
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew pentacyclic triterpenoids isolated from Leptopus chinensis and their hepatoprotective activities on tert-butyl hydroperoxide-induced oxidative injury†
Liping Longab,
Ye Yangab,
Tianliang Zhuab,
Xinxin Zhangab,
Shizhou Qiab,
Ting Liuab,
Kairu Songab,
Da Wang*c and
Huiyuan Gao *ab
*ab
aSchool of Traditional Chinese Materia Medica, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China. E-mail: sypugaohy@163.com
bKey Laboratory of Structure-Based Drug Design & Discovery of Ministry of Education, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China
cSchool of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, People's Republic of China
First published on 1st April 2021
Abstract
Eight unknown pentacyclic triterpenoids (1–4 and 8–11), along with eight known analogues (5–7 and 12–16) have been first isolated from the dried whole plant of Leptopus chinensis. The structures of the new compounds were determined by comprehensive spectroscopic methods, including 1D and 2D NMR, as well as HRESIMS measurements. Meanwhile, the hepatoprotective activities of the isolated compounds were preliminarily evaluated, and the results indicated that compounds 2, 5 and 16 possess potent protective effects on tert-butyl hydroperoxide (t-BHP)-induced oxidative injury in vitro, and further study revealed that 16 significantly alleviates t-BHP-induced hepatotoxicity by effectively improving cell viability and decreasing reactive oxygen species (ROS) generation and the cell apoptosis rate in HepG2 cells.
1. Introduction
Nowadays, multiple unfavorable life factors lead to the occurrence of various liver diseases such as fatty liver, hepatitis, liver fibrosis, liver cirrhosis and even liver cancer. These liver diseases lead to high morbidity and mortality in the public, affecting humans of all ages.1 In China, liver diseases affect approximately 300 million people and cause a great economic burden on society.2 During the search for new therapies to protect the liver from diverse harmful factors, natural plants have been considered as an important source to obtain potential lead compounds for the development of hepatoprotective drugs.3,4 Furthermore, a large number of biological compounds have been isolated and glycyrrhizic acid, as well as oleanolic acid, have been used in the clinic as representative drugs from natural plants.5,6Euphorbiaceae, one of the biggest families of natural plants, has been confirmed to possess significant hepatoprotective effects.7–9 Leptopus chinensis (Bunge) Pojark., a perennial herbaceous plant belonging to the Euphorbiaceae family, is widely distributed in most regions of China.10,11 This plant has been used as a type of folk medicine to treat viral hepatitis, nephritis and lung cancer.12 However, there are no in-depth phytochemical or pharmacological investigations into this plant, and few articles have reported the presence of several triterpenoids and sterols in L. chinensis.13,14 Thus, it is interesting to understand the material basis and further pharmacological effects of L. chinensis. As a result, eight new triterpenoids (1–4 and 8–11), together with eight known analogues (5–7 and 12–16) were obtained from the EtOAc extract of L. chinensis (Fig. 1). Based on the moderate hepatoprotective activity of the EtOAc fraction, these compounds were further evaluated for their hepatoprotective effect on tert-butyl hydroperoxide (t-BHP)-induced oxidative damage in vitro.
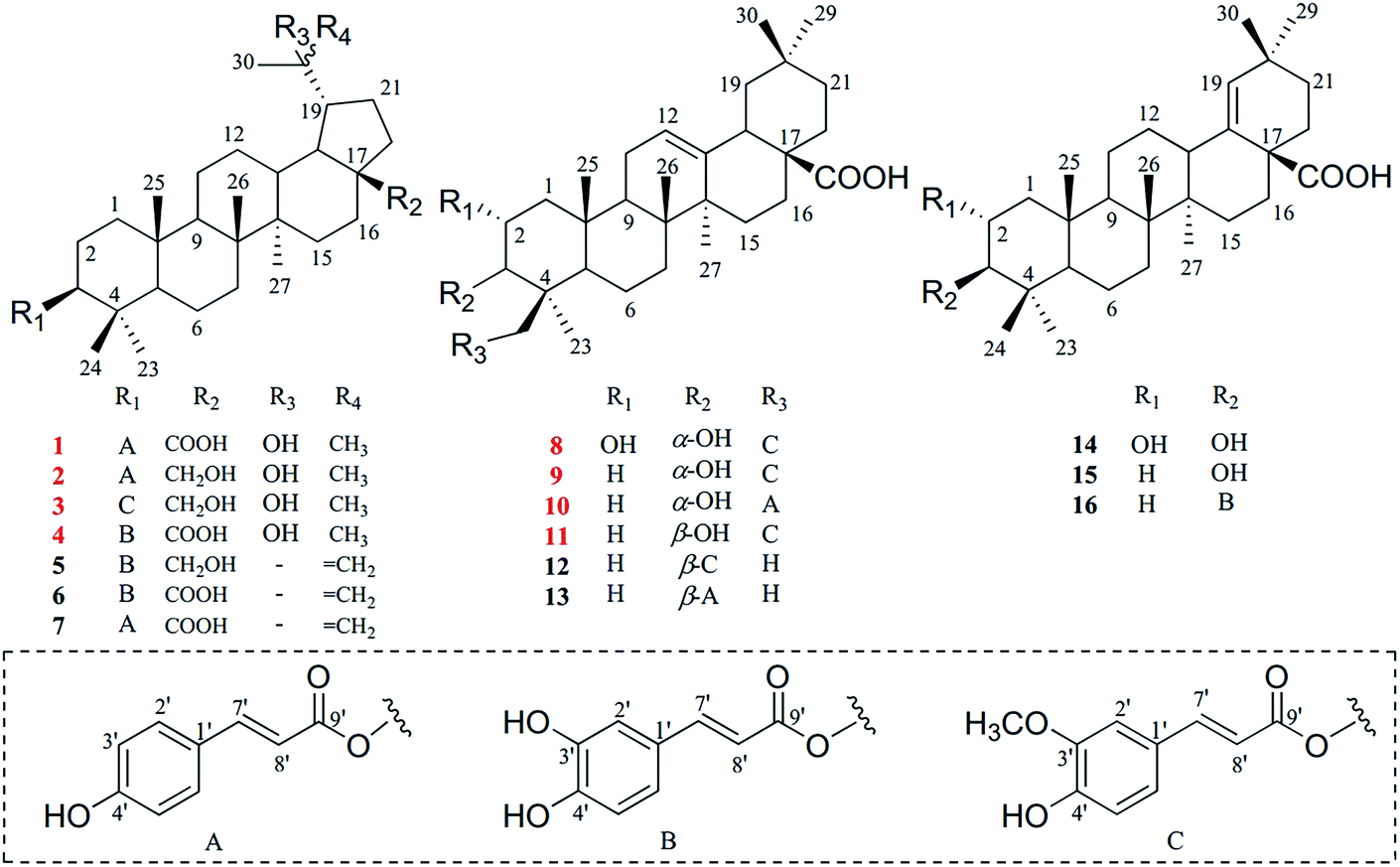 | ||
| Fig. 1 Structures of compounds 1–16. | ||
2. Experimental
2.1 General experimental procedures
Optical rotation values were recorded on a polarimeter (Anton-Paar MCP 200; Austria). Infrared (IR) spectra were obtained with a Fourier transform infrared spectrometer (IFS-55; Bruker). A Shimadzu UV-1700 spectrophotometer (Shimadzu, Kyoto, Japan) was used to obtain the ultraviolet (UV) spectra. The NMR spectra were recorded using ARX-400 and AV-600 spectrometers (Bruker) with TMS as an internal standard. HRESIMS data were recorded on an Agilent G6520 Q-TOF spectrometer (Santa Clara, CA, USA). Column chromatography was conducted using silica gel (100–200 mesh and 200–300 mesh, Qingdao, China) and ODS (50 μm, Aichi, Japan) as absorbents, and thin-layer chromatography (TLC) plates (GF254) were purchased from Qingdao Marine Chemical Co. Ltd. (Qingdao, China). High-performance liquid chromatography (HPLC) was performed using a Shimadzu LC-20AR instrument equipped with an SPD-20A UV detector (Shimadzu, Kyoto, Japan) and a YMC Rp-C18 column (5 μm, 10 × 250 mm) (YMC, Kyoto, Japan). Cell apoptosis and intracellular ROS measurements were analyzed using a Becton Dickinson FACS Calibur System. All solvents used for isolation were of analytical grade, and the reagents for HPLC were of chromatographic grade.2.2 Plant material
The dried whole plant of L. chinensis was purchased in August 2018 from Yunnan Yuancai Biotechnology Co. Ltd. (Kunming, Yunnan, China), and it was authenticated by Associate Prof. Jiu-zhi Yuan of Shenyang Pharmaceutical University. A voucher specimen (LCH-18-08-27) was deposited in the School of Traditional Chinese Medicine, Shenyang Pharmaceutical University, Shenyang, China.2.3 Extraction and isolation
The dried whole plant of L. chinensis (19.2 kg) was made into a powder and extracted with 75% aqueous EtOH three times under reflux. The filtered solutions were combined and evaporated under a vacuum condition to obtain a crude extract, which was suspended in water and further separated with petroleum ether, ethyl acetate and n-butanol, successively. The ethyl acetate extract (80.0 g) was separated using silica gel column chromatography eluted with CH2Cl2/MeOH (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 → 100:20) to yield fifteen fractions (Fr. 1 to Fr. 15). Fr. 3 (17.3 g) underwent further chromatography on a silica gel column and was eluted with CH2Cl2/MeOH (100:0 → 2:1) to afford six fractions (Fr. 3.1 to Fr. 3.6). Fr. 3.4 (2.0 g) was further subjected to an ODS column eluted with MeOH/H2O (40:60 → 95:5) to obtain seven more fractions (Fr. 3.4.1 to Fr. 3.4.7). Then, Fr. 3.4.3 (0.5 g) was separated by Rp-18 HPLC with MeOH/H2O (80:20) to obtain compounds 11 (3.0 mg, tR = 33.0 min), 12 (2.8 mg, tR = 53.5 min) and 13 (3.5 mg, tR = 41.8 min). Fr. 3.4.5 (1.5 g) was purified using semi-preparative Rp-18 HPLC with MeOH/H2O (75:25) to give compounds 5 (3.5 mg, tR = 126.8 min), 6 (7.2 mg, tR = 104.7 min) and 9 (5.9 mg, tR = 72.0 min). Furthermore, Fr. 3.4.6 (0.2 g) was isolated orderly by preparative and semi-preparative Rp-18 HPLC using CH3CN/H2O (65:35) to afford compounds 3 (1.5 mg, tR = 68.3 min) and 10 (1.8 mg, tR = 37.2 min). Fr. 5 (3.0 g) was divided into six parts (Fr. 5.1 to Fr. 5.6) using an ODS column eluted with MeOH/H2O (60:40 → 95:5). Fr. 5.3 (0.5 g) was further purified by Rp-18 HPLC with MeOH/H2O (75:25) to yield compounds 8 (3.8 mg, tR = 63.1 min), 14 (3.2 mg, tR = 91.4 min) and 16 (5.2 mg, tR = 88.3 min), whilst Fr. 5.4 (0.3 g) was separated by Rp-18 HPLC with MeOH/H2O (80:20) to obtain compounds 1 (5.4 mg, tR = 84.9 min), 2 (4.5 mg, tR = 75.1 min) and 15 (2.7 mg, tR = 53.2 min). Fr. 6 (1.0 g) was first separated into six fractions (Fr. 6.1 to Fr. 6.6) using an ODS column eluted with MeOH/H2O (60:40 → 95:5), then Fr. 6.5 (0.2 g) was also isolated using HPLC with CH3CN/H2O (75:25) to yield compounds 4 (1.7 mg, tR = 26.7 min) and 7 (3.0 mg, tR = 30.5 min).
ε) 226 (3.87), 311 (4.02) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 1; HRESIMS (negative-ion mode) m/z 619.3992 [M − H]− (calc. for C39H55O6, 619.3999).
:0 → 100:20) to yield fifteen fractions (Fr. 1 to Fr. 15). Fr. 3 (17.3 g) underwent further chromatography on a silica gel column and was eluted with CH2Cl2/MeOH (100:0 → 2:1) to afford six fractions (Fr. 3.1 to Fr. 3.6). Fr. 3.4 (2.0 g) was further subjected to an ODS column eluted with MeOH/H2O (40:60 → 95:5) to obtain seven more fractions (Fr. 3.4.1 to Fr. 3.4.7). Then, Fr. 3.4.3 (0.5 g) was separated by Rp-18 HPLC with MeOH/H2O (80:20) to obtain compounds 11 (3.0 mg, tR = 33.0 min), 12 (2.8 mg, tR = 53.5 min) and 13 (3.5 mg, tR = 41.8 min). Fr. 3.4.5 (1.5 g) was purified using semi-preparative Rp-18 HPLC with MeOH/H2O (75:25) to give compounds 5 (3.5 mg, tR = 126.8 min), 6 (7.2 mg, tR = 104.7 min) and 9 (5.9 mg, tR = 72.0 min). Furthermore, Fr. 3.4.6 (0.2 g) was isolated orderly by preparative and semi-preparative Rp-18 HPLC using CH3CN/H2O (65:35) to afford compounds 3 (1.5 mg, tR = 68.3 min) and 10 (1.8 mg, tR = 37.2 min). Fr. 5 (3.0 g) was divided into six parts (Fr. 5.1 to Fr. 5.6) using an ODS column eluted with MeOH/H2O (60:40 → 95:5). Fr. 5.3 (0.5 g) was further purified by Rp-18 HPLC with MeOH/H2O (75:25) to yield compounds 8 (3.8 mg, tR = 63.1 min), 14 (3.2 mg, tR = 91.4 min) and 16 (5.2 mg, tR = 88.3 min), whilst Fr. 5.4 (0.3 g) was separated by Rp-18 HPLC with MeOH/H2O (80:20) to obtain compounds 1 (5.4 mg, tR = 84.9 min), 2 (4.5 mg, tR = 75.1 min) and 15 (2.7 mg, tR = 53.2 min). Fr. 6 (1.0 g) was first separated into six fractions (Fr. 6.1 to Fr. 6.6) using an ODS column eluted with MeOH/H2O (60:40 → 95:5), then Fr. 6.5 (0.2 g) was also isolated using HPLC with CH3CN/H2O (75:25) to yield compounds 4 (1.7 mg, tR = 26.7 min) and 7 (3.0 mg, tR = 30.5 min).
ε) 226 (3.87), 311 (4.02) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 1; HRESIMS (negative-ion mode) m/z 619.3992 [M − H]− (calc. for C39H55O6, 619.3999).
| No. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δHa J (Hz) | δCb | δHa J (Hz) | δCb | δHc J (Hz) | δCd | δHa J (Hz) | δCb | |
| a Recorded at 600 MHz in DMSO-d6.b Recorded at 150 MHz in DMSO-d6.c Recorded at 600 MHz in CDCl3.d Recorded at 150 MHz in CDCl3 (“o” overlapped). | ||||||||
| 1 | 1.69 o, 1.01 m | 37.9 | 1.67 o, 0.99 m | 38.4 | 1.70 o, 1.04 m | 38.5 | 1.69 o, 0.99 m | 37.9 |
| 2 | 1.66 o, 1.58 m | 23.5 | 1.72 o, 1.30 m | 27.9 | 1.83 o, 0.93 m | 29.1 | 1.64 o, 1.58 m | 23.5 |
| 3 | 4.50 dd (11.6, 4.6) | 79.8 | 4.50 dd (11.4, 4.6) | 80.3 | 4.62 dd (11.2, 4.9) | 80.9 | 4.49 dd (11.6, 4.5) | 79.8 |
| 4 | — | 40.1 | — | 38.1 | — | 38.2 | — | 40.1 |
| 5 | 0.85 o | 54.6 | 0.85 o | 55.0 | 0.84 o | 55.4 | 0.85 o | 54.6 |
| 6 | 1.47 m, 1.38 o | 17.8 | 1.48 m, 1.40 o | 18.3 | 1.53 m, 1.41 o | 18.4 | 1.47 m, 1.38 o | 17.8 |
| 7 | 1.38 o, 1.34 m | 34.1 | 1.54 m, 1.41 o | 36.4 | 1.42 o, 1.04 m | 34.6 | 1.38 o, 1.33 m | 34.1 |
| 8 | — | 40.7 | — | 41.5 | — | 41.6 | — | 40.7 |
| 9 | 1.30 o | 49.8 | 1.31 o | 50.1 | 1.31 o | 50.3 | 1.31 o | 49.8 |
| 10 | — | 36.6 | — | 37.0 | — | 37.2 | — | 36.6 |
| 11 | 1.42 o, 1.15 m | 21.2 | 2.04 o, 0.88 o | 28.4 | 1.90 o, 1.39 m | 28.4 | 1.43 m, 1.16 m | 21.2 |
| 12 | 1.67 o, 1.45 m | 28.2 | 1.03 m, 0.92 m | 27.4 | 1.78 m, 1.07 m | 27.3 | 1.68 o, 1.45 m | 28.2 |
| 13 | 2.26 td (12.7, 3.3) | 37.6 | 1.39 o | 34.4 | 1.63 o | 36.3 | 2.26 td (12.6, 3.6) | 37.6 |
| 14 | — | 42.8 | — | 43.4 | — | 43.5 | — | 42.8 |
| 15 | 1.46 m, 1.07 m | 29.6 | 1.76 m, 0.72 m | 33.5 | 1.91 o, 1.80 m | 33.5 | 1.46 m, 1.10 m | 29.6 |
| 16 | 2.12 td (12.2, 3.2) | 31.9 | 1.86 dt (10.7, 2.0) | 30.1 | 1.92 o | 29.9 | 2.12 dd (12.2, 3.0) | 31.9 |
| 1.32 o | 1.05 o | 1.25 o | 1.31 o | |||||
| 17 | — | 57.9 | — | 49.0 | — | 49.3 | — | 57.9 |
| 18 | 1.48 o | 47.8 | 1.40 o | 48.6 | 1.56 o | 48.9 | 1.48 o | 47.8 |
| 19 | 2.01 td (10.5, 2.1) | 48.8 | 1.69 o | 50.2 | 1.83 o | 49.9 | 2.01 td (9.3, 1.8) | 48.8 |
| 20 | — | 71.3 | — | 71.8 | — | 73.7 | — | 71.3 |
| 21 | 1.75 o, 1.24 m | 28.5 | 1.65 o, 1.59 m | 24.0 | 1.69 o, 1.64 m | 24.0 | 1.75 o, 1.24 m | 28.5 |
| 22 | 1.59 m, 1.29 m | 36.2 | 1.39 m, 1.16 m | 21.4 | 1.49 m, 1.25 m | 21.5 | 1.60 m, 1.29 m | 36.2 |
| 23 | 0.82 s | 27.7 | 0.82 s | 28.2 | 0.90 s | 28.2 | 0.82 s | 27.7 |
| 24 | 0.87 s | 16.1 | 0.88 s | 17.1 | 0.92 s | 16.8 | 0.87 s | 16.1 |
| 25 | 0.84 s | 16.0 | 0.85 s | 16.4 | 0.89 s | 16.4 | 0.84 s | 16.0 |
| 26 | 0.88 s | 16.6 | 1.01 s | 16.3 | 1.00 s | 16.3 | 0.88 s | 16.6 |
| 27 | 0.96 s | 14.8 | 0.95 s | 15.5 | 1.06 s | 15.2 | 0.96 s | 14.8 |
| 28 | — | 177.7 | 3.57 dd (10.4, 5.4) | 58.9 | 3.86 d (10.8) | 61.0 | — | 177.7 |
| 3.04 dd (10.4, 5.4) | 3.32 d (10.8) | |||||||
| 29 | 1.08 s | 31.0 | 1.09 s | 32.2 | 1.14 s | 31.8 | 1.08 s | 30.9 |
| 30 | 1.00 s | 26.3 | 0.98 s | 25.4 | 1.24 s | 24.8 | 0.99 s | 26.3 |
| 1′ | — | 125.1 | — | 125.6 | — | 127.3 | — | 125.5 |
| 2′ | 7.54 d (8.6) | 130.3 | 7.54 d (8.6) | 130.8 | 7.03 d (1.8) | 109.4 | 7.03 d (1.8) | 114.8 |
| 3′ | 6.78 d (8.6) | 115.8 | 6.78 d (8.6) | 116.2 | — | 146.9 | — | 145.6 |
| 4′ | — | 159.8 | — | 160.2 | — | 148.0 | — | 148.3 |
| 5′ | 6.78 d (8.6) | 115.8 | 6.78 d (8.6) | 116.2 | 6.91 d (8.2) | 114.8 | 6.75 d (8.2) | 115.7 |
| 6′ | 7.54 d (8.6) | 130.3 | 7.54 d (8.6) | 130.8 | 7.07 dd (8.2, 1.8) | 123.2 | 6.99 dd (8.2, 1.8) | 121.3 |
| 7′ | 7.53 d (16.0) | 144.4 | 7.52 d (16.0) | 144.9 | 7.59 d (15.9) | 144.5 | 7.44 d (15.9) | 144.8 |
| 8′ | 6.36 d (16.0) | 114.6 | 6.36 d (16.0) | 115.1 | 6.29 d (15.9) | 116.4 | 6.23 d (15.9) | 114.3 |
| 9′ | — | 166.4 | — | 166.8 | — | 167.3 | — | 166.3 |
| 3′-OCH3 | — | — | — | — | 3.93 s | 56.1 | — | — |
| 28-COOH | 11.88 brs | — | — | — | — | — | 11.97 brs | — |
ε) 227 (3.81), 311 (4.05) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 1; HRESIMS (negative-ion mode) m/z 605.4202 [M − H]− (calc. for C39H57O5, 605.4206).ε) 232 (3.96), 324 (4.01) nm; for 1H and 13C-NMR data in CDCl3, see Table 1; HRESIMS (negative-ion mode) m/z 635.4321 [M − H]− (calc. for C40H59O6, 635.4312).ε) 218 (3.85), 320 (4.03) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 1; HRESIMS (negative-ion mode) m/z 635.3942 [M − H]− (calc. for C39H55O7, 635.3948).ε) 230 (3.93), 325 (3.90) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 2; HRESIMS (negative-ion mode) m/z 663.3891 [M − H]− (calc. for C40H55O8, 663.3897).
| No. | 8 | 9 | 10 | 11 | ||||
|---|---|---|---|---|---|---|---|---|
| δHa J (Hz) | δCb | δHa J (Hz) | δCb | δHa J (Hz) | δCb | δHa J (Hz) | δCb | |
| a Recorded at 600 MHz in DMSO-d6.b Recorded at 150 MHz in DMSO-d6 (“o” overlapped). | ||||||||
| 1 | 1.41 o, 1.18 o | 41.4 | 1.33 m, 1.14 m | 32.8 | 1.33 m, 1.13 m | 32.8 | 1.56 o, 0.97 m | 38.1 |
| 2 | 3.77 o | 64.4 | 1.83 o, 1.43 m | 25.0 | 1.83 o, 1.42 m | 25.0 | 1.56 o, 1.51 m | 26.9 |
| 3 | 3.50 dd (6.1, 2.8) | 72.7 | 3.52 dd (6.3, 2.7) | 68.5 | 3.51 dd (6.4, 2.5) | 68.5 | 3.16 dd (11.8, 4.9) | 76.9 |
| 4 | — | 42.5 | — | 41.3 | — | 41.3 | — | 41.6 |
| 5 | 1.31 o | 48.2 | 1.38 o | 48.8 | 1.38 o | 48.8 | 0.84 o | 55.0 |
| 6 | 1.50 m, 1.32 m | 17.9 | 1.50 m, 1.39 m | 18.0 | 1.50 m, 1.38 m | 18.0 | 1.71 o, 1.52 o | 19.5 |
| 7 | 1.86 o, 1.23 m | 32.6 | 1.43 m, 1.23 m | 32.6 | 1.42 m, 1.26 m | 32.6 | 1.36 m, 1.27 m | 33.0 |
| 8 | — | 39.0 | — | 39.0 | — | 39.0 | — | 38.9 |
| 9 | 1.61 o | 47.1 | 1.62 o | 47.0 | 1.62 o | 47.0 | 1.52 o | 47.2 |
| 10 | — | 37.6 | — | 36.5 | — | 36.5 | — | 36.6 |
| 11 | 1.85 o, 0.88 o | 23.1 | 1.84 o, 1.62 o | 23.0 | 1.84 o, 1.62 o | 23.0 | 1.82 o, 1.01 m | 23.0 |
| 12 | 5.18 brt (3.2) | 121.4 | 5.18 brt (3.4) | 121.5 | 5.17 brt (3.3) | 121.5 | 5.17 brt (3.4) | 121.5 |
| 13 | — | 143.9 | — | 143.8 | — | 143.8 | — | 143.8 |
| 14 | — | 41.3 | — | 41.2 | — | 41.2 | — | 41.3 |
| 15 | 1.66 m, 0.99 o | 27.1 | 1.65 o, 0.99 o | 27.1 | 1.65 o, 1.01 o | 27.1 | 1.63 o, 0.99 o | 27.2 |
| 16 | 1.91 td (13.5, 3.7) | 22.6 | 1.92 td (13.4, 3.7) | 22.7 | 1.92 td (13.3, 3.4) | 22.7 | 1.91 td (13.4, 3.7) | 22.6 |
| 1.48 o | 1.48 o | 1.49 o | 1.48 o | |||||
| 17 | — | 45.4 | — | 45.4 | — | 45.4 | — | 45.5 |
| 18 | 2.75 dd (13.2, 4.0) | 40.7 | 2.75 dd (13.2, 4.0) | 40.8 | 2.75 dd (13.8, 4.1) | 40.8 | 2.74 dd (13.7, 4.0) | 40.8 |
| 19 | 1.61 o, 1.06 m | 45.7 | 1.62 o, 1.05 m | 45.7 | 1.62 o, 1.07 m | 45.7 | 1.62 o, 1.06 m | 45.7 |
| 20 | — | 30.4 | — | 30.4 | — | 30.4 | — | 30.4 |
| 21 | 1.33 m, 1.13 m | 33.3 | 1.33 m, 1.14 m | 33.3 | 1.32 m, 1.15 m | 33.3 | 1.32 m, 1.14 m | 33.3 |
| 22 | 1.61 o, 1.44 m | 32.1 | 1.60 o, 1.43 m | 32.1 | 1.61 o, 1.45 m | 32.1 | 1.61 o, 1.44 m | 32.1 |
| 23 | 1.04 s | 22.9 | 0.99 s | 22.6 | 0.98 s | 22.6 | 1.09 s | 22.8 |
| 24 | 4.26 d (11.4) | 66.4 | 4.27 d (11.3) | 66.8 | 4.26 d (11.3) | 66.8 | 4.24 d (11.7) | 65.8 |
| 3.96 d (11.4) | 3.98 d (11.3) | 3.97 d (11.3) | 4.19 d (11.7) | |||||
| 25 | 0.92 s | 16.4 | 0.88 s | 15.2 | 0.88 s | 15.2 | 0.89 s | 14.8 |
| 26 | 0.70 s | 16.7 | 0.70 s | 16.7 | 0.71 s | 16.7 | 0.71 s | 16.6 |
| 27 | 1.11 s | 25.7 | 1.12 s | 25.7 | 1.12 s | 25.7 | 1.10 s | 25.5 |
| 28 | — | 178.6 | — | 178.6 | — | 178.6 | — | 178.6 |
| 29 | 0.88 s | 32.8 | 0.89 s | 32.7 | 0.88 s | 32.7 | 0.87 s | 32.8 |
| 30 | 0.88 s | 23.4 | 0.88 s | 23.4 | 0.88 s | 23.4 | 0.87 s | 23.4 |
| 1′ | — | 125.5 | — | 125.5 | — | 125.0 | — | 125.6 |
| 2′ | 7.31 d (1.7) | 111.2 | 7.31 d (1.8) | 111.2 | 7.54 d (8.6) | 130.3 | 7.28 d (1.8) | 111.2 |
| 3′ | — | 148.0 | — | 148.0 | 6.79 d (8.6) | 115.8 | — | 148.0 |
| 4′ | — | 149.4 | — | 149.3 | — | 159.8 | — | 149.3 |
| 5′ | 6.79 d (8.2) | 115.5 | 6.79 d (8.2) | 115.5 | 6.79 d (8.6) | 115.8 | 6.80 d (8.2) | 115.6 |
| 6′ | 7.11 dd (8.2, 1.7) | 123.2 | 7.10 dd (8.2, 1.8) | 123.2 | 6.79 d (8.6) | 130.3 | 7.08 dd (8.2, 1.8) | 123.1 |
| 7′ | 7.52 d (15.9) | 145.0 | 7.52 d (15.9) | 145.0 | 7.52 d (15.9) | 144.6 | 7.51 d (15.9) | 144.6 |
| 8′ | 6.45 d (15.9) | 114.5 | 6.44 d (15.9) | 114.6 | 6.36 d (15.9) | 114.2 | 6.40 d (15.9) | 115.0 |
| 9′ | — | 166.7 | — | 166.8 | — | 166.7 | — | 166.9 |
| 3′-OCH3 | 3.82 s | — | 3.82 s | 55.7 | — | — | 3.82 s | 55.8 |
| 28-COOH | 12.04 brs | — | 12.03 brs | — | 12.03 brs | — | 11.96 brs | — |
ε) 235 (3.85), 325 (3.91) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 2; HRESIMS (negative-ion mode) m/z 647.3955 [M − H]− (calc. for C40H55O7, 647.3948).ε) 225 (3.84), 312 (3.91) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 2; HRESIMS (negative-ion mode) m/z 617.3837 [M − H]− (calc. for C39H53O6, 617.3842).ε) 236 (3.88), 320 (3.90) nm; for 1H and 13C-NMR data in DMSO-d6, see Table 2; HRESIMS (negative-ion mode) m/z 647.3942 [M − H]− (calc. for C40H55O7, 647.3948).2.4 Cell culture
HepG2 cells were purchased from the Stem Cell Bank, Chinese Academy of Science, China. The cells were cultured in DMEM medium (Gibco, USA), supplemented with 10% fetal bovine serum (Gibco, USA) and 0.5% penicillin/streptomycin in a humidified atmosphere of 5% CO2 at 37 °C.2.5 Evaluation of hepatoprotective activity
The hepatoprotective activities of isolated compounds were measured using the MTT method. Briefly, the cells in the logarithmic growth phase were incubated in 96-well flat-bottomed cell culture at a density of 5 × 103 cells per well and were cultured overnight, before being pretreated with isolated compounds and a positive control (silymarin and ammonium glycyrrhetate) that was diluted to a specified concentration for 2 h. t-BHP was then added to a final concentration of 1 mM and the solutions were incubated for 2.5 h. Afterwards, MTT solution (5 mg mL−1) was added to each well and the solutions were incubated for another 3 h. The supernatant was removed, and the deposited formazan crystals were dissolved with DMSO, followed by the plates being shaken mildly to blend the mixture. The optical densities at 490 nm were measured using a Varioskan Flash Multimode Reader (Thermo Scientific). All experiments were performed at least three times.2.6 Intracellular ROS measurements
A DCFH-DA fluorescent probe was used for assessing the intracellular ROS production. Briefly, HepG2 cells were seeded in 6-well plates at a density of 4 × 105 cells per well overnight, and the plates were then incubated with DCFH-DA (10 μM) at 37 °C for 30 min and washed three times with PBS. Next, the cells were pretreated with different concentrations of compound 16 for 2 h, and were subsequently exposed to t-BHP (1 mM) for an additional 1 h. Cells were collected for analysis with laser scanning confocal microscopy and flow cytometry.2.7 Apoptosis assay
2.8 Statistical analysis
The results were expressed as mean values ± standard deviation. A one-way ANOVA test was used to evaluate the differences between the means of the groups using SPSS 19.0 software, and a p value less than 0.05 was considered to be significantly different.3. Results and discussion
3.1 Structure elucidation
Compound 1 was obtained as a white amorphous powder and the molecular formula of C39H56O6 was deduced from HRESIMS data at m/z 619.3992 [M − H]− (calc. for C39H55O6, 619.3999) and NMR data (Table 1). The 1H-NMR data of 1 showed signals of seven tertiary methyl singlets at δH 1.08, 1.00, 0.96, 0.88, 0.87, 0.84, and 0.82, as well as an oxygenated methine at δH 4.50 (dd, 11.6, 4.6 Hz, H-3). Moreover, the signals of two olefinic protons at δH 7.53 and 6.36 (each a d, 16.0 Hz, H-7′, 8′), and an aromatic proton spin system at δH 7.54 (d, 8.6 Hz, H-2′, 6′) and δH 6.78 (d, 8.6 Hz, H-3′, 5′) indicated the presence of a trans-p-coumaroyl unit. 39 Carbon signals were observed in the 13C-NMR (Table 1), including two carbonyl groups at δC 177.7 (C-28) and 166.4 (C-9′), a 1,4-disubstituted aromatic system at δC 159.8 (C-4′), 130.3 (C-2′, 6′), 125.1 (C-1′), and 115.8 (C-3′, 5′), two olefinic carbons at δC 144.4 (C-7′) and 114.6 (C-8′), and two oxygenated aliphatic signals at δC 79.8 (C-3) and 71.3 (C-20). Based on the 1D NMR, HSQC and HMBC data, compound 1 can be assigned as a triterpenoid skeleton linked with a trans-p-coumaroyl unit. Comparison of its 1D NMR data with the data of 3β-O-trans-coumaroyl betulinic acid indicated that they have a similar lupane-type triterpenoid skeleton,17 except for the presence of a hydroxyl unit at C-20 in 1. The key HMBC correlations of H-29/C-20, H-30/C-20, OH-20/C-20, and OH-20/C-30, combined with the signals of the double band unit, were not detected in the 13C-NMR spectrum, suggesting the connection of the hydroxyl unit to C-20. In addition, the correlation (Fig. 2A) of H-3 to C-9′ showed that the O-trans-p-coumaroyl moiety was attached to C-3 of the lupane framework. Furthermore, the NOESY correlations (Fig. 2B) from H-3 to H-5 and H-9, and the coupling constant of JH-2β/H-3α = 11.2 Hz, showed that H-3 is α-oriented. In conclusion, the structure of compound 1 was defined as 3β-O-trans-p-coumaroyl-20-hydroxy-lup-28-oic acid.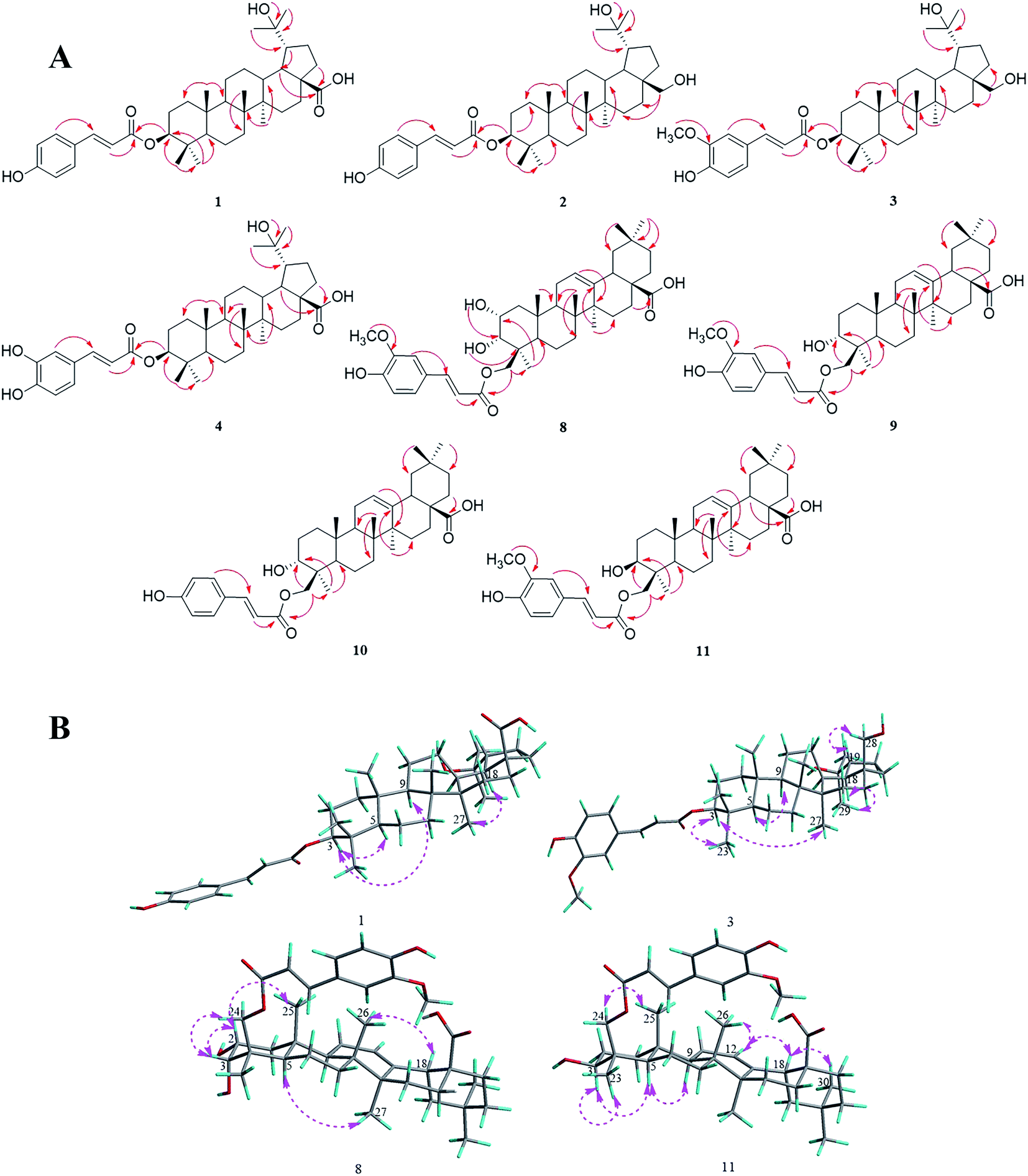 | ||
| Fig. 2 (A) Key HMBC correlations of compounds 1–4 and 8–11. (B) Major NOESY correlations within compounds 1, 3, 8 and 11. | ||
Compounds 2 and 3 were isolated as white amorphous powders, and their molecular formulas were identified from the HRESIMS data at m/z 605.4202 [M − H]− (calc. for C39H57O5, 605.4206) and 635.4321 [M − H]− (calc. for C40H59O6, 635.4312), respectively. The NMR spectrum of 2 was almost identical to that of 1 and suggested that the 28-carboxyl group present in 1 was replaced by a hydroxymethyl group in 2, and this was deduced from the proton signals of two oxygenated methylene groups at δH 3.57 and 3.04 (each a dd, 10.4, 5.4 Hz, H-28α, 28β) and the HMBC correlations (Fig. 2A) of H-28α and H-28β to C-16 (δC 30.1). Additionally, a comparison of the NMR data of 3 with that of 2 showed that the spectra were very similar, except for the presence of a trans-feruloyl unit in 3 rather than a trans-p-coumaroyl moiety in 2. On the basis of the HRESIMS and 1D NMR spectroscopic data, the trans-feruloyl moiety was identified from the proton signals at δH 7.07 (dd, 8.2, 1.8 Hz, H-6′), 7.03 (d, 1.8 Hz, H-2′) 6.91 (d, 8.2 Hz, H-5′) and 3.93 (s, 3′-OCH3), in accordance with the two oxygenated aromatic carbon signals at δC 146.9 (C-3′) and 148.0 (C-4′), and a methoxy signal at δC 56.1 (3′-OCH3). In addition, the HMBC spectra (Fig. 2A) of compounds 2 and 3 showed the key correlation from H-3 to C-9′, and the coupling constants of JH-2β/H-3α and NOESY correlations were identical to those of 1. Consequently, the structures of 2 and 3 were elucidated as 3β-O-trans-p-coumaroyl-20,28-dihydroxy-lupane and 3β-O-trans-feruloyl-20,28-dihydroxylupane, respectively.
Compound 4 was purified as a white amorphous powder with the molecular formula of C39H56O7, as determined by negative mode HRESIMS data at m/z 635.3942 ([M − H]−, calc. as 635.3948). According to its 1D NMR (Table 1), HSQC and HMBC data, 4 was found to show quite similar results to 1, with the main difference between them being that the trans-p-coumaroyl group of 1 was replaced with the trans-caffeoyl group of 4. This conclusion could be verified by the signals of a 1,3,4-trisubstituted aromatic proton spin system at δH 7.03 (d, 1.8 Hz, H-2′), 6.99 (dd, 8.2, 1.8 Hz, H-6′) and 6.75 (d, 8.2 Hz, H-5′), along with the two oxygenated aromatic carbon signals at δC 148.3 (C-4′) and 145.6 (C-3′). Moreover, the key HMBC correlation (Fig. 2A) from H-3 to C-9' (δC 166.3) indicated that an O-trans-caffeoyl moiety was located at C-3, and the NOESY correlations showed that 4 has the same relative configuration as 1. Thus, compound 4 was elucidated as 3β-O-trans-caffeoyl-20-hydroxy-lup-28-oic-acid.
Compound 8 was obtained as a white amorphous powder, and its molecular formula was identified as C40H56O8 on the basis of the 1D NMR and HRESIMS data with an [M − H]− ion at m/z 663.3891 (calc. for C40H55O8, 663.3897). The 1H-NMR spectrum (Table 2) of compound 8 showed proton signals of six tertiary methyl singlets at δH 0.70, 0.88, 0.88, 0.92, 1.04, and 1.11, one olefinic proton at δH 5.18 (brt, 3.2 Hz, H-12) and two oxygenated methines at δH 3.77 (overlap, H-2) and 3.50 (dd, 6.1, 2.8 Hz, H-3). Moreover, the trans-feruloyl unit was determined based on the signals of an aromatic proton spin system at δH 7.31 (d, 1.7 Hz, H-2′), 7.11 (dd, 8.2, 1.7 Hz, H-6′) and 6.79 (d, 8.2 Hz, H-5′), a double bond at δH 7.52 and 6.45 (each a d, 15.9 Hz, H-7′, 8′) and a methoxy singlet at δH 3.82 (s, 3′-OCH3). The 13C-NMR data (Table 2) revealed the occurrence of 40 carbon signals, including one double bond at δC 121.4 (C-11) and 143.9 (C-12), three oxygenated aliphatic carbons at δC 72.7 (C-3), 66.4 (C-24) and 64.4 (C-2), a carboxyl carbon at δC 178.6 (C-28), and eight sp2 hybridized carbon signals, implying that the structure of compound 8 was an oleanane-type triterpenoid skeleton attached to a trans-feruloyl moiety. Detailed observation of the 1D NMR data disclosed similarities to the spectrum of 23-trans-p-coumaroyloxy-2α,3β-dihydroxy-olean-12-en-28-oic acid,18 and further spectroscopic data analysis of the HSQC and HMBC spectra (Fig. 2A) showed the apparent differences. The trans-feruloyl moiety was attached to C-24, on account of the upfield shift observed for C-23 (δC 22.9) and the downfield shift observed for C-24 (δC 66.4) in the 13C-NMR spectrum, together with the HMBC correlations (Fig. 2A) of H-24α, 24β to C-3, C-4 and C-9′. Additionally, significant NOESY correlations (Fig. 2B) were observed from H-2 (δH 3.77) to H-25, demonstrating that H-2 was β-oriented, and the orientation of H-3β of 8 could be deduced from the coupling constant of JH-2β/H-3β = 6.1 Hz and the NOESY correlation of H-3 (δH 3.50) to H-24α and H-24β. Therefore, compound 8 was certified as 24-O-trans-feruloyl-2α,3α-dihydroxy-olean-12-en-28-oic acid.
Compounds 9 and 10 were isolated as white amorphous powders, and the HRESIMS negative ions at m/z 647.3955 [M − H]− and 617.3837 [M − H]− determined their molecular formulas to be C40H56O7 and C39H54O6, respectively. The NMR data of 9 showed a close similarity to the spectrum of 8, except for the absence of the hydroxyl group at C-2 in 9. The 1D NMR spectrum (Table 2) of 9 indicated one oxygenated methine at δH 3.52 (dd, 6.3, 2.7, H-3) and two oxygenated aliphatic carbons at δC 68.5 (C-3) and 66.8 (C-24). In addition, the NMR data of 10 was very similar to that of 9, except that the trans-feruloyl group in 9 was replaced by a trans-p-coumaroyl group in 10. Accordingly, compounds 9 and 10 were elucidated to be 24-O-trans-feruloyl-3α-hydroxy-olean-12-en-28-oic acid and 24-O-trans-p-coumaroyl-3α-hydroxy-olean-12-en-28-oic acid, respectively.
Compound 11 was obtained as a white amorphous powder, and it had the identical molecular formula of C40H56O7 as 9, and this was established via the HRESIMS negative ion at m/z 647.3942 [M − H]− (calc. as 647.3948). The NMR data (Table 2) of 11 closely resembled that of 9, indicating that the planar structures of these compounds were the same. Additionally, the NOESY correlations (Fig. 2B) from H-3 (δH 3.16) to H-5 and H-9, along with the coupling constant of JH-2β/H-3α = 11.8 Hz demonstrated that H-3 was α-oriented. Consequently, 11 was determined to be 24-O-trans-feruloyl-3β-hydroxy-olean-12-en-28-oic acid.
The other isolated eight compounds were identified as 3β-O-trans-caffeoyl-28-hydroxylupane (5),19 betulinic acid 3β-caffeate (6),20 3β-O-trans-coumaroyl betulinic acid (7),17 scaphopeta-lumate (12),21 3β-O-(Z)-coumaroyl oleanolic acid (13),22 2α,3β-dihydroxyolean-18-en-28-oic acid (14),23 morolic acid (15),24 and morolic acid 3-O-caffeate (16)25 through comparison of their NMR data with the reported spectroscopic data in the references.
3.2 Evaluation of hepatoprotective activity
t-BHP can be metabolized to harmful free radical intermediates, which are widely used to induce hepatotoxicity in HepG2 cells,26 and the cell viability can be determined by MTT assay. Thus, the protective effects of the isolated compounds (12.5 μM) on t-BHP-induced hepatotoxicity in HepG2 cells were investigated, and silymarin and ammonium glycyrrhetate were used as positive controls. Our results (Fig. 3A) indicated that compounds 2, 5 and 16 showed potent hepatoprotective activity with increased cell survival rates (CSRs) of 69.81 ± 5.61%, 71.04 ± 5.95% and 77.01 ± 3.05%, respectively, compared with the results of the model group (39.63 ± 1.2%).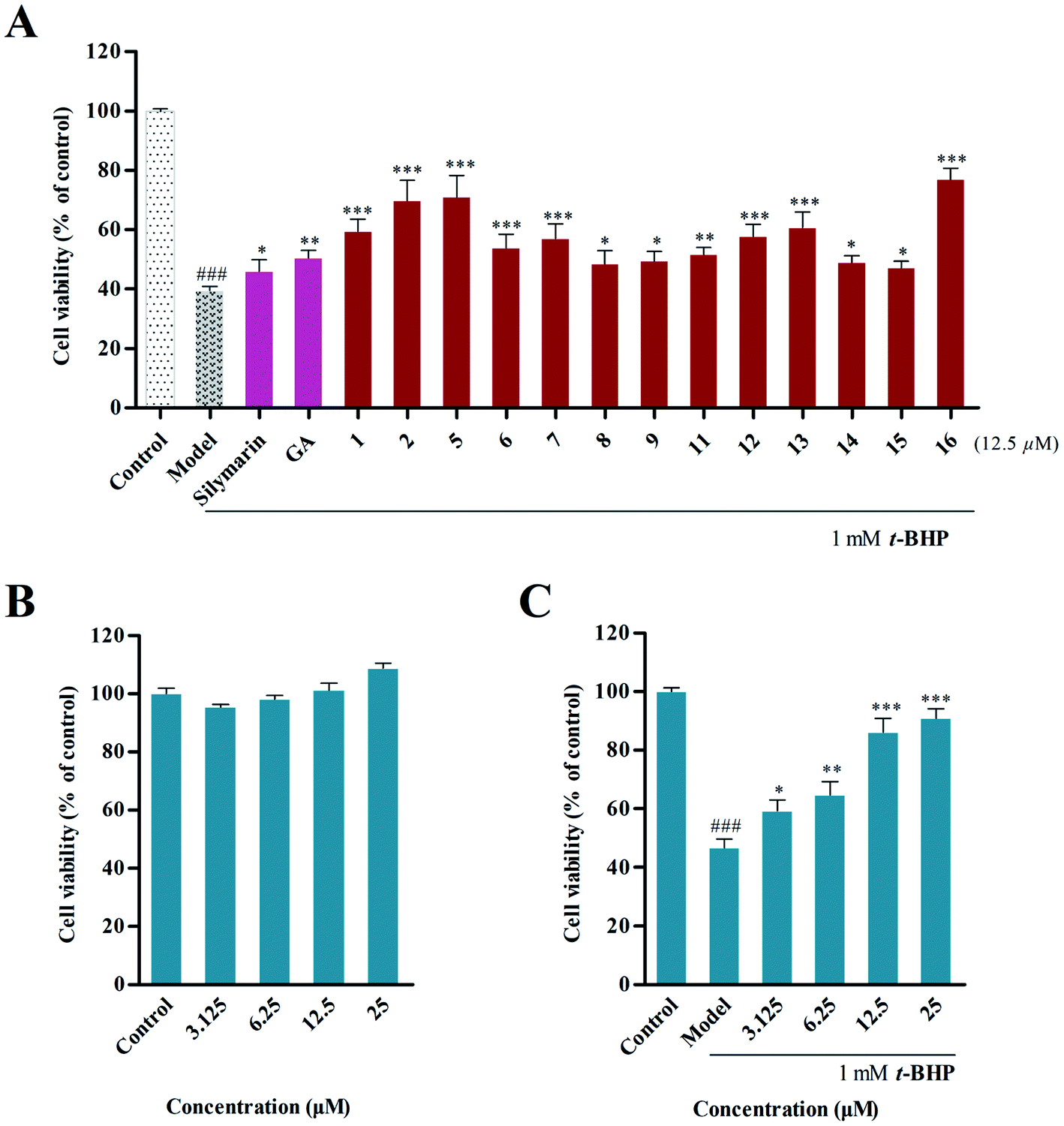 | ||
| Fig. 3 (A) Evaluation of the hepatoprotective effect of the isolated compounds (12.5 μM) on t-BHP-induced hepatotoxicity in HepG2 cells. (B) Cytotoxicity assay of compound 16 against HepG2 cells. (C) Hepatoprotective activity of 16 with concentrations of 3.125, 6.25, 12.5 and 25 μM. | ||
Subsequently, the ability of 16 to protect cells from t-BHP-induced oxidative injury was further examined. The cytotoxic assay revealed that 16 did not show cytotoxic activity on HepG2 cells at concentrations of 3.125, 6.25, 12.5 and 25 μM (Fig. 3B), and significant hepatoprotective activity was further observed in these concentrations in 16. In particular, pretreatment of 25 μM of 16 could improve cell survival with a rate of 90.95 ± 3.19%, compared with that of the model group (46.74 ± 2.95%) (Fig. 3C).
3.3 Preliminary structure–activity relationship
A preliminary structure–activity relationship of the isolated compounds was determined in vitro. Among them, compounds 5 and 16 exhibited the most obvious hepatoprotective activity (CSR = 71.04 ± 5.95% and 77.01 ± 3.05%), and these had the same substitution at C-3, suggesting that the trans-caffeoyl moiety possibly plays a positive role in the hepatoprotective activity. Meanwhile, comparison of the activities of 1, 2, 5 and 6 indicated that the hydroxymethyl group contributed more to the hepatoprotective activity than the carboxy group that is located at C-28. Furthermore, it was observed that compounds 12 and 13, which have a trans-p-coumaroyl or trans-feruloyl group at C-3, displayed more potent effects than compounds 8–9 and 11 that possessed this substituent at C-24, suggesting that different positions of substituent groups may affect their bioactivities. In addition, compound 8, having an additional α-hydroxyl group at C-2, was found to possess similar bioactivity as 9. This revealed that this hydroxyl group might have few effects on the activity. Additionally, the introduction of a trans-caffeoyl moiety resulted in increased activity by comparing the effects of 15 and 16.3.4 Inhibition of intracellular ROS accumulation
Overproduction of reactive oxygen species (ROS) could induce cellular oxidative damage, which is widely considered as a possible mechanism upstream of t-BHP-induced oxidative injury. In the present study, we found that t-BHP treatment significantly increased the amount of intracellular ROS compared with the control cells. However, pretreatment with compound 16 (2.78, 8.33, and 25.00 μM) inhibited the over-accumulation of the intracellular ROS level (Fig. 4A). At the same time, quantitative analysis also showed that 16 ameliorated t-BHP-induced intracellular ROS accumulation in HepG2 cells in a dose-dependent manner (Fig. 4B).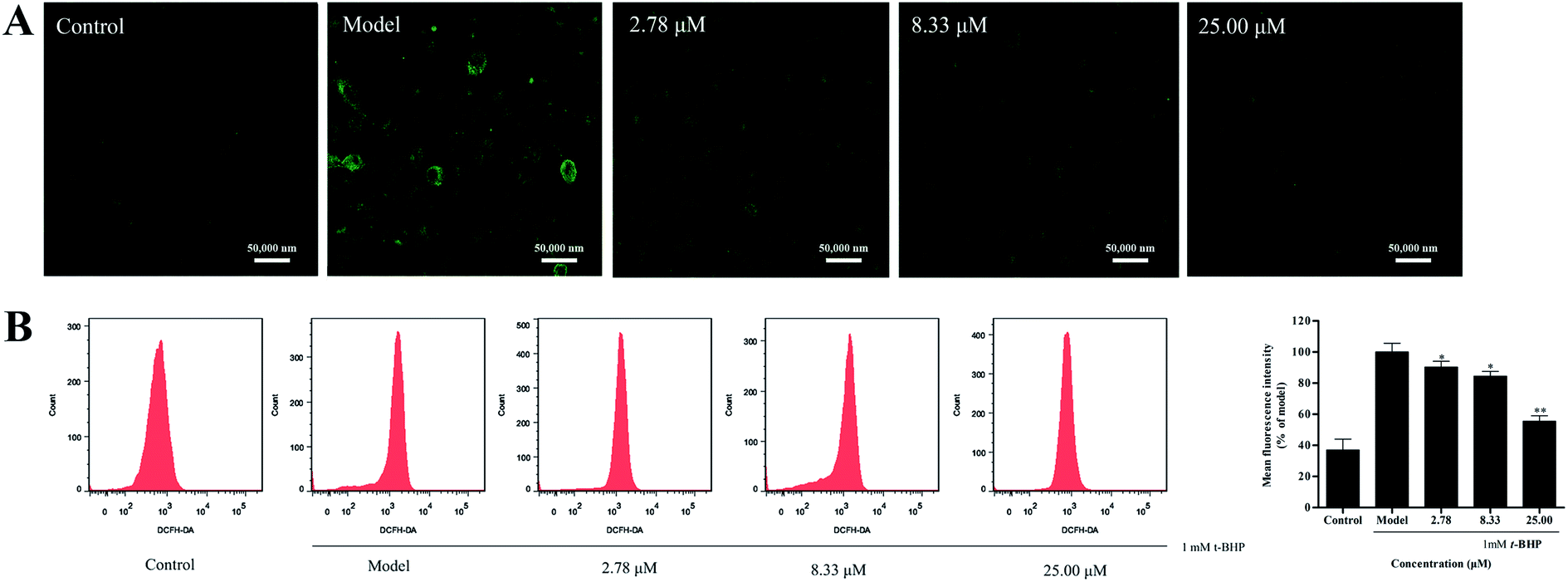 | ||
| Fig. 4 Effect of compound 16 on the intracellular ROS level of t-BHP-induced hepatotoxicity in HepG2 cells, and the intracellular ROS levels were measured by laser scanning confocal microscopy (A) and flow cytometry (B). | ||
3.5 Suppression effect of t-BHP-induced apoptosis
Excessive ROS may cause more extensive and irreparable cell injury, leading to the occurrence of cell death through apoptosis or necrosis.27 In addition, apoptosis has originally been described as morphological changes, with nuclear condensation and fragmentation being the most recognizable hallmarks. Therefore, we employed Hoechst 33258 staining to observe the morphology of cell nuclei. As illustrated in Fig. 5A, clearly condensed and fragmented cell nuclei were observed when they were treated with t-BHP alone, while the pretreatment of compound 16 reduced t-BHP-induced cell nuclei shrinkage and fragmentation, compared with that of the model group.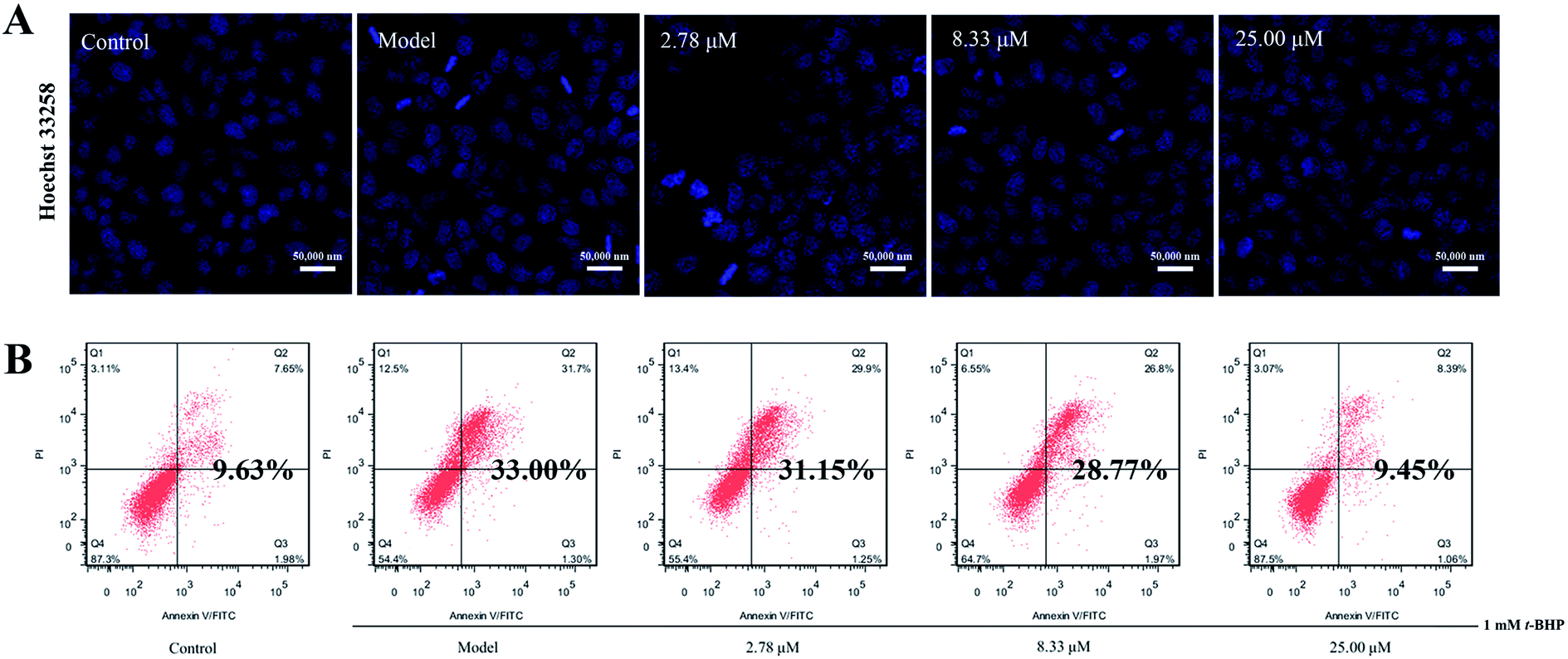 | ||
| Fig. 5 (A) HepG2 cell nuclei were stained with Hoechst 33258 and imaged using a laser scanning confocal microscope (scale bar 50 μm). (B) Apoptosis rates of t-BHP-induced hepatotoxicity in HepG2 cells were detected by annexin V-FITC/PI double staining using flow cytometry. | ||
In addition, the apoptosis rate was investigated via annexin V-FITC/PI double staining combined with flow cytometry, and the results revealed that t-BHP treatment clearly promoted the apoptosis rate from 9.63% for the control to 33.00%. However, pretreatment of 16 at concentrations of 2.78, 8.33, and 25.00 μM decreased the percentage of apoptosis in cells to 31.15%, 28.77% and 9.45%, respectively (Fig. 5B). These findings suggest that 16 suppresses t-BHP-induced apoptosis in HepG2 cells.
4. Conclusions
In this research, a systemic study to identify and assess the bioactive triterpenoids from the whole plant of Leptopus chinensis, in order to investigate their hepatoprotective activities, has been carried out. Ultimately, eight unknown pentacyclic triterpenoids (1–4 and 8–11) and eight known analogues (5–7 and 12–16) were obtained from this plant for the first time, and they have a common feature where the phenylpropanoid moiety is attached to a triterpenoid skeleton. Compounds 1–4 have an unusual skeleton with the transformation of the 20,29-double bond to a hydroxyl group located at C-20, and 8–11 feature a rare position of the phenylpropanoid moiety attached at C-24, with an α-orientation of the hydroxyl group at C-3. In addition, the in vitro measurements discovered that compounds 2, 5 and 16 display a significant protective effect on t-BHP-induced hepatotoxicity in HepG2 cells. Furthermore, 16 markedly ameliorates t-BHP-induced intracellular ROS accumulation and cell apoptosis in HepG2 cells. The discovery of these compounds enriches the structural diversity of pentacyclic triterpenoids and provides the potential for plant-derived natural products to be used for hepatoprotective drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the LiaoNing Revitalization Talents Program (No. XLYC 1905019) and the Natural Science Foundation of Liaoning Province (No. 201602691).Notes and references
- A. H. Zhang, H. Sun and X. J. Wang, Eur. J. Med. Chem., 2013, 63, 570–577 CrossRef CAS PubMed.
- F. S. Wang, J. G. Fan, Z. Zhang, B. Gao and H. Y. Wang, Hepatology, 2014, 60, 2099–2108 CrossRef PubMed.
- S. Shukla and A. Mehta, Braz. J. Bot., 2015, 38, 199–210 CrossRef.
- M. Gordaliza, Clin. Transl. Oncol., 2007, 9, 767–776 CrossRef CAS PubMed.
- G. B. Xu, Y. H. Xiao, Q. Y. Zhang, M. Zhou and S. G. Liao, Eur. J. Med. Chem., 2018, 145, 691–716 CrossRef CAS PubMed.
- A. S. Negi, J. K. Kumar, S. Luqman, K. Shanker, M. M. Gupta and S. P. S. Kbanuja, Med. Res. Rev., 2008, 28, 746–772 CrossRef CAS PubMed.
- J. R. Patel, P. Tripathi, V. Sharma, N. S. Chuhan and V. K. Dixit, J. Ethnopharmacol., 2011, 138, 286–313 CrossRef CAS PubMed.
- M. I. Ezzat, M. M. Okba, S. H. Ahmed, H. A. EI-Banna, A. Prince, S. O. Mohamed and S. M. Ezzat, PLoS One, 2020, 15, 1–23 CrossRef PubMed.
- J. B. Calixto, A. R. Santos, F. V. Cechinel and R. A. Yunes, Med. Res. Rev., 1998, 18, 225–258 CrossRef CAS.
- Editor Committee for Flora of China of Chinese Academy of Science, Flora of Chin, Science Press, 1994, vol. 44, p. 19 Search PubMed.
- Y. J. Chen, S. X. Cheng, Y. Long and Q. D. Hou, J. Zhengzhou Univ., Med. Sci., 2008, 43, 602–603 CAS.
- Z. Y. Wu, Xinhua Bencao Gangyao, Shanghai Science and Technology Press, 1991, p. 225 Search PubMed.
- L. B. Qu, X. L. Chen, J. S. Lu, J. W. Yuan and Y. F. Zhao, Chem. Nat. Compd., 2005, 41, 565–568 CrossRef CAS.
- J. S. Lu, X. L. Chen, Y. Long, Q. Z. Zhao, L. B. Qu, L. Jia and J. Y. Wei, Chin. Tradit. Herb. Drugs, 2001, 32, 112–113 CAS.
- Y. R. Yang, L. P. Long, X. X. Zhang, K. R. Song, D. Wang, X. Xiong, H. Y. Gao and L. P. Sha, RSC Adv., 2019, 9, 31758–31772 RSC.
- D. Wang, D. Su, X. Z. Li, D. Liu, R. G. Xi, H. Y. Gao and X. B. Wang, RSC Adv., 2016, 6, 27434–27446 RSC.
- N. Q. Chien, N. V. Hung, B. D. Santarsiero, A. D. Mesecar, N. M. Cuong, D. D. Soejarto, J. M. Pezzuto, H. H. Fong and G. T. Tan, J. Nat. Prod., 2004, 67, 994–998 CrossRef CAS PubMed.
- J. Q. Gu, E. J. Park, L. Luyengi, M. E. Hawthorne, R. G. Mehta, N. R. Farnsworth, J. M. Pezzuto and A. D. Kinghorn, Phytochemistry, 2001, 58, 121–127 CrossRef CAS.
- A. Patra, S. K. Chaudhuri and S. K. Panda, J. Nat. Prod., 1988, 51, 217–220 CrossRef CAS.
- H. Pan, L. N. Lundgren and R. Andersson, Phytochemistry, 1994, 37, 795–799 CrossRef CAS.
- J. C. Vardamides, A. G. B. Azebaze, A. E. Nkengfack, F. R. V. Heerden, Z. T. Fomum, T. M. Ngando, J. Conrad, B. Vogler and W. Kraus, Phytochemistry, 2003, 62, 647–650 CrossRef CAS PubMed.
- H. Takahashi, M. Iuchi, Y. Fujita, H. Minami and Y. Fukuyama, Phytochemistry, 1999, 51, 543–550 CrossRef CAS.
- J. Zhang, J. Liu, B. Xu, Y. Zhuang, M. Yosikawa and B. C. Yin, Asian J. Chem., 2014, 26, 4521–4522 CrossRef CAS.
- J. C. Ferreira Júnior, R. P. L. Lemos and L. M. Conserva, Biochem. Syst. Ecol., 2012, 44, 208–211 CrossRef.
- K. J. Park, L. Subedi, S. Y. Kim, S. U. Choi and K. R. Lee, Bioorg. Chem., 2018, 77, 527–533 CrossRef CAS PubMed.
- T. H. Do, O. Joonseok, M. K. Nguyen, T. D. Trong, V. D. Le, N. Q. D. Thi, M. L. Sang, S. J. Tae, J. Gilsaeng and N. Minkyun, J. Ethnopharmacol., 2013, 150, 875–885 CrossRef PubMed.
- J. Checa and J. M. Aran, J. Inflammation Res., 2020, 13, 1057–1073 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00962a |
| This journal is © The Royal Society of Chemistry 2021 |