
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkylamine screening and zinc doping of highly luminescent 2D tin-halide perovskites for LED lighting†
Yao
Liu‡
a,
Aifei
Wang‡§
 a,
Jiajing
Wu
a,
Chuying
Wang
a,
Ziliang
Li
b,
Guangcai
Hu
a,
Shiqi
Sui
a,
Jia-Xin
She
c,
Wen
Meng
a,
Weiqiang
Li
c and
Zhengtao
Deng
*a
a,
Jiajing
Wu
a,
Chuying
Wang
a,
Ziliang
Li
b,
Guangcai
Hu
a,
Shiqi
Sui
a,
Jia-Xin
She
c,
Wen
Meng
a,
Weiqiang
Li
c and
Zhengtao
Deng
*a
aCollege of Engineering and Applied Sciences, Nanjing University, Nanjing, Jiangsu 210023, China. E-mail: dengz@nju.edu.cn
bDepartment of Electrical and Computer Engineering, University of Toronto, 10 King's College Road, Toronto, Ontario M5S 3G4, Canada
cState Key Laboratory for Mineral Deposits Research, School of Earth Sciences and Engineering, Nanjing University, Nanjing, Jiangsu 210023, P. R. China
First published on 9th January 2021
Abstract
In recent years, two-dimensional (2D) layered tin halide perovskites have attracted much attention due to their excellent luminescence properties compared with their three-dimensional (3D) analogs. Here, we synthesized 2D perovskites of the form (RNH3)2SnBr4 through a facile hot-injection approach without the use of toxic tri-n-octylphosphine (TOP) as the solvent. We found that these compounds have strong photoluminescence (PL) emission with high PL quantum yields (QY) of 35% ((C6H13NH3)2SnBr4), 82% ((C8H17NH3)2SnBr4), 60% ((C12H25NH3)2SnBr4) and 50% ((C18H35NH3)2SnBr4). Among them, ((C6H13NH3)2SnBr4) was synthesized for the first time; however, (C18H36NH3)2SnBr4 perovskites showed the best stability with a PL QY decrease of less than 1% after 30 days under ambient air and humidity conditions. In addition, other 2D (C8H17NH3)2SnX4 (X = Br, I, or mixture) perovskites were prepared by adjusting the halide ratio, resulting in spectral tunability from yellow to red. Interestingly, through Zn2+ doping, the morphology became more uniform, and the PL stability was significantly improved. Finally, we used Zn2+-doped (C8H17NH3)2SnBr4 yellow phosphor to fabricate UV-pumped yellow LEDs. We expect these 2D perovskites to be promising phosphors in future solid-state lighting and display technologies.
Introduction
Lead halide perovskites (LHPs) have been used in many fields including light-emitting diode (LED) devices,1 solar cells2,3 and television displays4 due to their superior optical properties1,5 and low-cost preparation.6 Although many advances have been made in lead halide perovskites, their toxicity to humans and long-term pollution to the environment remain to be addressed.7,8 Thus, lead-free halide perovskites (LFHPs) with similar optoelectronic properties to LHPs have been broadly studied.9–14 Tin-halide perovskites have drawn a lot of attention for substitution of lead in all optoelectronic applications.15–17 However, owing to the lack of an inert pair effect as in Pb2+, divalent Sn is easily oxidized from the +2 to +4 state, causing deep defects in perovskites. The instability of tin-halide perovskites will seriously affect the device performance, even under trace amounts of water and oxygen in the glovebox.18–20A series of strategies to overcome the instability of tin-halide perovskites have been developed. Deng et al. successfully synthesized Cs2SnI6 nanocrystals and applied them to fabricate field effect transistors (FETs) via direct replacement of Sn2+ with Sn4+.21 Bi-doped Cs2SnCl6 bulk crystals with a PL QY near 80% were reported to emit deep-blue light, which also showed improved stability.22 In addition to direct substitution of Sn2+, introducing hydrophobic organic molecules into the crystal structure is another way to protect Sn2+ from water and oxygen. Recently, two-dimensional (2D) layered tin-halide perovskites with the formula of (R-NH3)2An−1SnnX3n+1 (R = alkyl chains; A = Cs+, MA+, FA+; X = Cl−, Br−, I−) have attracted much attention.10,23 Such 2D tin-halide perovskites showed good photoluminescence and electroluminescence as well as improved resistance to air and moisture10,15,24 compared with homologous three-dimensional (3D) ASnX3 perovskites. For instance, 2D (PEA)2SnI4 thin films were reported to exhibit superior photoluminescence and air stability over bulk CH3NH3SnI3 perovskites, which were used as emitting layers in LEDs.24 Ning's group reported hollow 2D layered (HMD)3SnBr8 crystals synthesized by the antisolvent method. These crystals showed bright yellow luminescence with a PL QY of 86% and were used to fabricate remarkably stable WLEDs by mixing with commercially blue phosphor.25 A (C18H35NH3)2SnBr4 2D perovskite film was applied to produce orange LEDs, showing improved device performance than the 2D analogues, but the usage of toxic and expensive tri-n-octylphosphine (TOP) during the synthesis limited its large-scale application.26 Recently, for the first time, our group synthesized highly emissive 2D tin-halide (OCTAm)2SnX4 perovskites, in aqueous solution with a PL QY near unity and high stability in air.27 We noted that Li's group reported the facile synthesis of 2D bulk tin-halide (RNH3)2SnBr4 perovskites and explored the effect of different carbon chains on 2D organic tin-halide perovskites.28
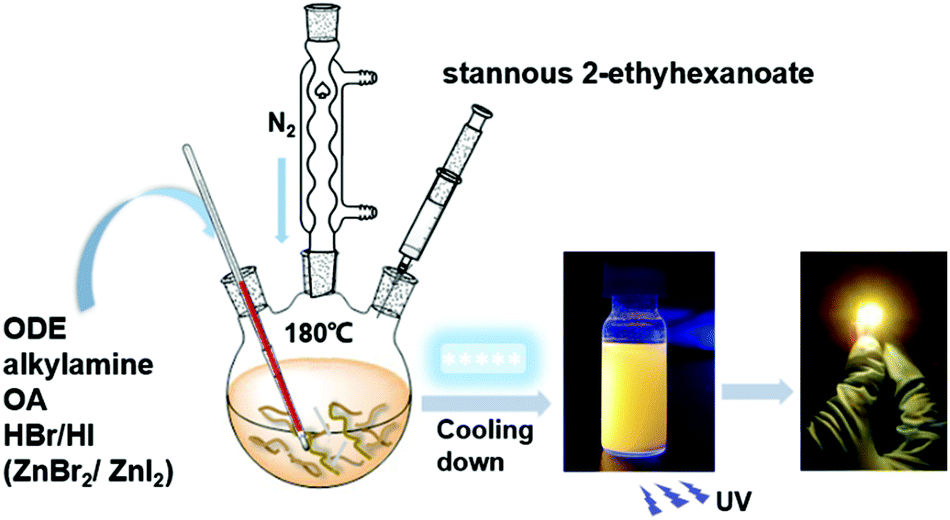 | ||
| Scheme 1 Schematic diagram of the synthesis of the 2D (RNH3)2SnX4 perovskites by a hot-injection method. | ||
The length of the organic amine chain will affect the transport of carriers, thereby affecting the device performance of 2D tin-halide perovskites.29 Herein, we synthesized 2D (RNH3)2SnX4 perovskites with high luminescence emission from yellow to red by a hot injection method. (RNH3) represents the long-chain amine cation: hexylamine cation (C6H13NH3+), octylamine cation (C8H17NH3+), dodecylamine cation (C12H25NH3+), and oleylamine cation (C18H35NH3+). For simplicity, we use C6, C8, C12, and C18 to represent (C6H13NH3)2SnBr4, (C8H17NH3)2SnBr4, (C12H25NH3)2SnBr4 and (C18H35NH3)2SnBr4, respectively, in the figures. Compared with room-temperature synthesis, perovskites synthesized by the hot injection method were more stable.30 In addition, we thoroughly discussed the effects of A-site organic cation substitution on their structural and optical properties and stability. The spectral tunability of 2D tin hybrid perovskites by adjusting the Br/I halide ratio was also explored. Furthermore, Zn2+ was introduced as a dopant into the 2D (C8H17NH3)2SnX4 (X = Br, I) perovskite lattice. Finally, we successfully used the Zn2+-doped (C8H17NH3)2SnBr4 perovskite as a yellow phosphor to fabricate UV-pumped LEDs. In comparison to previous reports, the differences and significance of the current work are as follows: (i) we used stannous 2-ethylhexanoate instead of the commonly used TOP–SnBr2 in the high temperature reaction, which could avoid the problem of toxicity and high cost; (ii) all the (RNH3)2SnBr4 samples showed bright emission from yellow to orange and exhibited good air stability; (iii) by adjusting the Br/I ratio, the PL emission peak could be tuned from yellow to red; (iv) better morphology and stability were achieved after Zn2+ doping into 2D (C8H17NH3)2SnX4 perovskites; (v) Zn2+-doped (C8H17NH3)2SnBr4 yellow phosphor with excellent optical properties was used to fabricate UV-pumped LEDs.
Results and discussion
We adopted a traditional hot-injection approach to synthesize the 2D (RNH3)2SnBr4 perovskites without utilizing the toxic TOP solvent (details are shown in the Experimental section). Typically, oleic acid (OA), alkylamine, and hydrobromic acid (HBr) were dissolved in octadecene (ODE) to form a RNH3+-halide-precursor solution; while the organic amine was partially protonated to RNH3+ through acidification, the rest of the amine and OA molecules acted as dual ligands. After the stannous 2-ethylhexanoate was injected at elevated temperatures, the luminescent 2D (RNH3)2SnBr4 perovskites were precipitated (Scheme 1).All the as-synthesized (RNH3)2SnBr4 perovskites showed periodic diffraction peaks in powder X-ray diffraction (PXRD) patterns (Fig. 1a), which were the characteristics of 2D perovskites.31,32 According to Bragg's law, the layer spacing of 2D (RNH3)2SnBr4 perovskites was calculated as 18.1 Å ((C6H13NH3)2SnBr4), 24.6 Å ((C8H17NH3)2SnBr4), 33.8 Å ((C12H25NH3)2SnBr4) and 39.6 Å ((C18H35NH3)2SnBr4). The schematic diagrams of the crystal structures of 2D (RNH3)2SnBr4 perovskites are shown in Fig. 1b. A single metal halide octahedral layer is ∼6.7 Å, so we can infer that two interleaving cation layers occupy spaces of ∼11.4 Å ((C6H13NH3)2SnBr4), ∼17.9 Å ((C8H17NH3)2SnBr4), ∼27.1 Å ((C12H25NH3)2SnBr4), and ∼32.9 Å ((C18H35NH3)2SnBr4).
 | ||
| Fig. 1 (a) Powder X-ray diffraction (PXRD) patterns and (b) schematic diagrams of crystal structures for 2D (RNH3)2SnBr4 perovskites. Scanning electron microscopy (SEM) images of 2D (RNH3)2SnBr4 perovskites: (c) C6, (d) C8, (e) C12, and (f) C18 (for simplicity, we use C6, C8, C12, and C18 to represent (C6H13NH3)2SnBr4, (C8H17NH3)2SnBr4, (C12H25NH3)2SnBr4, and (C18H35NH3)2SnBr4 perovskites, respectively). | ||
Fourier-transform infrared (FTIR) spectroscopy (Fig. S1, ESI†) also proved that the NH3+ characteristic peak at 3100 cm−1 existed in all the 2D (RNH3)2SnBr4 perovskites.33,34 X-Ray photoelectron spectroscopy (XPS) spectra of 2D (C8H17NH3)2SnBr4 perovskites are shown in Fig. S2 (ESI†). An obvious peak at 284 eV was related to C 1s; the peak at 400.9 eV was attributed to N 1s; the peaks located at 485.8 eV and 494.1 eV originated from Sn 3d of Sn2+ ions;26,28 the peak located at 67.5 eV was attributed to Br 3d.35 According to the integrated peak intensities, the elemental N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn:Br ratio of the 2D (C8H17NH3)2SnBr4 perovskites was 4.4:1:5.6. The total amount of nitrogen detected comes from alkylamine molecules from the perovskite structure and the surrounding organic protective shell.36 2D (RNH3)2SnBr4 perovskites with layered morphology were observed in scanning electron microscopy (SEM) (Fig. 1c–f and Fig. S3, ESI†), which agreed well with the results of XRD (Fig. 1a). The elements of nitrogen, tin, and bromide were distributed homogeneously in all the samples (Fig. S4, ESI†). The TEM and HAADF-STEM images in Fig. S5 (ESI†) also indicated the 2D layered structure of (C18H35NH3)2SnBr4 perovskites. The colloidal suspension of 2D (RNH3)2SnBr4 perovskites in hexane showed yellow to orange emission under 365 nm UV light as the alkylammonium ligand varies, among which the (C8H17NH3)2SnBr4 perovskites showed the strongest luminescence (Fig. 2a). The UV-vis absorption, PL excitation, and PL emission spectra of these (RNH3)2SnBr4 perovskites are shown in Fig. 2b. (RNH3)2SnBr4 perovskites with different alkylammonium cations showed absorption and PL peaks at different wavelengths, as summarized in Table 1. All these perovskites show broadband emission with extremely large full width at half maximum (FWHM) (134–138 nm). The [F(R)hν]2versus energy curves are shown in Fig. S6 (ESI†), showing that 2D (RNH3)2SnBr4 perovskites possess direct band gaps of 3.48 eV ((C6H13NH3)2SnBr4), 3.38 eV ((C8H17NH3)2SnBr4), 3.20 eV ((C12H25NH3)2SnBr4) and 3.36 eV ((C18H36NH3)2SnBr4). All the samples have relatively large Stokes shifts, i.e. >260 nm. The shifts of the emission peaks might be attributed to the transient elastic lattice distortions of octahedra which were affected by the different organic cations.37 The 2D (RNH3)2SnBr4 perovskites were further studied using three-dimensional excitation–emission matrix (EEM) fluorescence spectroscopy (Fig. 2c). With the change of the excitation wavelength, the emission peaks showed a little shift, indicating that there were no other impurities or additional energy levels in our samples. Fig. 2d illustrates that, with increasing alkyl chain length, the PL QY is enhanced from 35% ((C6H13NH3)2SnBr4) to 82% ((C8H17NH3)2SnBr4) initially, but then experienced further reduction to 60.2% ((C12H25NH3)2SnBr4) and 50.5% ((C18H35NH3)2SnBr4). The PL QY versus excitation wavelength curves of 2D (C6H13NH3)2SnBr4 (a), (C8H17NH3)2SnBr4 (b), (C12H25NH3)2SnBr4 (c), and (C18H35NH3)2SnBr4 (d) perovskites were also investigated (Fig. S7, ESI†). All these samples showed excitation dependent PL QY, achieving their highest values at 340 nm ((C6H13NH3)2SnBr4, (C8H17NH3)2SnBr4)) and 320 nm ((C12H25NH3)2SnBr4, (C18H35NH3)2SnBr4)).
:Sn:Br ratio of the 2D (C8H17NH3)2SnBr4 perovskites was 4.4:1:5.6. The total amount of nitrogen detected comes from alkylamine molecules from the perovskite structure and the surrounding organic protective shell.36 2D (RNH3)2SnBr4 perovskites with layered morphology were observed in scanning electron microscopy (SEM) (Fig. 1c–f and Fig. S3, ESI†), which agreed well with the results of XRD (Fig. 1a). The elements of nitrogen, tin, and bromide were distributed homogeneously in all the samples (Fig. S4, ESI†). The TEM and HAADF-STEM images in Fig. S5 (ESI†) also indicated the 2D layered structure of (C18H35NH3)2SnBr4 perovskites. The colloidal suspension of 2D (RNH3)2SnBr4 perovskites in hexane showed yellow to orange emission under 365 nm UV light as the alkylammonium ligand varies, among which the (C8H17NH3)2SnBr4 perovskites showed the strongest luminescence (Fig. 2a). The UV-vis absorption, PL excitation, and PL emission spectra of these (RNH3)2SnBr4 perovskites are shown in Fig. 2b. (RNH3)2SnBr4 perovskites with different alkylammonium cations showed absorption and PL peaks at different wavelengths, as summarized in Table 1. All these perovskites show broadband emission with extremely large full width at half maximum (FWHM) (134–138 nm). The [F(R)hν]2versus energy curves are shown in Fig. S6 (ESI†), showing that 2D (RNH3)2SnBr4 perovskites possess direct band gaps of 3.48 eV ((C6H13NH3)2SnBr4), 3.38 eV ((C8H17NH3)2SnBr4), 3.20 eV ((C12H25NH3)2SnBr4) and 3.36 eV ((C18H36NH3)2SnBr4). All the samples have relatively large Stokes shifts, i.e. >260 nm. The shifts of the emission peaks might be attributed to the transient elastic lattice distortions of octahedra which were affected by the different organic cations.37 The 2D (RNH3)2SnBr4 perovskites were further studied using three-dimensional excitation–emission matrix (EEM) fluorescence spectroscopy (Fig. 2c). With the change of the excitation wavelength, the emission peaks showed a little shift, indicating that there were no other impurities or additional energy levels in our samples. Fig. 2d illustrates that, with increasing alkyl chain length, the PL QY is enhanced from 35% ((C6H13NH3)2SnBr4) to 82% ((C8H17NH3)2SnBr4) initially, but then experienced further reduction to 60.2% ((C12H25NH3)2SnBr4) and 50.5% ((C18H35NH3)2SnBr4). The PL QY versus excitation wavelength curves of 2D (C6H13NH3)2SnBr4 (a), (C8H17NH3)2SnBr4 (b), (C12H25NH3)2SnBr4 (c), and (C18H35NH3)2SnBr4 (d) perovskites were also investigated (Fig. S7, ESI†). All these samples showed excitation dependent PL QY, achieving their highest values at 340 nm ((C6H13NH3)2SnBr4, (C8H17NH3)2SnBr4)) and 320 nm ((C12H25NH3)2SnBr4, (C18H35NH3)2SnBr4)).
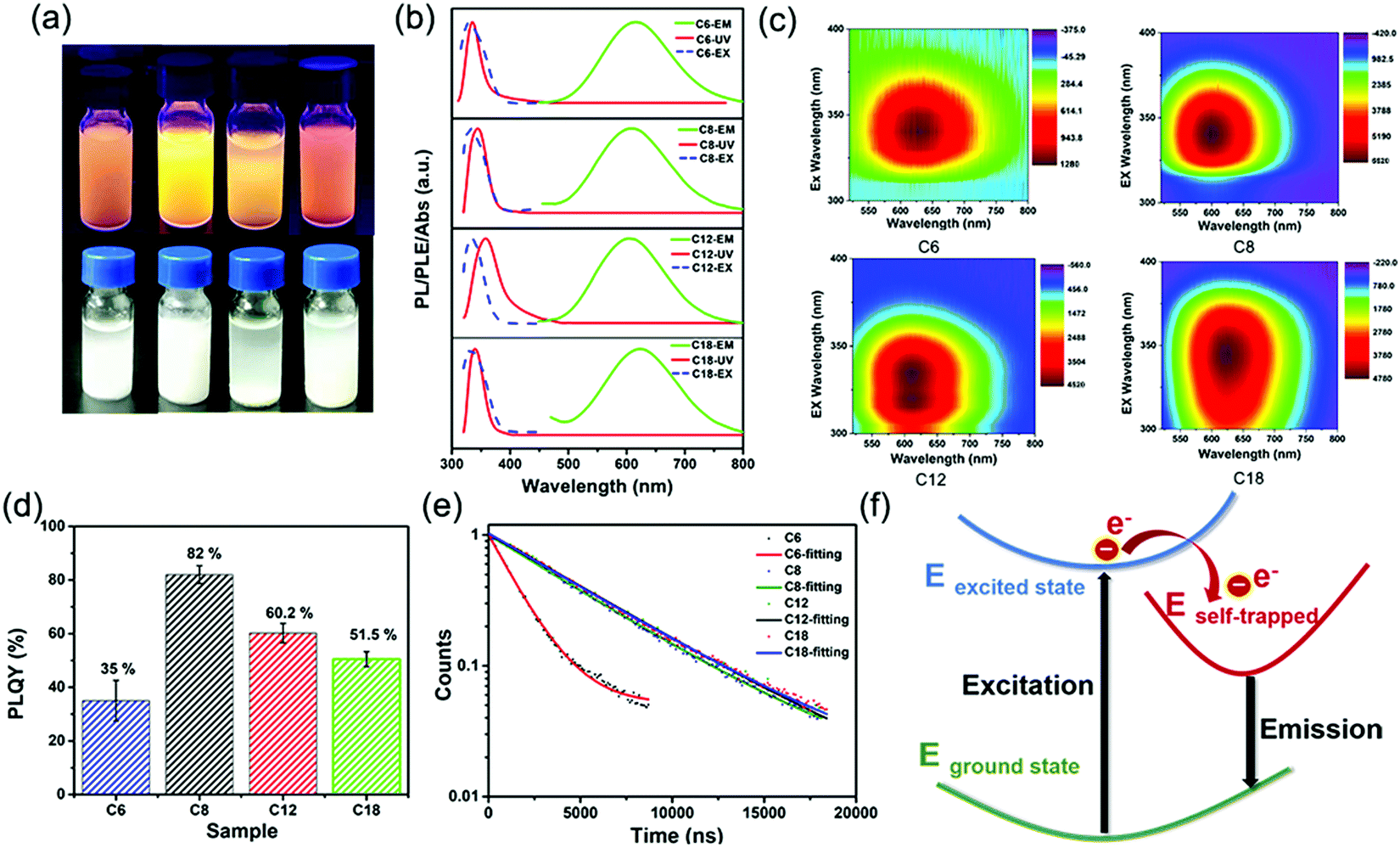 | ||
| Fig. 2 Optical properties of 2D (RNH3)2SnBr4 perovskites: (a) Photographs of the colloidal suspension at room temperature and under UV light. (b) UV-vis absorption, PL emission, and PL excitation spectra. (c) Three-dimensional excitation–emission matrix (EEM) fluorescence spectrum. (d) PL QY and (e) time-resolved PL decay spectra. (f) Schematic of exciton self-trapping in 2D (RNH3)2SnBr4 perovskites (for simplicity, we use C6, C8, C12, and C18 to represent (C6H13NH3)2SnBr4, (C8H17NH3)2SnBr4, (C12H25NH3)2SnBr4, and (C18H35NH3)2SnBr4 perovskites, respectively). | ||
| Samples | Absorption peak (nm) | PL peak (nm) | FWHM (nm) | Stokes shift (meV) | Absolute PL QY (%) | E g (eV) | τ ave (μs) |
|---|---|---|---|---|---|---|---|
| (C6H13NH3)2SnBr4 | 335 | 618 | 136 | 283 | 35.0 | 3.48 | 1.60 |
| (C8H17NH3)2SnBr4 | 344 | 610 | 137 | 266 | 82.0 | 3.38 | 4.99 |
| (C12H25NH3)2SnBr4 | 338 | 603 | 134 | 265 | 60.2 | 3.20 | 5.31 |
| (C18H35NH3)2SnBr4 | 349 | 623 | 138 | 274 | 51.5 | 3.36 | 5.21 |
The time-resolved PL decay curves of 2D (RNH3)2SnBr4 perovskites in Fig. 2e could be fitted by a single exponential function with a long lifetime of 1.60–5.31 μs. To illustrate the internal mechanism between the average PL lifetime and PLQY in various samples, we calculated the radiative (kr = PLQY/τavg) and nonradiative (knr = (1/τavg) − kr) decay rates based on the PLQY and τavg. As shown in Table S1 (ESI†), with increasing kr/knr, the nonradiative pathways were suppressed, and thus the PLQY of (RNH3)2SnBr4 increased.28,38–40 Table S1 (ESI†) shows that (C6H13NH3)2SnBr4 has the lowest kr/knr value of 0.5385. Therefore, we assumed that there were some non-ignorable non-radiative carrier trapping processes, which led to its lowest PLQY.28,41–43 The differences in PLQY and lifetime among these (RNH3)2SnBr4 indicated that the optical properties were influenced by the distortion of the structure.
In order to investigate the mechanism of broadband emission in our two-dimensional (2D) layered tin halide perovskites, we conducted a comprehensive review of 2D perovskites which also showed broadband emission, large Stokes shifts, and long lifetimes.28,44–47 Li et al. reported the fabrication of 2D (RNH3)2SnBr4 perovskites and their density functional theory (DFT) study revealed that these properties originated from a strong phonon coupling in the deformable lattice.28 Multi-layered lead iodide perovskites were investigated via temperature-dependent photoluminescence (PL) spectroscopy and transient absorption (TA) spectroscopy. The results proved that the broad emission was related to the electron–phonon coupling and structural deformation of the ground-state lattice arising from STEs.47 The optical properties obtained in this study were consistent with the literature; therefore, we inferred that the observed broad emission, large Stokes shift, and long lifetimes of our samples stemmed from STEs. As shown in Fig. 2f, when excitons are trapped, the distortion of STEs compared to the ground state broadens the emission. The highly distorted octahedron generates more STEs, resulting in longer PL lifetimes. Fig. S8 (ESI†) shows the stability curves of 2D (RNH3)2SnBr4 perovskites stored under ambient conditions for 30 days. All these samples exhibited good air stability. In particular, the PL QY of (C18H35NH3)2SnBr4 perovskites decreased less than 1% after 30 days. It was obvious that longer alkylammonium chains could protect the [SnBr6]− octahedral luminescence centre more effectively, which in turn enhanced the stability of the 2D (RNH3)2SnBr4 perovskites.29
Similar to other halide perovskites, their halide portion of 2D (RNH3)2SnBr4 perovskites can also be varied to tune the emission positions.48 Various compositions of (C8H17NH3)2SnX4 perovskites were synthesized by simply tuning the dosage of HBr and HI proportions. SEM images (Fig. 3a–c and Fig. S9, ESI†) showed that all the products possessed a 2D layered morphology. XRD patterns (Fig. 3d) of (C8H17NH3)2Sn(2Br/I)4 and (C8H17NH3)2SnI4 perovskites also implied periodic diffraction, which was an indication of a 2D layered structure and thus in agreement with the SEM results. In addition, we found that (003) planes showed a slight shift to lower angles from (C8H17NH3)2SnBr4 to (C8H17NH3)2SnI4 perovskites due to tin-halide bond expansion.27 The FTIR spectra of 2D (RNH3)2SnX4 perovskites are shown in Fig. S10 (ESI†), and it was pertinent to note that increasing I− anion did not affect the position and intensity of the functional groups. Similar to lead halide perovskites, when the bromide ion was gradually replaced by the I− anion, the PLE peak red-shifted from 332 to 370 nm and the PL peak red-shifted from 610 to 655 nm, accompanied by the luminescence change from yellow to red (Fig. 3e).48 The UV-vis absorption spectra of 2D (RNH3)2SnX4 perovskites showed that the absorption peak red-shifted from 344 nm to 370 nm as the I/Br ratio increased. The band gap values were estimated to be 3.38 eV ((C8H17NH3)2SnBr4), 2.89 eV ((C8H17NH3)2Sn(2Br/I)4) and 3.17 eV ((C8H17NH3)2SnI4), respectively (Fig. S11, ESI†). Time-resolved PL decay spectra of (RNH3)2SnX4 perovskites are shown in Fig. 3f. All the spectra were fitted with the single exponential decay function and the average PL lifetimes were as follows: 5 μs ((C8H17NH3)2SnBr4), 2.6 μs ((C8H17NH3)2Sn(2Br/I)4), and 1.41 μs ((C8H17NH3)2SnI4), which further verified their self-trapping emission mechanism. As expected, the PL QYs of these products were (C8H17NH3)2SnBr4 (82%), (C8H17NH3)2Sn(2Br/I)4 (69.2%), and (C8H17NH3)2SnI4 (33%), respectively, following the trend of the PL decay lifetime. The optical parameters of (RNH3)2SnX4 are summarized in Table 2.
 | ||
| Fig. 3 Scanning electron microscopy (SEM) images of C8 (a), C8-2Br/I (b), and C8-I (c) perovskites. (d) Powder X-ray diffraction (PXRD) patterns of (C8H17NH3)2SnX4 perovskites (X = Br, 2Br/I, I). (e) UV-vis absorption, PL emission, and PL excitation spectra of the as-prepared samples. (f) Time-resolved PL decay spectra of (C8H17NH3)2SnX4 perovskites (for simplicity, we use C8, C8-2Br/I and C8-I to represent (C8H17NH3)2SnBr4, (C8H17NH3)2Sn(2Br/I)4 and (C8H17NH3)2SnI4 perovskites, respectively). | ||
| Samples | Absorption peak (nm) | PL peak position (nm) | Absolute PL QY (%) | Stokes shift (nm) | τ ave (μs) |
|---|---|---|---|---|---|
| (C8H17NH3)2SnBr4 | 332 | 610 | 82.0 | 278 | 4.99 |
| (C8H17NH3)2Sn(2Br/I)4 | 363 | 638 | 69.2 | 275 | 2.60 |
| (C8H17NH3)2SnI4 | 370 | 655 | 33.0 | 285 | 1.41 |
Zn2+ doping has been considered as an effective method to achieve highly efficient perovskites.49–51 Here, we introduced Zn2+ ions into 2D (RNH3)2SnX4 (X = Br, I) perovskites for the first time. Fig. 4a and d show the UV-vis absorption, PL excitation, and PL emission spectra of Zn2+-doped (C8H17NH3)2SnBr4 and Zn2+-doped (C8H17NH3)2SnI4 perovskites. After doping, the PL peaks of (C8H17NH3)2SnBr4 and (C8H17NH3)2SnI4 obviously shifted from 610 nm to 580 nm and from 655 nm to 630 nm, respectively. The blue-shift of PL peaks might result from lattice contraction.52 The XRD patterns of Zn2+-doped (RNH3)2SnX4 (Fig. S12, ESI†) showed that, after doping, no new diffraction peaks appeared. In addition, the diffraction peaks slightly shifted to higher angles, indicating that the doping of zinc did not change the crystallinity of the perovskites but only contracted the lattice.53 We included the PL decay of both the undoped sample and doped samples for comparison as shown in Fig. 4b and e. The Zn2+-doped (C8H17NH3)2SnBr4 and Zn2+-doped (C8H17NH3)2SnI4 perovskites could also be fitted by a single exponential function with a long lifetime of 4.4 μs and 1.9 μs, respectively. The corresponding τavg and R2 values are listed in Table S2 (ESI†); compared with undoped samples, the lifetime of doped samples did not show any obvious change. Interestingly, both Zn2+-doped (C8H17NH3)2SnBr4 (82% to 85%) and (C8H17NH3)2SnI4 (33% to 50%) perovskites exhibited higher PL QYs compared with undoped ones, which might have resulted from fewer defects and suppressed nonradiative pathways in the doped perovskites.50,54–56 We calculated the radiative (kr = PLQY/τavg) and nonradiative (knr = (1/τavg) − kr) decay rates of undoped and doped samples based on the PLQY and τavg. The results are summarized in Table S3 (ESI†), and prove that, after zinc-doping, the nonradiative pathways were inhibited.38–40 Furthermore, the alloying significantly improved the stability of (C8H17NH3)2SnBr4 and (C8H17NH3)2SnI4 perovskites via lattice contraction (Fig. 4c).52 The SEM images (Fig. 4f) showed that Zn2+-doped (C8H17NH3)2SnI4 perovskites possessed uniform round sheet morphology with an average size of 690 nm. The real elemental composition of Zn2+/Sn2+ of Zn2+-doped (C8H17NH3)2SnBr4 and (C8H17NH3)2SnI4 perovskites were 3% and 4%, respectively, confirmed by inductively coupled plasma optical emission spectrometry (ICPOES).
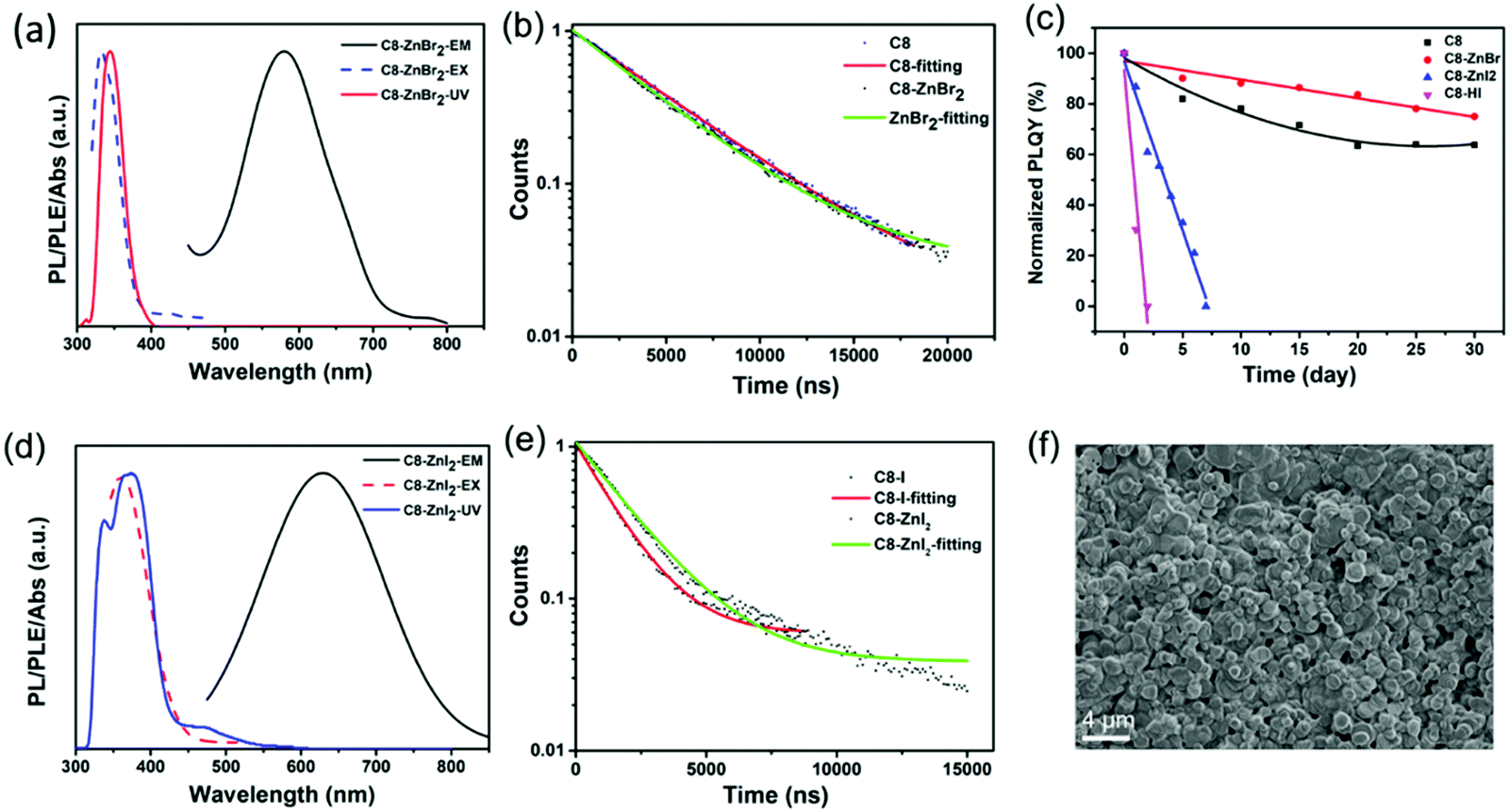 | ||
| Fig. 4 (a and d) UV-vis absorption, PL emission, and PL excitation spectra of the as-prepared C8 and C8-I doped with Zn2+. (b and e) Time-resolved PL decay and fitting curves. (c) Stability curves displayed by the PL QY of undoped and Zn2+-doped samples. (f) Scanning electron microscopy (SEM) images of Zn2+-doped C8-I samples (for simplicity, we use C8, C8-HI, C8-ZnBr2 and C8-ZnI2 to represent (C8H17NH3)2SnBr4, (C8H17NH3)2SnI4, Zn2+-doped (C8H17NH3)2SnBr4 and Zn2+-doped (C8H17NH3)2SnI4 perovskites, respectively). | ||
The Zn2+-doped (C8H17NH3)2SnBr4 perovskite was adopted as a yellow phosphor for UV pumped LED fabrication (Fig. 5). Both Zn2+-doped (C8H17NH3)2SnBr4 in the PS film (Fig. 5a) and the Zn2+-doped (C8H17NH3)2SnBr4 LED device (Fig. 5b) showed bright yellow emission. As shown in Fig. 5d, the CIE coordinates, colour rendering index (CRI) and correlated color temperature (CCT) of the LED were measured to be (0.5060, 0.4542), 75 and 2455 K, respectively. In order to test the stability of the yellow LED, we recorded a series of PL spectral curves by tuning the driving voltage (Fig. 5c). It is worth noting that the PL intensity increased with driving voltage changing from 3 V to 9 V, without an obvious PL peak shift, indicating its good colour stability. These results motivated us to explore further applications including in display backlights.
 | ||
| Fig. 5 (a) Images of Zn2+-doped (C8H17NH3)2SnBr4 yellow phosphor embedded in the PS film under sunlight (top) and 365 nm UV light (bottom). (b) 365 nm UV light pumped yellow LED device. (c) PL spectra of a yellow LED obtained by varying the driving voltage. (d) Chromaticity coordinates of Zn2+-doped (C8H17NH3)2SnBr4 yellow phosphor plotted on the CIE1931 chromaticity chart. | ||
Conclusions
In summary, we have demonstrated the synthesis of the lead-free (C6H13NH3)2SnBr4, (C8H17NH3)2SnBr4, (C12H25NH3)2SnBr4 and (C18H35NH3)2SnBr4 2D perovskites with bright PL emission properties through a facile hot-injection method using the low-cost and environment-friendly hydrobromic acid and stannous 2-ethylhexanoate as the precursors. Among them, (C8H17NH3)2SnBr4 perovskites with broad and bright yellow emission centered at 610 nm achieved the highest PL QY (82%). Interestingly, we found that the length of organic amine cations could strongly affect the emission peak, which might be attributed to the transient elastic lattice distortions. Through the incorporation of Zn2+ ions, the 2D (C8H17NH3)2SnBr4 showed an enhanced stability and improved morphology uniformity. A yellow-emitting LED was fabricated using the Zn2+-doped (C8H17NH3)2SnBr4 perovskite. These 2D perovskites are promising phosphors with potential applications in future solid-state lighting and display technologies.Experiments
Chemicals
1-Octadecene (ODE, Aladdin, GC >90%), oleic acid (OA, Aladdin, AR, 85%), hexylamine (Aladdin, AR, 99%), n-octylamine (Aladdin, AR, 99%), dodecylamine (Aladdin, AR, 98%), oleylamine (Aladdin, AR, 80–90%), hydrobromic acid (HBr, Macklin, AR, 48 wt% in H2O), hydroiodic acid (HI, Aladdin, AR, 55.0–58.0% with ≤1.5% H3PO2), zinc bromide (Macklin, AR), zinc iodide (Aladdin, AR, ≥98%), stannous 2-ethylhexanoate (Aladdin, AR, 95%), n-hexane (Macklin, AR, 97%), and polystyrene (PS, Aladdin) were purchased and used without further purification.Synthesis of (C8H17NH3)2SnX4
To prepare the stannous 2-ethylhexanoate/ODE solution, 370 μL stannous 2-ethylhexanoate was added into 740 μL ODE, and the solution was mixed well under ultrasonication. In a typical synthesis, ODE (20 mL), OA (0.4 mL), n-octylamine (0.4 mL) and HBr (163 μL) were added to a 50 mL three-neck round bottom flask and degassed at 120 °C for 2 h. The temperature was increased to 180 °C under a N2 atmosphere. Then 780 μL of the stannous 2-ethylhexanoate/ODE solution was swiftly injected, and the flask was immersed in an ice bath to stop the reaction after 10 s. The crude solution was dispersed in hexane and centrifuged at 10000 rpm for 2 min, and the supernatant was discarded. Finally, a white precipitate of 2D the (C8H17NH3)2SnBr4 perovskite was obtained with strong yellow fluorescence under UV light. For the synthesis of (C8H17NH3)2Sn(2Br/I)4 or (C8H17NH3)2SnI4, the variable compositions were achieved by controlling the ratio of HBr/HI.
Synthesis of (RNH3)2SnBr4
To synthesize other (RNH3)2SnBr4 perovskites, octylamine was replaced by another organic amine chain with the same molar ratio.Synthesis of Zn2+-doped (C8H17NH3)2SnX4
To prepare Zn2+-doped (C8H17NH3)2SnX4, 10% ZnBr2 or ZnI2 (by the molar ratio of Sn) was added into a mixed solution of ODE (20 mL), OA (0.4 mL), n-octylamine (0.4 mL) and HBr (163 μL). The subsequent steps were the same as in the synthesis of (C8H17NH3)2SnBr4.Fabrication of LEDs
Yellow Zn2+-doped (C8H17NH3)2SnBr4 phosphor was well mixed with 25% polystyrene (PS)–dichloromethane solution. The mixed phosphor of PS paste was dried on a 365 nm ultraviolet LED chip (1 W, 365 nm) in air to fabricate a yellow LED lamp.Characterization
Powder X-ray diffraction (PXRD) was carried out using a Bruker AXS D8 X-ray diffractometer (USA) equipped with monochromated Cu Kα radiation (λ = 1.5418 Å). The diffraction patterns were scanned in the 2θ range of 10–40 degrees with a step size of 0.06 at room temperature. X-Ray photoelectron spectroscopy (XPS) was performed using an achromatic Al Kα source (1486.6 eV) and a double pass cylindrical mirror analyzer (ULVAC-PHI 5000 VersaProbe) (Japan). Fourier transform infrared (FTIR) spectroscopy was conducted using a Fourier transform infrared spectrometer (Nicolet NEXUS 870) (USA). The UV-vis absorption spectra were measured using a Shimadzu UV-3600 Plus spectrophotometer (Japan) under ambient conditions. Photoluminescence (PL) measurements were conducted using a Horiba PTI QuantaMaster 400 steady-state fluorescence system (Japan) under ambient conditions. Scanning electron microscopy (SEM) was performed on a Zeiss ULTRA55 electron microscope. The absolute fluorescence quantum yields (PLQYs) were measured using a Horiba PTI QuantaMaster 400 steady-state fluorescence system with an integrated sphere and double-checked with a Hamamatsu Photonics Quantaurus-QY (model: C11347-11) under ambient conditions. Transmission electron microscopy (TEM) was carried out on an FEI TECNAI G2 F20 and an electron microscope operating at 200 kV. The available spectral resolution is about 0.1 nm. Time-resolved PL emission decay curves were collected at room temperature and detected using a low temperature time-resolved fluorescence system (Edinburgh, FLS920) (UK) with the samples being prepared by depositing different perovskites on glass substrates directly. Elemental concentrations of Zn2+/Sn2+ were analyzed via inductively coupled plasma optical emission spectrometry (ICP-OES) using a Skyray® ICP-3000 instrument.Author contributions
Y. L. synthesized the samples, conducted the measurements, and wrote the first draft of the manuscript. A. W. helped with the measurement of the samples and co-drafted the manuscript. J. W., C. W., W. M. and Z. L. revised the manuscript. G. H and S. S. helped with the measurement of the PL decay. J. S. and W. L. helped with the ICP measurements. Z. D. supervised the project. All authors analysed the data and contributed to the discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (22075129) and the Natural Science Foundation of Jiangsu Province (Grant No. BK20180339 and BZ2018008).Notes and references
- Z. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS.
- H. Huang, A. S. Susha, S. V. Kershaw, T. F. Hung and A. L. Rogach, Adv. Sci., 2015, 2, 1500194 CrossRef PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- W. Liu, Q. Lin, H. Li, K. Wu, I. Robel, J. M. Pietryga and V. I. Klimov, J. Am. Chem. Soc., 2016, 138, 14954–14961 CrossRef CAS PubMed.
- X. Yuan, S. Ji, M. C. De Siena, L. Fei, Z. Zhao, Y. Wang, H. Li, J. Zhao and D. R. Gamelin, Chem. Mater., 2017, 29, 8003–8011 CrossRef CAS.
- K. P. Marshall, R. I. Walton and R. A. Hatton, J. Mater. Chem. A, 2015, 3, 11631–11640 RSC.
- C. Huo, B. Cai, Z. Yuan, B. Ma and H. Zeng, Small Methods, 2017, 1, 1600018 CrossRef.
- T. C. Jellicoe, J. M. Richter, H. F. J. Glass, M. Tabachnyk, R. Brady, S. E. Dutton, A. Rao, R. H. Friend, D. Credgington, N. C. Greenham and M. L. Böhm, J. Am. Chem. Soc., 2016, 138, 2941–2944 CrossRef CAS PubMed.
- B. Yang, F. Hong, J. Chen, Y. Tang, L. Yang, Y. Sang, X. Xia, J. Guo, H. He, S. Yang, W. Deng and K. Han, Angew. Chem., Int. Ed., 2019, 58, 2278–2283 CrossRef CAS PubMed.
- B. Yang, X. Mao, F. Hong, W. Meng, Y. Tang, X. Xia, S. Yang, W. Deng and K. Han, J. Am. Chem. Soc., 2018, 140, 17001–17006 CrossRef CAS PubMed.
- S. Liu, B. Yang, J. Chen, D. Wei, D. Zheng, Q. Kong, W. Deng and K. Han, Angew. Chem., Int. Ed., 2020, 59, 21925–21929 CrossRef CAS PubMed.
- W. Yang, F. Igbari, Y. Lou, Z. Wang and L. Liao, Adv. Energy Mater., 2020, 10, 1902584 CrossRef CAS.
- G. Xing, M. H. Kumar, W. K. Chong, X. Liu, Y. Cai, H. Ding, M. Asta, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Adv. Mater., 2016, 28, 8191–8196 CrossRef CAS PubMed.
- H. Fang, S. Adjokatse, S. Shao, J. Even and M. A. Loi, Nat. Commun., 2018, 9, 243 CrossRef PubMed.
- F. Wang, J. Ma, F. Xie, L. Li, J. Chen, J. Fan and N. Zhao, Adv. Funct. Mater., 2016, 26, 3417–3423 CrossRef CAS.
- H. Liang, F. Yuan, A. Johnston, C. Gao, H. Choubisa, Y. Gao, Y. Wang, L. K. Sagar, B. Sun, P. Li, G. Bappi, B. Chen, J. Li, Y. Wang, Y. Dong, D. Ma, Y. Gao, Y. Liu, M. Yuan, M. I. Saidaminov, S. Hoogland, Z.-H. Lu and E. H. Sargent, Adv. Sci., 2020, 7, 1903213 CrossRef CAS PubMed.
- C. Gao, Y. Jiang, C. Sun, J. Han, T. He, Y. Huang, K. Yao, M. Han, X. Wang, Y. Wang, Y. Gao, Y. Liu, M. Yuan and H. Liang, ACS Photonics, 2020, 7, 1915–1922 CrossRef CAS.
- A. Wang, X. Yan, M. Zhang, S. Sun, M. Yang, W. Shen, P. Wang and Z. Deng, Chem. Mater., 2016, 28, 8132–8140 CrossRef CAS.
- Z. Tan, J. Li, C. Zhang, Z. Li, Q. Hu, Z. Xiao, T. Kamiya, H. Hosono, G. Niu, E. Lifshitz, Y. Cheng and J. Tang, Adv. Funct. Mater., 2018, 28, 1801131 CrossRef.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- L. Lanzetta, J. M. Marin-Beloqui, I. Sanchez-Molina, D. Ding and S. A. Haque, ACS Energy Lett., 2017, 2, 1662–1668 CrossRef CAS.
- P. Fu, M. Huang, Y. Shang, N. Yu, H. Zhou, Y. Zhang, S. Chen, J. Gong and Z. Ning, ACS Appl. Mater. Interfaces, 2018, 10, 34363–34369 CrossRef CAS PubMed.
- X. Zhang, C. Wang, Y. Zhang, X. Zhang, S. Wang, M. Lu, H. Cui, S. V. Kershaw, W. W. Yu and A. L. Rogach, ACS Energy Lett., 2019, 4, 242–248 CrossRef CAS.
- A. Wang, Y. Guo, Z. Zhou, X. Niu, Y. Wang, F. Muhammad, H. Li, T. Zhang, J. Wang, S. Nie and Z. Deng, Chem. Sci., 2019, 10, 4573–4579 RSC.
- L. Hou, Y. Zhu, J. Zhu and C. Li, J. Phys. Chem. C, 2019, 123, 31279–31285 CrossRef CAS.
- F. M. Li, Y. Xie, Y. Hu, M. Long, Y. Zhang, J. B. Xu, M. Qin, X. Lu and M. Liu, ACS Energy Lett., 2020, 5, 1422–1429 CrossRef CAS.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, Angew. Chem., Int. Ed., 2018, 57, 9083–9087 CrossRef CAS PubMed.
- C. C. Stoumpos, D. H. Cao, D. J. Clark, J. Young, J. M. Rondinelli, J. I. Jang, J. T. Hupp and M. G. Kanatzidis, Chem. Mater., 2016, 28, 2852–2867 CrossRef CAS.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS PubMed.
- S. Mourdikoudis and L. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- W. Xu and T. Wang, Langmuir, 2017, 33, 82–90 CrossRef CAS PubMed.
- Y. Dang, Y. Zhou, X. Liu, D. Ju, S. Xia, H. Xia and X. Tao, Angew. Chem., Int. Ed., 2016, 55, 3447–3450 CrossRef CAS PubMed.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- C. Quarti, N. Marchal and D. Beljonne, J. Phys. Chem. Lett., 2018, 9, 3416–3424 CrossRef CAS PubMed.
- B. Yang, J. Chen, F. Hong, X. Mao, K. Zheng, S. Yang, Y. Li, T. Pullerits, W. Deng and K. Han, Angew. Chem., Int. Ed., 2017, 56, 12471–12475 CrossRef CAS PubMed.
- B. Yang and K. Han, Acc. Chem. Res., 2019, 52, 3188–3198 CrossRef CAS PubMed.
- B. Yang, J. Chen, S. Yang, F. Hong, L. Sun, P. Han, T. Pullerits, W. Deng and K. Han, Angew. Chem., Int. Ed., 2018, 130, 5457–5461 CrossRef.
- J. Tong, J. Wu, W. Shen, Y. Zhang, Y. Liu, T. Zhang, S. Nie, Z. Deng, J. Tong, J. Wu, W. Shen, Y. Zhang, Y. Liu, T. Zhang, S. Nie and Z. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 9317–9325 CrossRef CAS PubMed.
- G. Li, J. Huang, H. Zhu, Y. Li, J. Tang and Y. Jiang, Chem. Mater., 2018, 30, 6099–6107 CrossRef CAS.
- Z. Liu, Y. Zhang, Y. Fan, Z. Chen, Z. Tang, J. Zhao, Y. Lv, J. Lin, X. Guo, J. Zhang and X. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 13053–13061 CrossRef CAS PubMed.
- M. D. Smith and H. I. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS PubMed.
- L. Mao, Y. Wu, C. C. Stoumpos, B. Traore, C. Katan, J. Even, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 11956–11963 CrossRef CAS PubMed.
- K. Thirumal, W. K. Chong, W. Xie, R. Ganguly, S. K. Muduli, M. Sherburne, M. Asta, S. Mhaisalkar, T. C. Sum, H. S. Soo and N. Mathews, Chem. Mater., 2017, 29, 3947–3953 CrossRef CAS.
- W. Paritmongkol, E. R. Powers, N. S. Dahod and W. A. Tisdale, J. Phys. Chem. Lett., 2020, 11, 8565–8572 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- M. Que, W. Chen, P. Chen, J. Liu, X. Yin, B. Gao and W. Que, Chem. Phys., 2019, 517, 80–84 CrossRef CAS.
- J. Li, J. Chen, L. Xu, S. Liu, S. Lan, X. Li and J. Song, Mater. Chem. Front., 2020, 4, 1444–1453 RSC.
- R. Ren, Z. Wang, X. Meng, C. Xu, J. Qiao, W. Sun and K. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 23959–23967 CrossRef CAS PubMed.
- X. Shen, Y. Zhang, S. V. Kershaw, T. Li, C. Wang, X. Zhang, W. Wang, D. Li, Y. Wang, M. Lu, L. Zhang, C. Sun, D. Zhao, G. Qin, X. Bai, W. W. Yu and A. L. Rogach, Nano Lett., 2019, 19, 1552–1559 CrossRef CAS.
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005–8011 CrossRef CAS PubMed.
- F. Li, Y. Liu, H. Wang, Q. Zhan, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 8546–8554 CrossRef CAS.
- W. Zhao, D. Yang, Z. Yang and S. Liu, Mater. Today Energy, 2017, 5, 205–213 CrossRef.
- W. van der Stam, J. J. Geuchies, T. Altantzis, K. H. W. van den Bos, J. D. Meeldijk, S. Van Aert, S. Bals, D. Vanmaekelbergh and C. de Mello Donega, J. Am. Chem. Soc., 2017, 139, 4087–4097 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S12 and Tables S1–S3. CCDC. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00845a |
| ‡ These two authors contributed equally to this work. |
| § Current address: Institute of Advanced Materials (IAM), Nanjing Tech University (NJ Tech), 5 Xinmofan Road, Nanjing 210009, P. R. China. |
| This journal is © The Royal Society of Chemistry 2021 |