
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of a covalent binder to the oncoprotein gankyrin using a NIR-Based OBOC screening method†
Marianne E. Maresh,
Breanna L. Zerfas,
Brice S. Wuthrich and
Darci J. Trader *
*
Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: dtrader@purdue.edu
First published on 1st April 2021
Abstract
Despite huge advancements in the process of synthesizing small molecules as part of one-bead-one-compound (OBOC) libraries, progress lags in the ability to screen these libraries against proteins of interest. Recently, we developed a method to screen OBOC libraries in which a target protein is labeled with a near-infrared (NIR) range fluorophore. The labeled protein incubates with beads of a library in a 96-well plate, then the plate is imaged for fluorescence. Fluorescence intensities produced by the labeled protein binding the bead can be quantitated and provide a basis to rank hits. Here, we present an application of this technique by screening the oncoprotein gankyrin against a 343-member peptoid library. The library was composed of four positions occupied by one of seven amines. In the third position, an amine that facilitates covalent binding via a sulfonyl fluoride moiety was incorporated. After screening for gankyrin binders twice, ten structures showed overlap in the types of amines present at each position. These initial hits were validated with an in-gel fluorescence assay in which the labeled ligands covalently interacted with purified gankyrin. Excitingly, one peptoid was validated from this analysis. This hit was also shown to bind gankyrin in the presence of HEK 293T lysate. Results from this study demonstrate successful use of our screening method to quickly identify quality binders to a target protein of interest.
Introduction
Development of one-bead-one-compound (OBOC) libraries has expedited the process of synthesizing thousands of small molecules.1 Although significant progress has been made in expanding the types of small molecules that can be synthesized on-bead, progress lags in the development of simple techniques to efficiently screen these libraries against a biological target of interest. A common method to screen OBOC libraries uses magnetic beads and antibodies to link the resin bead to the magnetic bead.2,3 A strong magnet is used to physically separate hit beads. Although this method has been successfully used to screen large OBOC libraries against protein targets, it has several drawbacks.2 For example, it requires several binding events, increasing the risk of identifying false positives, as nonspecific binding between the library beads and antibodies or magnetic beads is possible. Similarly, the primary hits must bind the protein of interest significantly as a strong interaction between the protein and resin is required to withstand the multiple wash and binding steps. Finally, hits cannot be readily prioritized from this method, as there is no indication of the strength of the binding between the target and the ligand until the re-synthesis of the ligand from the resin bead followed by additional experiments to quantitate the binding interaction. This slows the validation process, as all ligands from the screen must be further tested to evaluate their binding to a target protein. While the development and screening of DNA encoded libraries has greatly advanced the abilities to screen large ligand libraries and with some methods can prioritize hits, significant expertise in the decoding of the hits and the data analysis is required.4–6In an effort to overcome the limitations of screening with magnetic beads and to develop a ligand identification method broadly applicable to non-screening experts, attention has been turned to the incorporation of fluorophores in OBOC screening methods. Recently, methods utilizing quantum dots conjugated to streptavidin have undergone development.7 The target protein labeled with biotin is allowed to interact with a library bead, then streptavidin-conjugated quantum dots bind the target protein. Fluorescence produced by the quantum dot can be visualized under a microscope to evaluate binding of the protein to a ligand on bead. Other methods that utilize automated confocal nanoscanning and other fluorescent-based imaging techniques have been developed in an effort to make OBOC screening techniques more quantitative.8–11 Similarly, methods to print chemical libraries on glass slides and allowing fluorescently labeled protein to bind have also been developed, but are limited in the types of small molecules that can be screened.12 Although these methods overcome some of the limitations of magnetic beads, the emission of fluorophores utilized by these techniques often overlap with autofluorescence produced by the biological target or the intrinsic fluorescence produced by the bead. To overcome this, other groups have investigated the use of fluorophores that emit at long wavelengths to reduce autofluorescence produced by the protein and bead.13 In an effort to build upon currently available screening methods, we recently reported an OBOC screening technique that utilizes proteins conjugated to near-infrared range (NIR) fluorophores.14 Briefly, beads of an OBOC library are individually separated into the wells of a 96-well plate and blocked with protein blocking buffer. Purified target protein is then labeled with a NIR fluorophore and incubated with library beads. Beads are then rinsed, and the plate is imaged for NIR fluorescence. The fluorescence intensities resulting from the labeled protein binding the small molecule on-bead can be quantified and provide a basis to rank hits (Fig. 1). We validated our screening conditions by comparing ligands of carbonic anhydrase with varying binding affinities. The amount of fluorescence observed on the bead with our method correlated with the known KD values of the ligands in solution.14,15
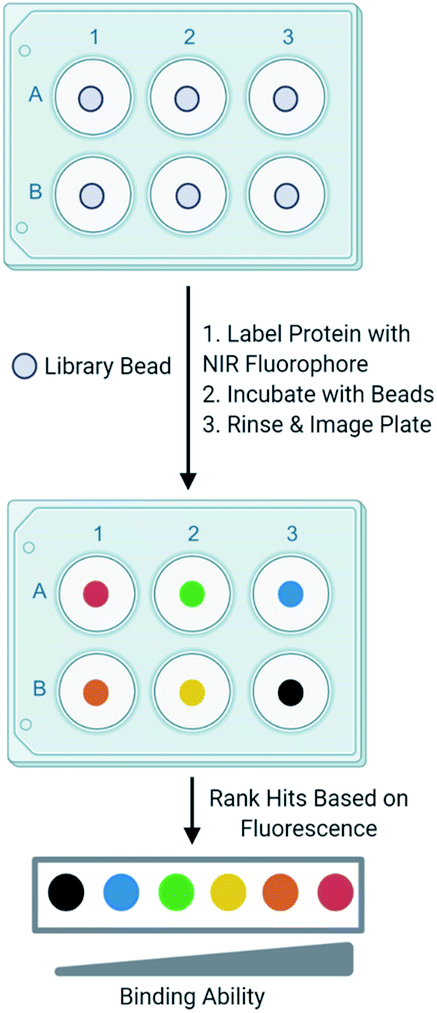 | ||
| Fig. 1 Screening scheme. Our screening method involves individually separating OBOC library beads into the wells of a 96-well plate. The target protein is then labeled with a NIR fluorophore and incubates with library beads. The plate is then imaged for fluorescence. Fluorescence intensities produced by the protein binding the bead are quantitated and provide a basis to rank hits. | ||
Here, we report the successful application of this method to discover a new ligand to the protein gankyrin by screening a 343-member OBOC covalent peptoid library. Gankyrin is known to be overexpressed in several cancers including types of breast, liver, and colorectal.16–18 When gankyrin is overexpressed, it interacts with the E3 ligase MDM2 and promotes ubiquitination and subsequent degradation of p53, a key cell cycle regulating protein.19 Overexpression of gankyrin also promotes degradation of retinoblastoma protein, leading to cell cycle dysregulation and uncontrolled proliferation.20 A small molecule inhibitor of gankyrin was recently described and was shown to stabilize p53.21
In an effort to expand upon the small molecule probes available to study the role of gankyrin, we screened the protein against a peptoid library that contained a sulfonyl fluoride group using our OBOC screening technique. The screen against gankyrin produced ten initial hits. In-gel fluorescence validation experiments produced one validated hit. Excitingly, this hit bound gankyrin in HEK 293T cell lysate, indicating it could be further developed into a probe to monitor gankyrin expression in cells. These results indicate our screening method could be used to identify small molecule binders to many different target proteins. Since the activity of the protein does not need to be known, the pool of proteins that can be screened is vast. Additionally, prioritization of hits allows for expedited validation, as only small molecules that provide high fluorescence intensities can be readily selected, decreasing the resources required to confirm primary hits.
Experimental details
Library synthesis
90 μm TentaGel resin was swollen in dimethylformamide (DMF) followed by dichloromethane (DCM) and a linker structure was synthesized on the resin prior to creating the library to establish physical space between the bead and the small molecule and to assist in identification of the peptoids (see ESI† for the detailed protocol) (Fig. 2A).2 Standard solid phase peptide and peptoid chemistry was used to synthesize both the linker and the OBOC library.22,23 The split and pool method was utilized to synthesize the library in which, after activation with a 2 M bromoacetic acid (BAA) and 1 M N,N′-diisopropylcarbodiimide (DIC) solution, resin was roughly equally divided into seven fritted syringes. 1 M stocks of position one amines were prepared in dry DMF and added to one of the syringes (Fig. 2B). Amines coupled for one hour at 37 °C. Resin was then washed with dry DMF and combined back into one syringe and activated for the next amine. This process was repeated for all listed amines. For position three, resin was not split into separate syringes, as only one amine was used. The amines in position four were coupled as described above. When synthesis of the library was complete, the Pbf group on the arginine of the linker was removed by agitating the resin in a 95% trifluoroacetic acid (TFA), 2.5% triisoproylsilane (TIPS), 2.5% DCM solution for one hour. The resin was then washed three times with DMF followed by three washes with DCM. The resin was stored at 4 °C until use (see ESI† for full procedure).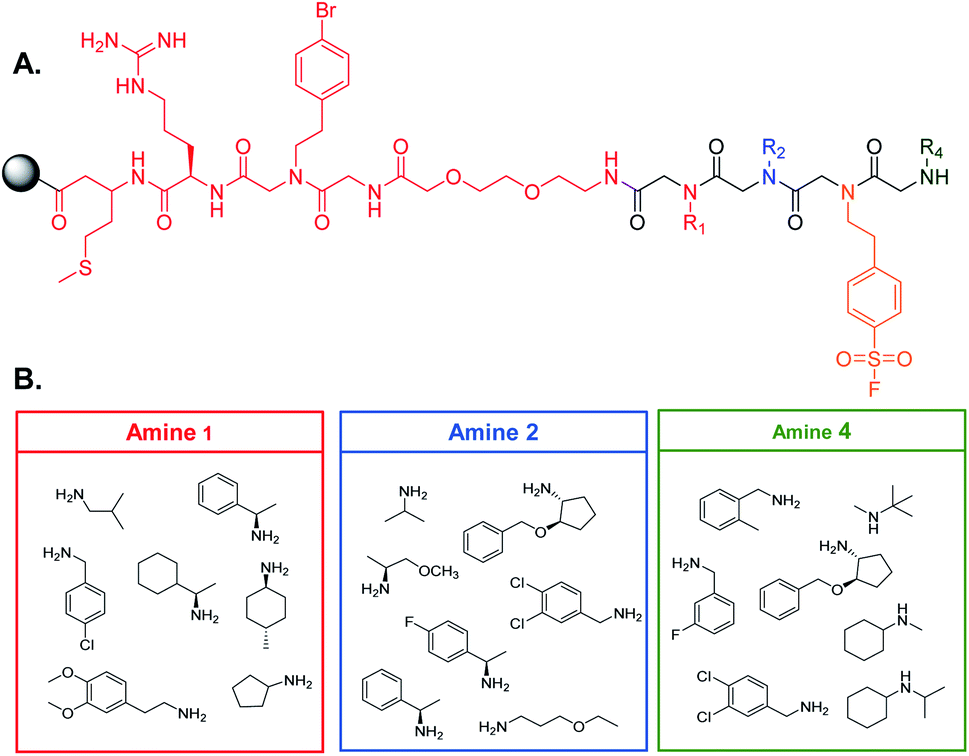 | ||
| Fig. 2 OBOC library design. (A) Structure of the linker (red) and peptoid scaffold. The arginine residue on the linker provides the necessary charge to ionize peptoids during MALDI-TOF analysis. The bromophenethylamine provides a unique isotope pattern for identification of peptoid fragments by MALDI-TOF. (B) Amines selected for each position of the peptoid library. Only one amine, 4-(2-aminoethyl)benzensulfonyl fluoride, was coupled at position three to facilitate covalent binding of the peptoid to the target. | ||
Gankyirn purification and labeling with a NIR fluorophore
The pQTEV-PSMD10 was a gift from Konrad Buessow (Addgene plasmid # 31332; http://n2t.net/addgene:31332; RRID:Addgene_31332).24 This plasmid for bacterial expression of His6-labeled gankyrin was transformed into a Rosetta competent bacteria cell line for subsequent expression and purification. The plasmid was acquired from Addgene. A starter culture of bacteria was grown overnight, then added to 1 L of sterilized Luria-Bertani (LB) broth with ampicillin and chloramphenicol. Bacteria grew until an OD600 of 0.6–0.8 was reached and protein expression was induced with the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG). Bacteria were lysed and His-tagged gankyrin was bound to HisPur Ni-NTA resin then eluted with an imidazole gradient. Fractions of the gradient were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and the gel was subsequently stained with Coomassie. Fractions containing pure gankyrin were collected and dialyzed overnight in phosphate-buffered saline (PBS) at 4 °C. Protein was concentrated to 1 mg mL−1 and stored at 4 °C for up to three days before use.1 mg of gankyrin was labeled with the NIR fluorophore by using an IRDye 800CW Protein Labeling Kit purchased from LI-COR. Protein in PBS buffer was brought to a pH of 8.5 by adding 1 M potassium phosphate, pH 9.0. A vial of NIR fluorophore was then thawed and dissolved in ultrapure water. The correct amount of dye was calculated through the manufacturer's protocol and added to the protein. This incubated for 2 hours at 4 °C and excess dye was removed by passing the labeled protein through a desalting column. Absorbance of the labeled protein at 280 and 780 nm was determined to calculate the protein![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fluorophore ratio. The protein was then aliquoted and stored at −20 °C and thawed on ice before use.
:fluorophore ratio. The protein was then aliquoted and stored at −20 °C and thawed on ice before use.
General screening scheme
Beads of the OBOC library were swollen in PBS (see ESI† for full procedure) then individually separated into the wells of a black 96-well plate with a clear bottom. Beads were blocked with protein blocking buffer for 30 minutes at room temperature. Labeled gankyrin was thawed and diluted in protein blocking buffer and 0.6 ng was added to each well containing a library bead. The final volume in each well was 50 μL. The protein incubated with the beads overnight at 4 °C with gentle agitation, protected from light. The following day, the protein solution was removed, and the beads were washed with PBS to remove excess protein. The beads were then resuspended in PBS and the plate was imaged for NIR fluorescence with a LI-COR Odyssey CLx imaging system. Fluorescence intensity from each bead was quantified using ImageStudio software from LI-COR.Two screens of 424 beads of the 343 compound OBOC library were conducted against gankyrin. The top 5% of fluorescence intensities from each screen were considered hits. These beads were separated into the wells of chemical resistant 96-well plate. Bound protein was removed by adding increasingly organic solvents to the beads and allowing them to agitate at room temperature (see ESI† for full procedure). The peptoids were cleaved from the beads by agitating with a cyanogen bromide cocktail overnight. The plate was then dried and the cleaved product was resuspended in a water/acetonitrile solution and spotted with matrix on a plate for MALDI-TOF analysis. Structures of the hits from both screens were identified and compiled. Overlapping structures in which similar amines were present at similar positions between the two screens were selected for further study. This analysis lead to ten primary hits that required validation.
Peptoid synthesis for validation experiments
An in-gel fluorescence assay was used for validation in which fluorescently labeled ligands incubated with purified gankyrin. A linker containing a monomethyoxytrityl (MMT)-protected cysteine residue for later attachment of fluorescein via a maleimide group was synthesized on high loading rink amide resin (see ESI† for full procedure). Hit structures were synthesized on this linker using the procedure described in the library synthesis section. Briefly, resin was activated by adding a premixed solution of 2 M BAA and 1 M DIC and allowing this to agitate for 15 minutes at 37 °C. The first amine of the structure was then diluted in dry DMF to achieve a 1 M concentration and this was added to the resin after draining the activation solution. The amine was coupled to the resin for 1 hour at 37 °C. The resin was then washed with dry DMF and the cycle of adding the activating solution followed by the amine was repeated with all remaining amines. After synthesis was complete, the MMT was removed from the cysteine to couple the fluorescein by agitating resin in a 2% TFA solution in DCM. The pH of the resin was then neutralized by washing with a 10% N,N-diisopropylethylamine (DIPEA) solution in DCM. Next, fluorescein maleimide was dissolved in DMF and added to the resin. This coupled for 2 hours at room temperature, protected from light. The resin was then rinsed with DMF followed by DCM. Resin was stored at 4 °C, protected from light until purification.The fluorescently labeled peptoid was cleaved from the resin by adding a 95% TFA cocktail (see ESI† for full procedure). The solution was collected and TFA was evaporated by blowing argon on the product. The product was then resuspended in 50/50 water/acetonitrile with 0.1% TFA and purified by reverse phase high performance liquid chromatography (HPLC). Fractions were run on an Agilent single quadrupole liquid chromatography mass spectrometry (LC/MS) system to confirm purity and the peptoid's identity. Fractions from the HPLC containing pure product were then combine and lyophilized to obtain powder. The product was stored at −20 °C protected from light until use.
In-gel fluorescence validation assay
A small amount of purified fluorescently labeled negative control or hit ligand were scrapped from the vial and dissolved in dimethyl sulfoxide (DMSO). A fraction of this was diluted 1:100 in PBS and the concentration was determined by measuring the absorbance of the solution at 494 nm and using Beer's Law with the molar extinction coefficient of fluorescein. Ligands were then further diluted in DMSO to achieve stocks such that the final concentrations of the negative control or test ligand would be 20 μM, 8 μM, 1 μM, or 0.1 μM in the sample. Gankyrin was purified as described above and diluted in PBS to achieve the desired concentration. 1 μL of each ligand at each concentration was added to 49 μL of diluted gankyrin. The final amount of gankyrin in each sample was 500 ng. Samples incubated for 3 hours at 4 °C with gentle rotation, protected from light. The binding between gankyrin and the ligands was stopped by adding 20 μL of a 4× Laemmli buffer and heating at 95 °C for 5 minutes. 50 μL of the samples were then subjected SDS-PAGE. Gels were washed three times with ultrapure water then imaged for fluorescence with an Azure Sapphire Biomolecular Imager system. Fluorescence intensities that resulted from the labeled ligand or negative control binding gankyrin were quantified using ImageStudio software from LI-COR.
Pulldown of gankyrin in spiked HEK 293T cell lysate
Human Embryonic Kidney (HEK) 293T cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) at 37 °C with 5% CO2. Cells were pelleted at 1000 × g and washed once with PBS. Cells were lysed with Mammalian Protein Extraction Reagent (MPER) supplemented with HALT protease inhibitor (see ESI† for full protocol) and centrifuged at 4 °C to clarify the lysate. Protein concentration of the lysate was determined by measuring absorbance at 280 nm with a NanoDrop One system. Lysate was diluted in PBS and purified gankyrin was added to a final concentration of 1% of the total protein. The final amount of gankyrin in the sample was 500 ng with a total protein amount of 50000 ng per sample. A small amount of fluorescently labeled negative control or ligand was scrapped from the vial and dissolved in DMSO. The concentration of the ligands was determined as described above and the ligands were further diluted in DMSO such that the final concentration would be 8 μM in the sample. 1 μL of the fluorescently labeled negative control or test ligand was added to the gankyrin-spiked lysate. The final volume of each sample was 50 μL. Samples were incubated at 4 °C for 3 hours with gentle rotation, protected from light. 20 μL of 4× Laemmli buffer was then added to each sample and samples were heated at 95 °C for 5 minutes. Samples were subjected to SDS-PAGE and imaged for fluorescence with an Azure Sapphire Biomolecular Imager. Bands with the highest fluorescence intensities were excised with a clean razor blade and the gel was rescanned to ensure the bands were completely removed. These samples were sent to the Purdue Proteomics Facility for analysis to determine the identity of the protein bound to the ligand.
Results
Screening gankyrin against an OBOC covalent library
With our screening method previously optimized,14 we turned our attention to screening a protein of interest against an OBOC library. The oncoprotein gankyrin was selected as our target protein. When gankyrin is overexpressed, it forms abnormal protein–protein interactions which leads to degradation of cell cycle regulating proteins.25 It was also chosen as a target because it has several serine and tyrosine residues throughout its sequence that could covalently bind a peptoid library that contained the sulfonyl fluoride moiety. A 343-member OBOC peptoid library was synthesized as describe above using standard solid phase peptoid synthesis.22 A linker structure was first synthesized on the resin to aid in identification of the peptoids by MALDI-TOF (Fig. 2A).2 The library was composed of three positions occupied by one of seven amines. Amines were selected at random for positions one, two, and four (Fig. 2B). The third position contained a sulfonyl fluoride moiety, which can bind covalently to amino acid residues such as threonine and serine.26Gankyrin was purified and labeled with a NIR-emitting fluorophore (ESI Fig. S1†). Beads of the OBOC library were individually separated into the wells of black 96-well plate with a clear bottom followed by 0.6 ng of labeled protein diluted in blocking buffer was added to each well. While our protein of interest was gankyrin, the labeling of a protein with a NIR-dye should be broadly applicable to other proteins of interest. Beads were washed to remove excess protein and plates were imaged for NIR-fluorescence with a LI-COR Odyssey CLx imaging system (Fig. 3). Importantly, the resolution of the instrument was set to 81 μm to facilitate better visualization of individual beads in each well, as the beads are an average of 90 μm in diameter. Clear differences in the fluorescence intensities of the beads were observed, indicating that gankyrin bound the small molecules attached to the resin beads to different extents (Fig. 3). Some wells contained beads that resulted in very low fluorescence intensities after interacting with labeled gankyrin, indicating weak binding between the protein and small molecule. However, some beads resulted in very high fluorescence intensities, indicating a significant amount of gankyrin bound the small molecule on-bead. The best potential ligands will be able to bind multiple gankyrin proteins, leading to an increase in fluorescent signal. The fluorescence intensities that resulted from the labeled protein binding the small molecule on-bead were quantified and ranked. Two screens of 424 beads of the library against gankyrin were performed. Fluorescent signal from the beads were ranked, and the top 5% were moved forward to identify their structures.
 | ||
| Fig. 3 Screening an OBOC Library Against NIR-Labeled Gankyrin. 424 beads of the OBOC were separated in the wells of a 96-well plate and blocked with protein blocking buffer. Purified gankyrin was conjugated to the NIR fluorophore, diluted in blocking buffer and added to each well. The beads were then rinsed with PBS and imaged as shown above. Yellow circles represent the outline of individual wells of a 96-well plate. Zoomed in images of beads with small molecules representing a weak, good, and excellent binder are shown. Fluorescence intensities that resulted from labeled protein binding the bead were quantified and ranked. The screen was performed twice. | ||
Hit beads were removed from their corresponding wells in the screening plates and transferred to chemical resistant plate to cleave the peptoid from the resin bead. Structures were identified using a MALDI TOF/TOF as described above (ESI Appendix S1†). Structures that were successfully identified from both screens were compiled and analyzed. We noticed several overlapping structures between the two screens in which similar amines were present at the same position. This indicated that these amines were likely important to binding gankyrin. A total of ten overlapping structures between the two screens were identified and pursued for further validation using an in-gel fluorescence assay. By limiting the primary hit pool to only those beads that produced a significant fluorescent signal in the replicate screenings and analyzing which amine monomers were present the most in the hits, we only needed to re-synthesize and validate ten molecules.
Validation of hits with an in-gel fluorescence assay
For validation, we selected an in-gel fluorescence assay in which fluorescently labeled ligands were incubated with purified gankyrin. We assumed that the best binders to gankyrin were through a covalent linkage via the sulfonyl fluoride moiety in the third position of each ligand and hypothesized the binding would withstand SDS-PAGE analysis. The gel could then be imaged for fluorescence and the amount of binding could be quantitated based on the gel band signal. All ten hit structures were resynthesized on resin with a linker conjugated to fluorescein (Fig. 4A). As a negative control, a peptoid was synthesized containing amines that were present in library but were not observed in any of the hit structures (Fig. 4A). Importantly, the negative control also had the sulfonyl fluoride in the third position, so we could determine how much, if any, of the binding was due to a non-specific covalent interaction. All ligands and the negative control were purified by HPLC prior to use (ESI Appendix S2†).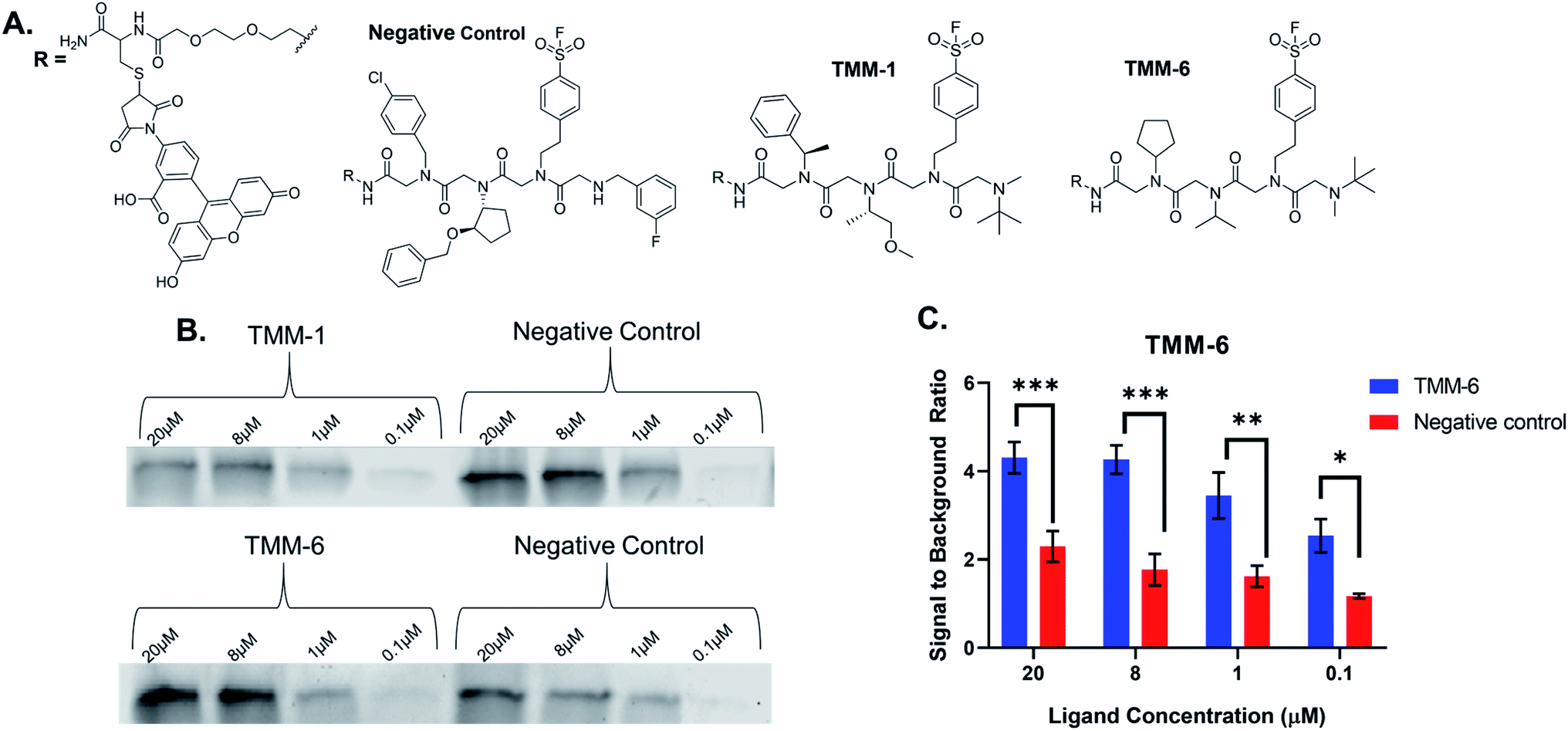 | ||
| Fig. 4 Validation of hits from the Initial Screen. (A) Structures of the negative control and examples of hits from the initial screen, TMM-1 and TMM-6. Each peptoid was synthesized on a linker in which fluorescein was coupled to fluorescently label the ligands. The negative control was composed of amines that were present in the initial screen, but not identified in any of the hit structures. The amine that facilitates covalent binding was retained in the third position of the negative control. (B) Varying concentration of each labeled ligand or the negative control were incubated with 500 ng of purified gankyrin for 3 hours at 4 °C. Since the interaction between the ligands and gankyrin is covalent, it can withstand SDS-PAGE analysis. Gels were then imaged, revealing fluorescing protein bands which demonstrated that the ligands had bound gankyrin. TMM-1 was not a valid hit, as the bands produced by the ligand binding gankyrin are less intense than those produced by the negative control. TMM-6 produced more intense bands than the negative control, indicating that it is a true hit. (C) The in-gel fluorescence experiment was performed in experimental triplicate with TMM-6. The signal of the fluorescent protein bands was quantified at each concentration of ligand and normalized to a DMSO control sample. This data was then compiled and graphed as shown. TMM-6 was identified as a true hit because it produced higher fluorescence intensities after binding gankyrin than the negative control at every tested concentration. 2-way ANOVA analysis: *p < 0.05, **p < 0.01, ***p < 0.005. | ||
Fluorescently labeled ligands were diluted such that the final concentration in each sample would range from 20–0.1 μM and incubated with purified gankyrin. After incubation, the reaction was quenched by adding laemmli buffer and heating. Samples were then subjected to SDS-PAGE and the gels were imaged for fluorescence (Fig. 4B). We expected to find that validated hits would produce fluorescent protein bands with higher intensities than the negative control at each concentration tested. This would indicate that the amines were playing an important role in the binding interaction.
Many of our ligands were not validated as hits, as the fluorescence intensities that resulted from the labeled ligand binding the protein were not greater than those produced by binding of the negative control (Fig. 4B & ESI Fig. S2†). However, one peptoid did produce significantly higher fluorescence intensities than the negative control, TMM-6. The in-gel fluorescence experiment with TMM-6 was conducted in experimental triplicate to validate this initial result (ESI Fig. S3†). Fluorescence intensities of each protein band produced by TMM-6 binding gankyrin were quantified. This was then normalized for each replicate of the experiment by dividing the fluorescent signal from each concentration by the signal produced by incubating DMSO with gankyrin. The normalized data for each concentration of the three replicates was graphed, revealing that TMM-6 produced significantly higher signal than the negative control at every concentration tested (Fig. 4C). TMM-6 was the only peptoid of the 10 hits that produced a significantly higher signal than the negative control, validating it as a binder.
To ensure the fluorescein tag was not responsible for binding of the TMM-6 to the protein, a nonfluorescent version of the ligand was synthesized and purified. A competition experiment was conducted in which either labeled or unlabeled TMM-6 was incubated with purified gankyrin. The second ligand was then added and allowed to react. Samples were subjected to SDS-PAGE and the gel was imaged for fluorescence (ESI Fig. S4†). We observed that binding of the fluorescent TMM-6 to gankyrin was significantly lower when the unlabeled ligand was added first, indicating that binding between the ligand and gankyrin is not dependent on the fluorescein moiety. As an additional control to ensure that binding observed by SDS-PAGE was the result of a covalent linkage of the ligand to the protein and not other nonspecific interactions, a fluorescently labeled version of TMM-6 without the sulfonyl fluoride moiety was synthesized by exchanging the 4-(2-aminoethyl)benzensulfonyl fluoride for 2-phenylethylamine. 2-Phenylethylamine is very similar in structure to 4-(2-aminoethyl)benzensulfonyl fluoride, but the sulfonyl fluoride is not present, therefore no covalent interaction between this version of TMM-6 and gankyrin can occur. The noncovalent or covalent ligand were incubated with purified gankyrin and subjected to SDS-PAGE (ESI Fig. S5†). Each sample was prepared in triplicate and the fluorescence intensities of the resulting protein bands were quantified. As expected, protein incubated with TMM-6 containing the 2-phenylethylamine produced very low fluorescence intensities, indicating that nonspecific interactions between TMM-6 and gankyrin were not responsible for the observed fluorescent bands. Conversely, samples in which protein was incubated with the fluorescently labeled TMM-6 with the amine that facilitates covalent binding showed significantly higher fluorescence intensities than incubation with the noncovalent versions (ESI Fig. S5†). Taken together, this data indicates that covalent binding of the ligand to the gankyrin after interacting with the sulfonyl fluoride moiety.
Detecting gankyrin in spiked HEK 293T cell lysate
After identifying TMM-6 as our lead compound, we next sought to determine the specificity of this ligand to gankyrin. To explore this, lysate from HEK 293T cells was spiked with purified gankyrin. Gankyrin was added to the lysate such that it was 1% of the total protein concentration. Then, fluorescently labeled TMM-6 was added to the spiked lysate to a final concentration of 8 μM. As a control, 8 μM of the fluorescently labeled negative control peptoid was also added to samples of spiked lysate. After incubation, samples were subjected to SDS-PAGE and the gel was imaged for fluorescence (Fig. 5A). Two intense fluorescent bands were observed after the addition of TMM-6, indicating that it had strongly bound two proteins in the spiked lysate sample. Both sets of bands were excised from the gel for proteomics analysis to determine the identity of the proteins. Excitingly, proteomics analysis revealed that both bands were predominately composed of gankyrin (Fig. 5B). In addition to having moderate sequence coverage of gankyrin in each set of bands, each displayed high MS/MS counts of 43 and 45, respectively, indicating that gankyrin was the most abundant protein in the excised samples. The larger molecular weight band is likely to be a ubiquitinated variant of gankyrin or gankyrin contained within a protein–protein complex that did not completely denature on the SDS-PAGE gel. Other less intense bands were observed in samples incubated with both TMM-6 and the negative control, indicating that some nonspecific binding to other proteins in the lysate occurred. However, the most prominent fluorescing bands in the TMM-6 samples were most abundantly composed of gankyrin and these bands were not as intense in the negative control sample. Some nonspecific binding of TMM-6 to other proteins was anticipated, as the sulfonyl fluoride will facilitate covalent linkage to serine and threonine residues present in other proteins in the sample. However, the extent of nonspecific binding of TMM-6 was moderately low compared to its binding to gankyrin, indicating moderate specificity of TMM-6 to the oncoprotein.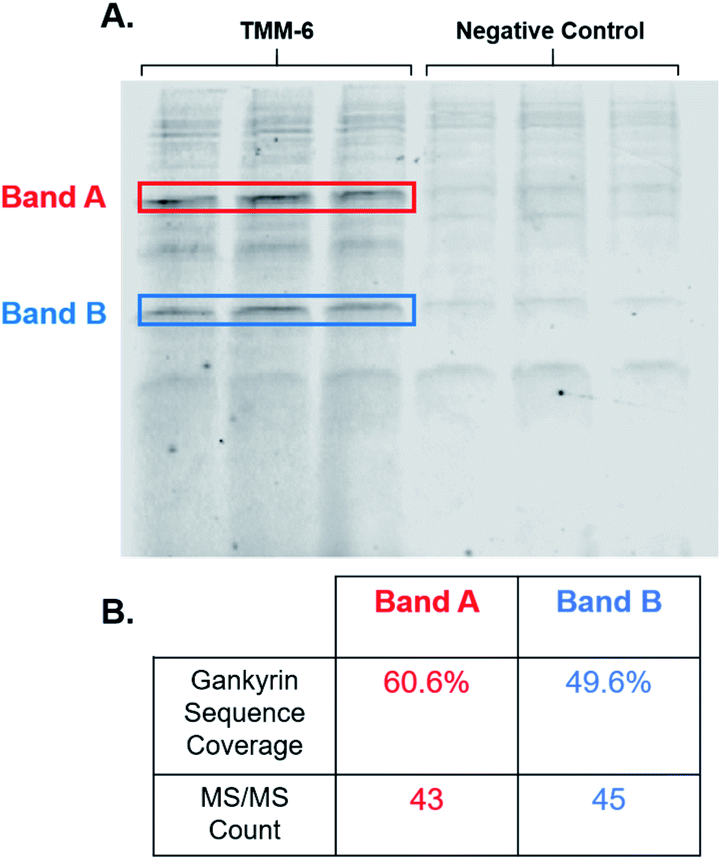 | ||
| Fig. 5 TMM-6 predominately binds gankyrin in spiked HEK 293T cell lysate. Purified gankyirn was added to HEK 293T cell lysate such that it was 1% of the total protein concentration. Fluorescently labeled TMM-6 or negative control was added to the spiked lysate samples to a final concentration of 8 μM. Ligands reacted with lysate proteins for 3 hours at 4 °C. (A) Samples were then subjected to SDS-PAGE and the gel was imaged for fluorescence. Two intense bands resulted from the addition of TMM-6, indicating the ligand strongly bound two proteins. These bands were not as pronounced in the samples dosed with the negative control. (B) The two highlighted bands were excised from the gel and proteomics analysis was performed to determine the identity of the proteins. Gankyrin was the most predominate protein in both bands. 60.6% and 49.6% of gankyrin's sequence as identified in the bands, respectively. Both bands had high MS/MS counts corresponding to gankyrin. | ||
Conclusions
We have recently developed the framework to screen OBOC libraries against biological targets of interest in which hits can be prioritized from the initial screen. Here, we demonstrate successful application of this technology by screening the oncoprotein gankyrin against a 343-member peptoid library bearing an amine that facilitates covalent linkage of the ligand to a protein. Our results demonstrate that our developed OBOC screening and prioritization method can be used as a rapid way to identify novel small molecule binders of target proteins. After screening the library twice and analyzing the amine monomers at each position, we had ten primary hits. From these ten primary hits, one validated to bind gankyrin selectively. We did attempt to tag endogenously produced gankyrin in live HEK 293T cells, but since gankyrin is rapidly degraded we could not detect it despite using proteasome inhibitors.To the best of our knowledge, this is the first reported covalent binder to gankyrin. Gankyrin was selected as a target not only because it is an oncoprotein that forms novel protein–protein interactions when over expressed, but it also does not have an enzymatic function. Our screening method does not require a measurable functional output of a protein, making gankyrin an interesting target to validate our technique. The successful identification of TMM-6 from our screen indicates that the types of proteins that could be screened by this method are vast. We believe this screening method can be used to rapidly identify binders to other proteins implicated in disease that do not have a known function or have a function that is difficult to monitor. This technology could also be further expanded to identify novel ligands on the surface of cancer cells or overexpressed antibodies from serum. Expansion of this screening technique will increase the types of biological targets that can be screened against OBOC libraries and can provide new small molecule probes to evaluate these targets.
Conflicts of interest
Prof. Trader is a scientific advisor and shareholder of Booster Therapeutics GmbH. Other authors declare no conflicts.Acknowledgements
This work was supported through a start-up package from Purdue University School of Pharmacy, the Purdue University Center for Cancer Research NIH grant (P30 CA023168), the American Cancer Society Institutional Research Grant (IRG-14-190-56) to the Purdue University Center for Cancer Research, and a grant from the NIH-NIGMS (R21GM131206). Proteomic experiments including sample preparation, mass spectrometry data collection and analysis was performed at the Purdue Proteomics Facility, Bindley Bioscience Center, Purdue University. Fig. 1 of this manuscript was created with BioRender.com. MEM was supported by a fellowship from the Purdue Cancer Center.References
- K. S. Lam, M. Lebl and V. Krchňák, The “One-Bead-One-Compound” Combinatorial Library Method, Chem. Rev., 1997, 97(2), 411–448, DOI:10.1021/cr9600114.
- D. J. Trader, S. Simanski and T. Kodadek, A Reversible and Highly Selective Inhibitor of the Proteasomal Ubiquitin Receptor Rpn13 Is Toxic To Multiple Myeloma Cells, J. Am. Chem. Soc., 2015, 137(19), 6312, DOI:10.1021/jacs.5b02069.
- Y. Gao, S. Amar, S. Pahwa, G. Fields and T. Kodadek, Rapid Lead Discovery Through Iterative Screening of One Bead One Compound Libraries, ACS Comb. Sci., 2015, 17(1), 49–59, DOI:10.1021/co500154e.
- W. G. Cochrane, M. L. Malone, V. Q. Dang, V. Cavett, A. L. Satz and B. M. Paegel, Activity-Based DNA-Encoded Library Screening, ACS Comb. Sci., 2019, 21(5), 425–435, DOI:10.1021/acscombsci.9b00037.
- A. B. MacConnell, P. J. McEnaney, V. J. Cavett and B. M. Paegel, DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis, ACS Comb. Sci., 2015, 17(9), 518–534, DOI:10.1021/acscombsci.5b00106.
- K. E. Denton and C. J. Krusemark, Crosslinking of DNA-Linked Ligands to Target Proteins for Enrichment from DNA-Encoded Libraries, RSC Med. Chem., 2016, 7(10), 2020–2027, 10.1039/c6md00288a.
- H. J. Olivos, K. Bachhawat-Sikder and T. Kodadek, Quantum Dots As A Visual Aid For Screening Bead-Bound Combinatorial Libraries, ChemBioChem, 2003, 4(11), 1242–1245, DOI:10.1002/cbic.200300712.
- W. Heusermann, B. Ludin, N. T. Pham, M. Auer, T. Weidemann and M. Hintersteiner, A Wide-Field Fluorescence Microscope Extension for Ultrafast Screening of One-Bead One-Compound Libraries Using a Spectral Image Subtraction Approach, ACS Comb. Sci., 2016, 18(5), 209–219, DOI:10.1021/acscombsci.5b00175.
- M. Hintersteiner, T. Kimmerlin, F. Kalthoff, M. Stoeckli, G. Garavel, J.-M. Seifert, N.-C. Meisner, V. Uhl, C. Buehler, T. Weidemann and M. Auer, Single Bead Labeling Method for Combining Confocal Fluorescence On-Bead Screening and Solution Validation of Tagged One-Bead One-Compound Libraries, Chem. Biol., 2009, 16(7), 724–735, DOI:10.1016/j.chembiol.2009.06.011.
- M. Hintersteiner and M. Auer, A Two-Channel Detection Method for Autofluorescence Correction and Efficient on-Bead Screening of One-Bead One-Compound Combinatorial Libraries Using the COPAS Fluorescence Activated Bead Sorting System, Methods Appl. Fluoresc., 2013, 1(1), 017001, DOI:10.1088/2050-6120/1/1/017001.
- M. M. Marani, M. C. Martínez Ceron, S. L. Giudicessi, E. de Oliveira, S. Côté, R. Erra-Balsells, F. Albericio, O. Cascone and S. A. Camperi, Screening of One-Bead-One-Peptide Combinatorial Library Using Red Fluorescent Dyes. Presence of Positive and False Positive Beads, J. Comb. Chem., 2009, 11(1), 146–150, DOI:10.1021/cc800145c.
- P. J. Hergenrother, K. M. Depew and S. L. Schreiber, Small-Molecule Microarrays: Covalent Attachment and Screening of Alcohol-Containing Small Molecules on Glass Slides, J. Am. Chem. Soc., 2000, 122(32), 7849–7850, DOI:10.1021/ja0014032.
- K. Mendes, J. M. Ndungu, L. F. Clark and T. Kodadek, Optimization of the Magnetic Recovery of Hits from One-Bead–One-Compound Library Screens, ACS Comb. Sci., 2015, 17(9), 506–517, DOI:10.1021/acscombsci.5b00090.
- M. E. Maresh and D. J. Trader, Development of a Method To Prioritize Protein–Ligand Pairs on Beads Using Protein Conjugated to a Near-IR Dye, ACS Comb. Sci., 2019, 21(3), 223–228, DOI:10.1021/acscombsci.8b00165.
- K. E. Denton and C. J. Krusemark, Crosslinking of DNA-Linked Ligands to Target Proteins for Enrichment from DNA-Encoded Libraries, RSC Med. Chem., 2016, 7(10), 2020–2027, 10.1039/c6md00288a.
- L. Gao, H. Xie, L. Dong, J. Zou, J. Fu, X. Gao, L. Ou, S. Xiang and H. Song, Gankyrin Is Essential for Hypoxia Enhanced Metastatic Potential in Breast Cancer Cells, Mol. Med. Rep., 2014, 9(3), 1032–1036, DOI:10.3892/mmr.2013.1860.
- Y. Sun, Y. Tan, Z. Lu, B. Li, C. Sun, T. Li, L. Zhao, Z. Liu, G. Zhang, J. Yao and J. Li, Arctigenin Inhibits Liver Cancer Tumorigenesis by Inhibiting Gankyrin Expression via C/EBPα and PPARα, Front. Pharmacol., 2018, 9, 268 CrossRef PubMed.
- Z. Bai, Y. Tai, W. Li, C. Zhen, W. Gu, Z. Jian, Q. Wang, J. E. Lin, Q. Zhao, W. Gong, B. Liang, C. Wang and T. Zhou, Gankyrin Activates IL-8 to Promote Hepatic Metastasis of Colorectal Cancer, Cancer Res., 2013, 73(14), 4548–4558, DOI:10.1158/0008-5472.can-12-4586.
- H. Higashitsuji, H. Higashitsuji, K. Itoh, T. Sakurai, T. Nagao, H. Sumitomo, T. Masuda, S. Dawson, Y. Shimada, R. J. Mayer and J. Fujita, The Oncoprotein Gankyrin Binds to MDM2/HDM2, Enhancing Ubiquitylation and Degradation of P53, Cancer Cell, 2005, 8(1), 75–87, DOI:10.1016/j.ccr.2005.06.006.
- H. Higashitsuji, K. Itoh, T. Nagao, S. Dawson, K. Nonoguchi, T. Kido, R. J. Mayer, S. Arii and J. Fujita, Reduced Stability of Retinoblastoma Protein by Gankyrin, an Oncogenic Ankyrin-Repeat Protein Overexpressed in Hepatomas, Nat. Med., 2000, 6(1), 96–99, DOI:10.1038/71600.
- A. Chattopadhyay, C. J. O'Connor, F. Zhang, C. Galvagnion, W. R. J. D. Galloway, Y. S. Tan, J. E. Stokes, T. Rahman, C. Verma, D. R. Spring and L. S. Itzhaki, Discovery of a Small-Molecule Binder of the Oncoprotein Gankyrin That Modulates Gankyrin Activity in the Cell, Sci. Rep., 2016, 6, 23732, DOI:10.1038/srep23732.
- A. S. Culf and R. J. Ouellette, Solid-Phase Synthesis of N-Substituted Glycine Oligomers (A-Peptoids) and Derivatives, Molecules, 2010, 15(8), 5282–5335, DOI:10.3390/molecules15085282.
- G. M. Figliozzi, R. Goldsmith, S. C. Ng, S. C. Banville and R. N. Zuckermann, Synthesis of N-Substituted Glycine Peptoid Libraries, Methods Enzymol., 1996, 267, 437–447, DOI:10.1016/s0076-6879(96)67027-x.
- K. Büssow, C. Scheich, V. Sievert, U. Harttig, J. Schultz, B. Simon, P. Bork, H. Lehrach and U. Heinemann, Structural Genomics of Human ProteinsTarget Selection and Generation of a Public Catalogue of Expression Clones, Microb. Cell Fact., 2005, 4, 21, DOI:10.1186/1475-2859-4-21.
- P. P. Nanaware, M. P. Ramteke, A. K. Somavarapu and P. Venkatraman, Discovery of Multiple Interacting Partners of Gankyrin, a Proteasomal Chaperone and an Oncoprotein-Evidence for a Common Hot Spot Site at the Interface and Its Functional Relevance, Proteins, 2014, 82(7), 1283–1300, DOI:10.1002/prot.24494.
- A. Narayanan and L. H. Jones, Sulfonyl Fluorides as Privileged Warheads in Chemical Biology, Chem. Sci., 2015, 6(5), 2650–2659, 10.1039/c5sc00408j.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10976b |
| This journal is © The Royal Society of Chemistry 2021 |