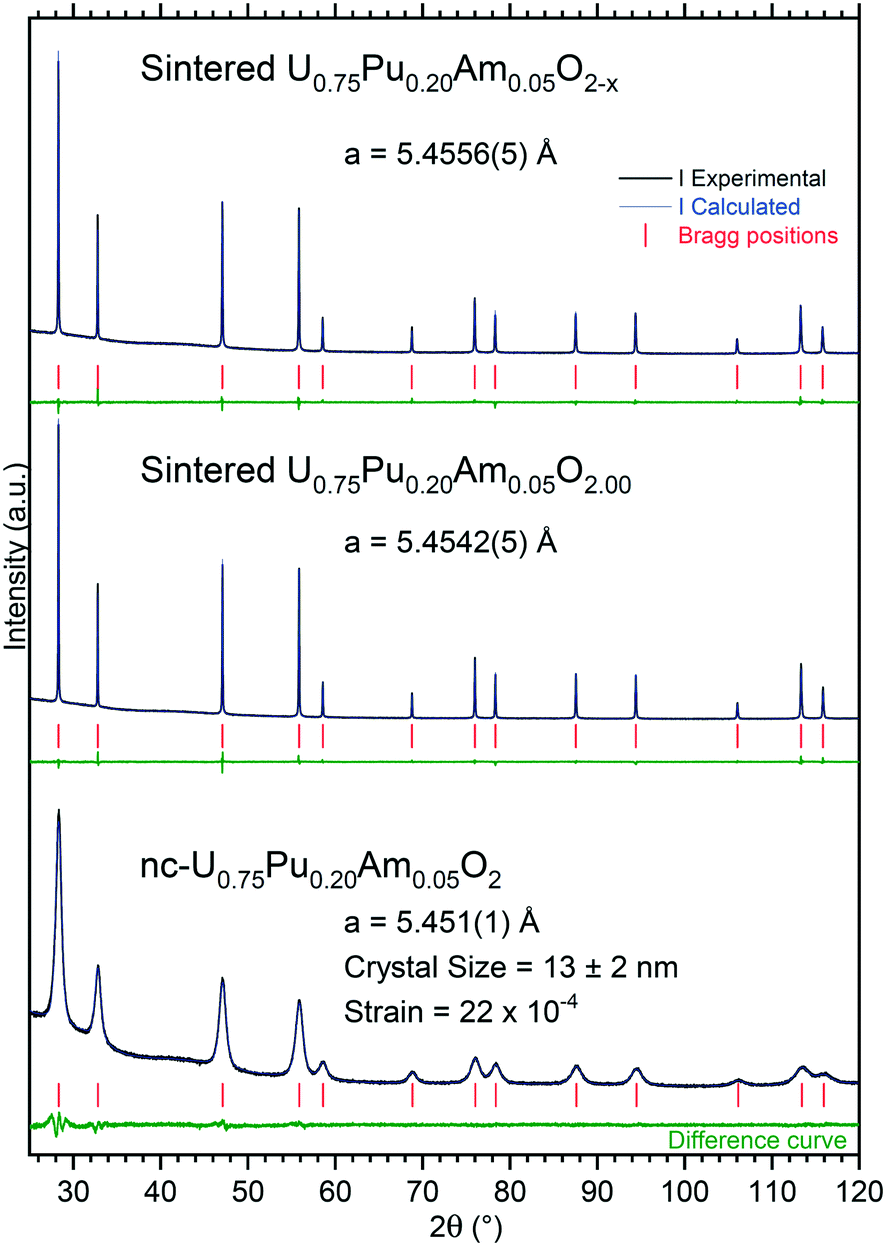
Due to the size of the nanoparticles in the original powder, the SEM cannot resolve them individually, and only the agglomerates were observed in the micrographs of Fig. 3, with their characteristic round morphology and sizes ranging from <500 nm to 2 μm. TEM analyses shed more light on the morphology and composition. The agglomerates that could be observed in the SEM micrographs were clearly distinguishable and made out of smaller nanoparticles, with sizes of 5–20 nm. Electron diffraction patterns obtained from these agglomerates showed the FCC structure of the nanoparticles, compatible with the lattice parameter calculated from XRD. EELS analyses proved the composition of the nanocrystallites to be close to the original composition, obtaining results with slight variations around 75–80% at. U, 18–20% at. Pu and about 5% at. Am.
 | ||
| Fig. 3 nc-U0.75Pu0.20Am0.05O2: detail of the (U,Pu,Am)O2 nanocrystals forming the agglomerates in a SEM image (left). TEM image showing two agglomerations of nanoparticles, with the corresponding polycrystalline electron diffraction (center). The EELS spectrum showing the white lines of the M4 and M5 edges for both uranium and plutonium and the M5 edge for americium, together with the integration areas used for the semi-quantification (right). | ||
The analysed specimen of stoichiometric U0.75Pu0.20Am0.05O2.00 pellet showed a large fraction of pores homogeneously distributed over the whole sample, in line with the geometrical density measurements which showed 72–79% TD (see Table S1†). The scanning electron microscopy revealed no further information on this sample, as the examples in the Fig. 4. TEM analyses showed micrometric grains with no defects or inhomogeneities. The semi-quantitative analyses showed the ratio between the actinides to be in good agreement with those of the fabrication. The structure seems to be preserved as FCC as proven by the electron diffractions obtained on several grains.
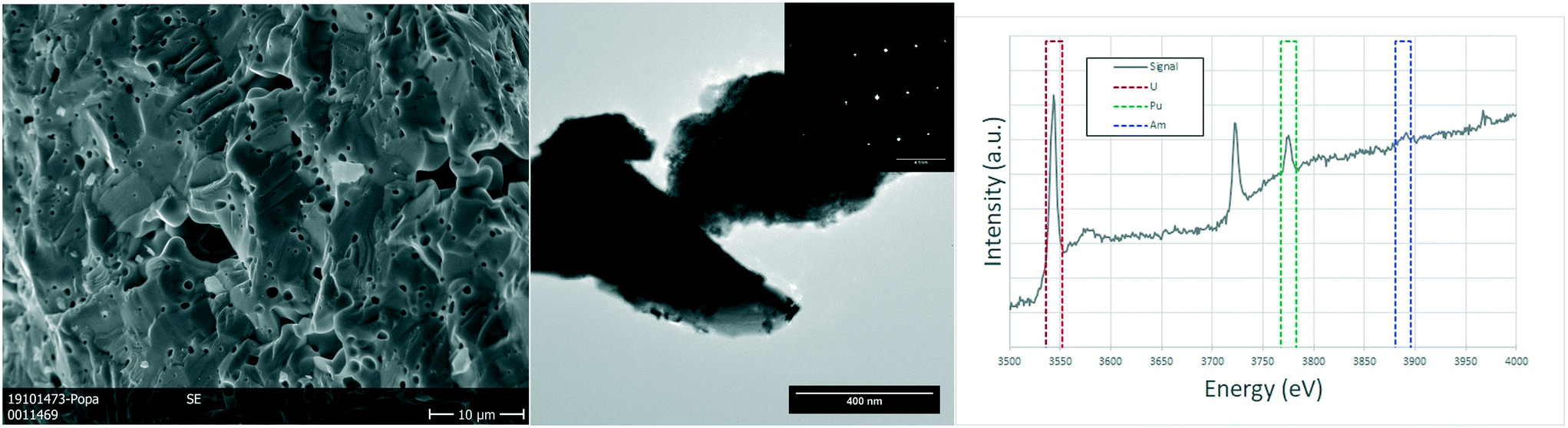 | ||
| Fig. 4 Sintered U0.75Pu0.20Am0.05O2.00: detailed SEM image on the porosity of the sample (left). TEM image showing a typical view of the sample with the electron diffraction illustrating a single crystal (center). EELS spectrum showing the edges for uranium and plutonium and the areas used to estimate the ratios between U, Am and Pu (right). | ||
In the case of the oxygen sub-stoichiometric specimen (U0.75Pu0.20Am0.05O2−x), the porosity seemed to be on average lower than that of the stoichiometric sample (in line with the geometrical density measurements, 83–87% TD). However, some regions showed an increased number and size of pores (Fig. 5), which led to the creation of a channelling effect, connecting large pores into continuous channels separating individual grains. The TEM study showed once again no defects or relevant details observed on the crystals which have an FCC structure (as seen on the electron diffraction proving the [111] orientation of the crystal) and large grains.
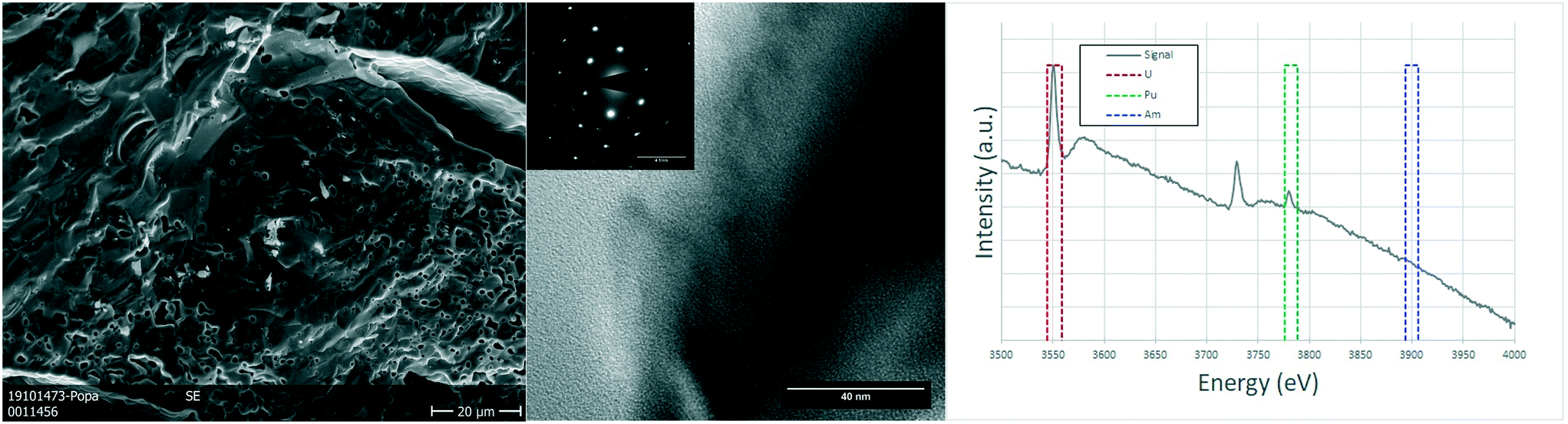 | ||
| Fig. 5 Sintered U0.75Pu0.20Am0.05O2−x: SEM micrograph showing the variations on the porosity in different regions of the sample (left). TEM image revealing no defects (center) and electron diffraction showing the monocrystalline nature of the region, coherent with a [1 1 1] orientation of a FCC structure (inset). EELS spectrum of the same region used for the semi-quantification of the actinides (right). | ||
Note that this limited direct sintering behaviour is contrasting with recent results on UO2+x powders obtained by the same method for which a much higher density was reached,26 and very likely originates from the extremely high free volume in the as-synthesized material. Thus, in further trials, a new heating stage (1 h at 400 °C under argon) was introduced in the workflow (prior to pressing the powder into discs or pellets), in order to pre-densify the powders and to vaporise the water and to release the trapped gases. The result of this stage was beneficial, as it can be observed from the data summarised in Table S1.†
 | ||
| Fig. 6 XRD patterns of the nanocrystalline and sintered (U,Am)O2 specimens recorded instantly after synthesis/conditioning. | ||
As in the previous case, the sintering process induced a sharpening of the diffraction peaks in the XRD measurements due to crystal growth. Under moisturised Ar/H2 sintering atmosphere, the materials are expected to be stoichiometric U1−xAmxO2.00. The lattice parameters obtained in this case were 5.4659(5) Å for U0.90Am0.10O2.00 and 5.4676(5) Å for U0.80Am0.20O2.00, respectively.
Lattice parameter of the (U,Am)O2 mixed oxides are difficult to interpret due to the occurrence of complex charge distribution in this system.27,28 Lattice parameters of nanocrystalline mixed oxides and sintered specimens obtained in this study are presented in Fig. 7 together with other experimental values present in the literature,27–31 and models proposed by Nishi et al.31 the expected trend for fully reduced material (U4+1−yAm3+y)O2−y/2 containing only uranium at the oxidation state IV and americium at the oxidation state III is presented by the curve at the top of the graph. This trend was experimentally observed in different mixed oxides.32–34 It corresponds to the progressive change from the tetravalent fluorite M4+O2 dioxide to trivalent bixbyite-like M3+2O3 sesquioxide end-members. However, it seems that this level of reduction is never reached for (U,Am)O2 mixed oxides, even after reductive sintering. The curve resulting from Vegard's law between the UO2.00 to AmO2.00 end-members is shown at the bottom part of the graphs. However, it was well established that the Vegard's law is irrelevant for this system due to charge transfer occurring between uranium and americium in the mixed oxide.27,28,31 In stoichiometric mixed (U,Am)O2.00 oxides, Am(III) and U(V) were reported to be present in equivalent proportions.28 Since (U4+1−2yU5+yAm3+y)O2.00 has in average larger ionic radii than the hypothetical (U4+1−yAm4+y)O2.00 with the same U/Am composition, lattice parameters of stoichiometric mixed oxides (U,Am)O2.00 show systematically larger lattice parameters than those suggested by Vegard's law. Experimental values obtained by Lebreton29 are also reported in Fig. 7 with the higher values corresponding to mixed oxides freshly reduced material (1 hour, 1100 °C, Ar/H2 (4%)), and the lower values corresponding the same product after spontaneous oxidation under storage condition (room temperature, air) occurring from few hours to few weeks after reductive treatment. This shows that at high Am content, a broad range of lattice parameters (i.e. O/M ratios) can be achieved but with a high sensitivity to oxidation, while at low Am content, mixed oxide seems close to stoichiometry from fresh reduction to long air storage. From the results presented in Fig. 7, the lattice parameters for the materials in this work suggest a composition close to stoichiometry (O/M≈2) for U0.90Am0.10O2 while U0.80Am0.20O2 seems to have a significantly higher lattice parameter than the expected trend, which suggest a significant substoichiometry (O/M < 2).
 | ||
| Fig. 7 Variation of lattice parameters in the (U,Am)O2(−δ) system: full range of composition (a) and closer view of the compositions of interest (b). | ||
We have checked the limit of the Ce(III) incorporation (as a surrogate for Am(III)) in the (U,Ce)O2 solid solutions by using the hydrothermal decomposition of mixed oxalates method over the full compositional range, i.e. to answer the question until which cerium concentration we can apply this synthesis route without the formation of undesired micro-crystalline Ce(CO3)OH as observed in pure cerium oxalate hydrothermal decomposition.11 The results showed that the nanocrystalline solid solutions FCC structure formed up to a composition of U0.5Ce0.5O2, which indicates that this method might be applied for materials with an americium content up to 50%; above this concentration, the charge compensation needs further oxidation of uranium into hexavalent form. Note that nanocrystalline solid solutions with FCC structure can form over the full compositional range in the uranium–cerium system by using an alternative precursor36 (hydrothermal treatment of mixed hydroxides).
3.4 Cationic charge distribution in sintered and nc-U0.80Am0.20O2
HR-XANES spectroscopy data at the U M4 edge for as-synthesised nc-U0.80Am0.20O2 revealed the presence of mixed U(IV) and U(V) oxidation states, with majority of the U atoms accommodating U(V) oxidation state. The energy position of feature B in the UVBiO4 spectrum37 located at about 3727.3 eV is characteristic of the main absorption intensity of U(V) (cf.Fig. 8a).16,35 Note that the time from synthesis to measurement was 6 months for the nc-U0.80Am0.20O2. As previously described for (U,Ce)O2 nanoparticles,36 the oxidation of the material occurs under storage conditions due to the high sensitivity for oxidation of these materials due to their high specific surface area. Given the size of the nanocrystals (10 ± 2 nm) in nc-U0.80Am0.20O2, and the storage condition (nitrogen atmosphere with up to 1% O2), it can be expected that this majority of U(V) in the nanocrystalline samples is not the result of the synthesis route, but the result of the oxidation of the material before the HR-XANES measurement. We reported such oxidation also previously for UxPu1−xO2 nanosized powders.16 A small amount of U(V) in the bulk UO2 reference is due to oxidation as UO2 was kept in air. U M4 edge HR-XANES data acquired 76–78 days after sintering clearly indicate reduction of uranium toward U(IV) for sintered material. The post-edge region of the spectra exhibits two broad peaks typical for the FCC cubic structure (see Fig. S4†). Prior to sintering these shape-resonances in the range from 3740 eV to 3770 eV have rather low intensity, whereas after sintering those gain intensity and are better resolved. This result is in accordance with the increased particle size and crystallinity reported by XRD. Similar trend has been reported earlier for nanosized and bulk PuO2.38 | ||
| Fig. 8 HR-XANES spectra acquired at U M4 (a) and Am M5 (b) edges for nc-U0.80Am0.20O2, nc-U0.80Am0.20O2 after sintering and UO2, BiUO4, AmVO4, AmO2 reference materials. | ||
The Am M5 edge spectra of nc-U0.80Am0.20O2 and the sintered U0.80Am0.20O2 are depicted in Fig. 8b. The energy positions of the first intense peaks and the main peak of the Am(III) reference coincide (line C in Fig. 8b). This suggests that Am(III) dominates in both samples and is in agreement with previous reports that Am(III) stabilizes along with U(V). It is evident that the shoulder of the spectrum is positioned at the energy position of the main spectral maximum of AmIVO2 (line D). This shoulder is also present for Am(III) in AmVO4 thus its existence does not necessary indicate Am(IV) in the two samples. However, after sintering the intensity of the shoulder decreases and this suggests reduction of Am(IV) to Am(III). This result is an evidence for minor amount of Am(IV) stabilized in the as prepared nc-U0.80Am0.20O2 crystals. The presence of Am(IV) in the nanomaterial nc-U0.80Am0.20O2 with 20% Am content is a new finding since americium is found to be in its trivalent oxidation state in the U1−xAmxO2±y (x = 0 to 0.5) bulk system in previous studies,27–31 even after calcination under air.39,40 We assume that Am is partially oxidised on the surface of the nanoparticles.
It must be noted that, despite this new observation suggesting a minor amount of Am(IV) in nanosized material, the overall valence composition of uranium and americium in the nanoparticles is very similar to the one reported for U1−xAmxO2±y sintered materials oxidized in air up to 1200 °C by Epifano et al.39,40 for similar U/Am compositions. However, the results of these authors suggests that, for this degree of oxidation, the cubic phase of U0.80Am0.20O2±y should have a lattice parameter of about 5.433 Å, which is very much smaller than the value of 5.466(1) Å measured in our case on nc-U0.80Am0.20O2 soon after synthesis. This result tends to confirm that the oxidation of the nanocrystals occurred under storage conditions before the HR-XANES measurement, due to the very high sensitivity of such materials against oxidation, as it was observed previously.36
3.5 Lattice expansion of (U,Pu,Am)O2 and (U,Am)O2
The behaviour of the different mixed oxides under self-irradiation was monitored by XRD (Fig. 9). The expansion of lattice parameters follows an exponential trend with the equation:| a = a0 + Δa(max) × [1 − exp(−D/B)] | (1) |
| ΔV/V0 = A × [1 − exp(−D/B)] | (2) |
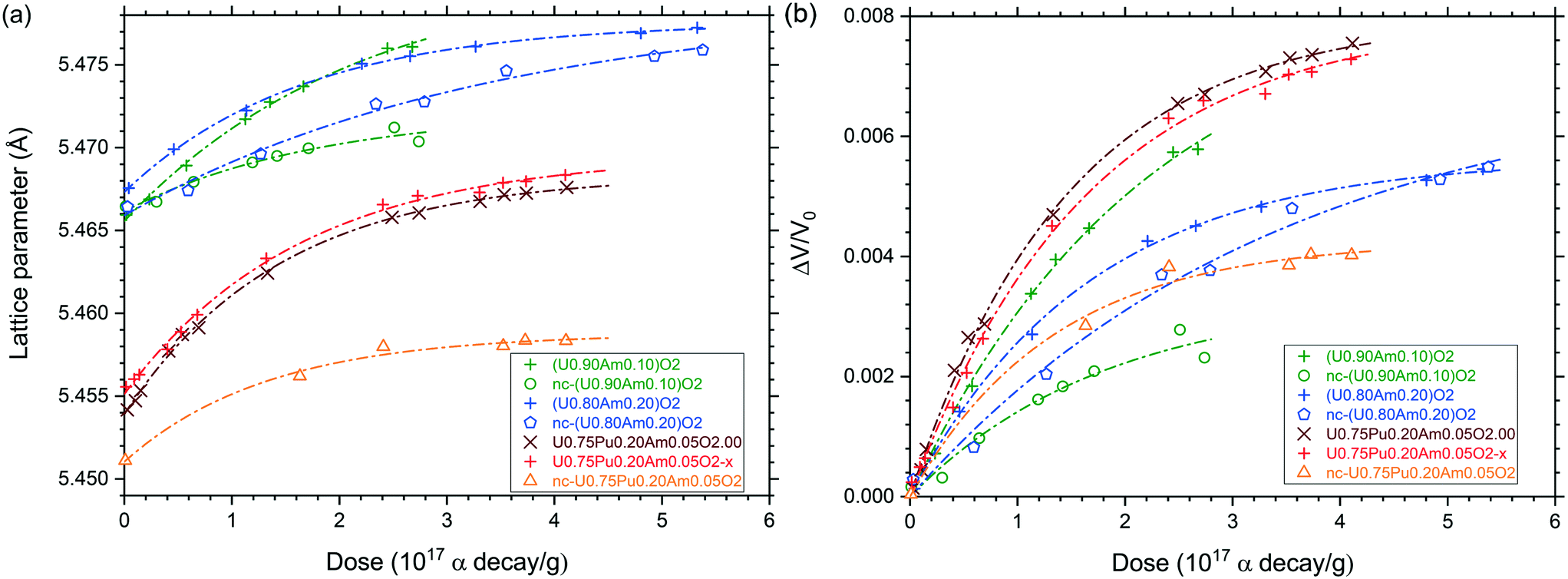 | ||
| Fig. 9 Variation of the lattice parameter (a) and relative lattice volume (b) of (U,Pu,Am)O2 (nano), sub- and stoichiometric and (U, Am)O2 10 and 20%, nano and stoichiometric as function of the alpha dose. | ||
Lattice parameter values and lattice volume expansion presented in Fig. 9 were fitted to eqn (1) and (2) in order to obtain the constants a0, Δa(max), B and A (Table 2). One can see that the specific dose of the exponential trend (B) was about 2 × 1017 α/g and the maximal volume swelling was about 0.8% at saturation, which is very similar to what was already described for pure PuO2,41 AmO2,42 (U,Pu)O2 (ref. 43) or (U,Am)O2 (ref. 30) mixed oxides. In case of all nano-crystalline materials and of sintered U0.80Am0.20O2, the volume swelling at saturation was significantly lower. For the nano-crystalline materials, this may be explained by the fact that alpha irradiation is causing disorder to phases which are already significantly disordered compared to sintered specimens, or by the shorter diffusion path of alpha particles to the grain boundaries. In case of the U0.80Am0.20O2 sintered sample, the low swelling might be explained by a significant substoichiometry (O/M < 2), as suggested by the initially large lattice parameter, in combination with a moderate swelling under alpha self-irradiation. Alternatively, the lattice variation observed could result from the superposition of damage accumulation (volume increase) and oxidation (volume decrease). However, oxidation is not the privileged explanation here, since (i) the typical exponential trend observed during the time of measurement would suggest that oxidation and alpha damage swelling occurred along similar time frame which seems unlikely, and (ii) a strong oxidation of the nc-U0.80Am0.20O2 as observed with HR-XANES would bring much more pronounced lattice shrinkage down to about 5.433 Å according to the work of Epifano et al.39,40 therefore, the epoxy resin used for XRD sample preparation has probably a protective effect against oxidation.
| a = a0 + Δa(max) × [1 – exp(−D/B)] | A, % | |||
|---|---|---|---|---|
| ΔV/V0 = A[1 – exp(−D/B)] | ||||
| a 0 (Å) | Δa(max) (Å) | B (α/g) | ||
| U0.75Pu0.20Am0.05O2.00 | 5.4539 | 0.014 | 1.46 × 1017 | 0.80 |
| U0.75Pu0.20Am0.05O2−x | 5.4551 | 0.014 | 1.64 × 1017 | 0.80 |
| nc-U0.75Pu0.20Am0.05O2 | 5.4510 | 0.008 | 1.33 × 1017 | 0.43 |
| U0.90Am0.10O2 | 5.4656 | 0.015 | 2.18 × 1017 | 0.84 |
| nc-U0.90Am0.10O2 | 5.4662 | 0.006 | 1.81 × 1017 | 0.33 |
| U0.80Am0.20O2 | 5.4673 | 0.010 | 1.65 × 1017 | 0.56 |
| nc-U0.80Am0.20O2 | 5.4659 | 0.013 | 3.45 × 1017 | 0.71 |
Note that the effect of alpha self-irradiation on particle size and strain of the nanocrystalline materials has also been followed. However, none of the variation observed seems significant (Table S3 and Fig. S5†).
4. Conclusions
We present here the first synthesis of homogeneous, nanocrystalline (U,Am)O2 and (U,Pu,Am)O2 solid solutions. The results confirm the feasibility of the oxalate decomposition under hot compressed water method for production of Am-containing oxides. Experiments performed using cerium surrogate indicates that nanocrystalline solid solutions with FCC structure is forming up to x = 0.5 in U1−xCexO2, suggesting that this method might be extended up to an americium content of 50%. This demonstrates that this practice can be applied for synthesis of pure tetravalent actinide oxides (as in the cases of Th, U, Np and Pu) and mixed oxides (as demonstrated for U–Th, U–Pu, U–Am and U–Pu–Am oxide systems). Conventional sintering was tested and gives promising results, especially by introducing of pre-densification heating stage in the workflow. Both uranium and americium appear in mixed oxidation states in the as-prepared and sintered U0.80Am0.20O2 nanocrystals but the fluorite FCC structure is preserved. Uranium is mostly oxidised to its pentavalent form and Am(III) is mainly present in the U0.80Am0.20O2 nanocrystals, but large amount of U(V) is reducing to U(IV) during sintering. We show that part of Am is in tetravalent form in the as-prepared U0.80Am0.20O2 nanocrystals.Author contributions
J.-F. V. conceptualisation; formal analysis (XRD); investigation (XRD); methodology; validation; writing – original draft; writing – review & editing. D. F. project administration; resources; writing – review & editing. O. W. methodology; writing – original draft. O. D. B. investigation (SEM, TEM); writing – original draft. D. B. investigation (XRD); writing – original draft. E. Z. supervision (chemical analysis); writing – original draft. N. P. formal analysis (XANES); investigation (XANES); methodology (XANES); validation (XANES); writing – original draft; writing – review & editing. T. V. formal analysis (XANES); funding acquisition (XANES); project administration (XANES); supervision (XANES); validation; writing – original draft; writing – review & editing. R. K. project administration; resources; supervision; writing – review & editing. K. P. conceptualisation; investigation (XRD); methodology; supervision; validation; writing – original draft; writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Analytical Service of the JRC-Karlsruhe for the chemical and isotopic characterization of the samples. The technical support of Co Boshoven, Sebastien Gardeur, Antony Guiot, Patrick Lajarge, Tim Prüßmann, Octavian Vălu is highly appreciated. N. P. and T. V. gratefully acknowledge funding from the European Research Council (ERC) Consolidator Grant 2020 under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 101003292). We thank the Institute for Beam Physics and Technology (IBPT) for the operation of the storage ring, the Karlsruhe Research Accelerator (KARA).References
- C. Behar, “Technology roadmap update for generation IV nuclear energy systems”, in OECD Nuclear Energy Agency for the Generation IV International Forum, vol. 17, 2014 Search PubMed.
- J. Somers, Energy Procedia, 2011, 7, 169–176 CrossRef CAS.
- E. D'Agata, P. R. Hania, D. Freis, J. Somers, S. Bejaoui, F. F. Charpin, P. J. Baas, R. A. F. Okel, S. van Til, M. Lapetite and F. Delage, Nucl. Eng. Des., 2017, 311, 131–141 CrossRef.
- D. Greneche, “RED-IMPACT: impact of partitioning, transmutation and waste reduction technologies on the final nuclear waste disposal; synthesis report”, Forschungszentrum Jülich, vol. 15, 2008 Search PubMed.
- Council Directive 2011/70/Euratom of 19 July 2011 establishing a Community framework for the responsible and safe management of spent fuel and radioactive waste.
- F. Delage, J. Carmack, C. B. Lee, T. Mizuno, M. Pelletier and J. Somers, J. Nucl. Mater., 2013, 441, 515–519 CrossRef CAS.
- C. Tamain, B. Arab-Chapelet, M. Rivenet, X. F. Legoff, G. Loubert, S. Grandjean and F. Abraham, Inorg. Chem., 2016, 55, 51–61 CrossRef CAS PubMed.
- C. Tamain, B. Arab-Chapelet, M. Rivenet, S. Grandjean and F. Abraham, J. Solid State Chem., 2016, 236, 246–256 CrossRef CAS.
- V. Tyrpekl, J.-F. Vigier, D. Manara, T. Wiss, O. Dieste Blanco and J. Somers, J. Nucl. Mater., 2015, 460, 200–208 CrossRef CAS.
- O. Walter, K. Popa and O. Dieste Blanco, Open Chem., 2016, 14, 170–174 CAS.
- K. Popa, O. Walter, O. Dieste Blanco, A. Guiot, D. Bouëxière, J.-Y. Colle, L. Martel, M. Naji and D. Manara, CrystEngComm, 2018, 20, 4614–4622 RSC.
- D. Bouëxière, K. Popa, O. Walter and M. Cologna, RSC Adv., 2019, 9, 6542–6547 RSC.
- E. De Bona, O. Walter, H. Störmer, T. Wiss, G. Baldinozzi, M. Cologna and K. Popa, J. Am. Ceram. Soc., 2019, 102, 3814–3818 CrossRef CAS.
- E. De Bona, L. Balice, L. Cognini, M. Holzhäuser, K. Popa, O. Walter, M. Cologna, D. Prieur, T. Wiss and G. Baldinozzi, J. Eur. Ceram. Soc., 2021, 41, 3655–3663 CrossRef CAS.
- L. Balice, D. Bouëxière, M. Cologna, A. Cambriani, J.-F. Vigier, E. De Bona, G. D. Sorarù, C. Kübel, O. Walter and K. Popa, J. Nucl. Mater., 2018, 498, 307–313 CrossRef CAS.
- G. Kauric, O. Walter, A. Beck, B. Schacherl, O. Dieste Blanco, J. F. Vigier, E. Zuleger, T. Vitova and K. Popa, Mater. Today Adv., 2020, 8, 100105 CrossRef.
- M. Romanowski, S. Franzini and L. Koch, Proceedings of the 8th Annual ESARDA Symposium, London, 1987, pp. 111–113 Search PubMed.
- A. Morgenstern, C. Apostolidis, R. Carlos-Marquez, K. Mayer and R. Molinet, Radiochim. Acta, 2002, 90, 81–85 CrossRef CAS.
- R. Buda, R. Carlos-Marquez, E. Dahms, F. d'Amati, L. Emblico, J. Horta-Domenech, A. LeTerrier, K. Lützenkirchen, S. Millet, S. Morel, A. Mühleisen, A. Nichol, V. Piron, D. Poublan, M. Ramos-Pascual, A. M. Sanchez-Hernandez, F. Sarli, P. Schwalbach, A. Terrasi, P. van Belle, M. Vargas-Zuniga, J. Zsigrai and E. Zuleger, Comparative assessment of the Pu content of MOX samples by different techniques, ESARDA Bulletin, 2013, 49, 1–7 Search PubMed.
- H. Ottmar, S. Abousahl, A. Morgenstern and M. C. Vincent, Proceedings of the 25th Annual ESARDA Symposium, Stockholm, 2003 Search PubMed.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr., 2014, 229, 345–352 Search PubMed.
- A. Zimina, K. Dardenne, M. A. Denecke, D. E. Doronkin, E. Huttel, H. Lichenberg, S. Mangold, T. Pruessmann, J. Rothe, T. Spangenberg, S. Steininger, T. Vitova, H. Geckeis and J.-D. Grunwaldt, Rev. Sci. Instrum., 2017, 88, 113113 CrossRef CAS PubMed.
- T. Vitova, I. Pidchenko, D. Fellhauer, P. S. Bagus, Y. Joly, T. Pruessmann, S. Bahl, E. Gonzalez-Robles, J. Rothe, M. Altmaier, M. A. Denecke and H. Geckeis, Nat. Commun., 2017, 8, 16053 CrossRef CAS PubMed.
- G. K. Williamson and W. H. Hall, Acta Metall., 1953, 1, 22–31 CrossRef CAS.
- C. Duriez, J.-P. Alessandri, T. Gervais and Y. Philipponneau, J. Nucl. Mater., 2000, 277, 143–158 CrossRef CAS.
- J. Manaud, R. Podor, X. F. Le Goff, J. Maynadié, N. Dacheux and N. Clavier, J. Eur. Ceram. Soc., 2021, 41, 6697–6707 CrossRef CAS.
- E. Epifano, M. Naji, D. Manara, A. C. Scheinost, C. Hennig, J. Lechelle, R. J. M. Konings, C. Guéneau, D. Prieur, T. Vitova, K. Dardenne, J. Rothe and P. M. Martin, Commun. Chem., 2019, 2, 59 CrossRef.
- D. Prieur, P. M. Martin, A. Jankowiak, E. Gavilan, A. C. Scheinost, N. Herlet, P. Dehaudt and P. Blanchart, Inorg. Chem., 2011, 50, 12437–12445 CrossRef CAS PubMed.
- F. Lebreton, Synthesis and characterisation of uranium-americium mixed oxides, PhD Thesis, University of Limoges, 2014 Search PubMed.
- D. Horlait, F. Lebreton, P. Roussel and T. Delahaye, Inorg. Chem., 2013, 52, 14196–14204 CrossRef CAS PubMed.
- T. Nishi, M. Nakada and M. Hirata, J. Solid State Chem., 2017, 256, 252–255 CrossRef CAS.
- A. Nakamura, Solid State Ionics, 2010, 181, 1543–1564 CrossRef CAS.
- D. Horlait, L. Claparede, N. Clavier, S. Szenknect, N. Dacheux, J. Ravaux and R. Podor, Inorg. Chem., 2011, 50, 7150–7161 CrossRef CAS PubMed.
- D. Prieur, J. F. Vigier, J. Rothe and J. Somers, J. Nucl. Mater., 2014, 448, 4–7 CrossRef CAS.
- I. Pidchenko, K. O. Kvashnina, T. Yokosawa, N. Finck, S. Bahl, D. Schild, R. Polly, E. Bohnert, A. Rossberg, J. Gottlicher, K. Dardenne, J. Rothe, T. Schafer, H. Geckeis and T. Vitova, Environ. Sci. Technol., 2017, 51, 2217–2225 CrossRef CAS PubMed.
- D. Prieur, J.-F. Vigier, K. Popa, O. Walter, O. Dieste, Z. Varga, A. Beck, T. Vitova, A. C. Scheinost and P. M. Martin, Inorg. Chem., 2021, 60, 14550–14556 CrossRef CAS PubMed.
- K. Popa, D. Prieur, D. Manara, M. Naji, J.-F. Vigier, P. M. Martin, O. Dieste Blanco, A. C. Scheinost, T. Prüβmann, T. Vitova, P. E. Raison, J. Somers and R. J. M. Konings, Dalton Trans., 2016, 45, 7847–7855 RSC.
- T. Vitova, I. Pidchenko, D. Fellhauer, T. Pruessmann, S. Bahl, K. Dardenne, T. Yokosawa, B. Schimmelpfennig, M. Altmaier, M. Denecke, J. Rothe and H. Geckeis, Chem. Commun., 2018, 54, 12824–12827 RSC.
- E. Epifano, R. Vauchy, F. Lebreton, R. Lauwerier, A. Joly, A. Scheinost, C. Guéneau, C. Valot and P. M. Martin, J. Nucl. Mater., 2020, 531, 151986 CrossRef CAS.
- E. Epifano, R. Vauchy, F. Lebreton, A. Joly, C. Guéneau, C. Valot and P. M. Martin, J. Nucl. Mater., 2020, 531, 151991 CrossRef CAS.
- M. Noe and J. Fuger, Inorg. Nucl. Chem. Lett., 1974, 10, 7–19 CrossRef CAS.
- T. D. Chikalla and L. Eyring, J. Inorg. Nucl. Chem., 1968, 30, 133–145 CrossRef CAS.
- M. Kato, A. Komeno, H. Uno, H. Sugata, N. Nakae, K. Konashi and M. Kashimura, J. Nucl. Mater., 2009, 393, 134–140 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ce00527a |
| This journal is © The Royal Society of Chemistry 2022 |