
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInduction of cell cycle arrest and apoptosis by copper complex Cu(SBCM)2 towards oestrogen-receptor positive MCF-7 breast cancer cells†
Jhi Biau Foo *ab,
Li Shan Nga,
Ji Hui Lima,
Pau Xien Tana,
Yan Zhi Lora,
Jason Siau Ee Loob,
May Lee Lowc,
Lee Chin Chand,
Chaw Yee Behe,
Sze Wei Leongd,
Latifah Saiful Yazangh,
Yin Sim Tor*f and
Chee Wun Howa
*ab,
Li Shan Nga,
Ji Hui Lima,
Pau Xien Tana,
Yan Zhi Lora,
Jason Siau Ee Loob,
May Lee Lowc,
Lee Chin Chand,
Chaw Yee Behe,
Sze Wei Leongd,
Latifah Saiful Yazangh,
Yin Sim Tor*f and
Chee Wun Howa
aFaculty of Pharmacy, MAHSA University, Jalan SP2, Bandar Saujana Putra, 42610 Jenjarom, Kuala Langat, Selangor, Malaysia. E-mail: foojhibiau@gmail.com
bSchool of Pharmacy, Faculty of Health & Medical Sciences, Taylor's University, No. 1 Jalan Taylor's, 47500 Subang Jaya, Selangor Darul Ehsan, Malaysia. E-mail: JhiBiau.Foo@taylors.edu.my
cDepartment of Pharmaceutical Chemistry, School of Pharmacy, International Medical University, No. 126, Jalan Jalil Perkasa 19, 57000 Bukit Jalil, Kuala Lumpur, Malaysia
dVirology Lab 1, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia (UPM), 43400 Serdang, Selangor, Malaysia
eLaboratory of Vaccines and Immunotherapeutics, Institute of Bioscience, Universiti Putra Malaysia (UPM), 43400 Serdang, Selangor, Malaysia
fSchool of Biosciences, Faculty of Health & Medical Sciences, Taylor's University, No. 1 Jalan Taylor's, 47500 Subang Jaya, Selangor Darul Ehsan, Malaysia. E-mail: YinSim.Tor@taylors.edu.my
gLaboratory of Molecular Biomedicine, Institute of Bioscience, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia
hDepartment of Biomedical Sciences, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia (UPM), 43400 Serdang, Selangor, Malaysia
First published on 11th June 2019
Abstract
Copper complexes have the potential to be developed as targeted therapy for cancer because cancer cells take up larger amounts of copper than normal cells. Copper complex Cu(SBCM)2 has been reported to induce cell cycle arrest and apoptosis towards triple-negative breast cancer cells. Nevertheless, its effect towards other breast cancer subtypes has not been explored. Therefore, the present study was conducted to investigate the effect of Cu(SBCM)2 towards oestrogen-receptor positive MCF-7 breast cancer cells. Growth inhibition of Cu(SBCM)2 towards MCF-7 and human non-cancerous MCF-10A breast cells was determined by MTT assay. Morphological changes of Cu(SBCM)2-treated-MCF-7 cells were observed under an inverted microscope. Annexin V/PI apoptosis assay and cell cycle analysis were evaluated by flow cytometry. The expression of wild-type p53 protein was evaluated by Western blot analysis. The intracellular ROS levels of MCF-7 treated with Cu(SBCM)2 were detected using DCFH-DA under a fluorescence microscope. The cells were then co-treated with Cu(SBCM)2 and antioxidants to evaluate the involvement of ROS in the cytotoxicity of Cu(SBCM)2. Docking studies of Cu(SBCM)2 with DNA, DNA topoisomerase I, and human ribonucleotide reductase were also performed. The growth of MCF-7 cells was inhibited by Cu(SBCM)2 in a dose-dependent manner with less toxicity towards MCF-10A cells. It was found that Cu(SBCM)2 induced G2/M cell cycle arrest and apoptosis in MCF-7 cells, possibly via a p53 pathway. Induction of intracellular ROS was not detected in MCF-7 cells. Interestingly, antioxidants enhance the cytotoxicity of Cu(SBCM)2 towards MCF-7 cells. DNA topoisomerase I may be the most likely target that accounts for the cytotoxicity of Cu(SBCM)2.
1. Introduction
Cancer is a polygenic and multifactorial complex series of disease characterised by an uncontrolled proliferation of cells.1,2 For the past 50 years, drug treatment for oestrogen-receptor (ER) positive breast cancer has shown great clinical advances and has served as a paradigm for the development of cancer targeted therapies. ER positive is the major subtype of breast cancer, and it has been proven that five years of therapy with tamoxifen reduces the recurrence of breast cancer and improves the overall survival of women with early-stage ER positive breast cancer.3,4Despite the standard regimen with tamoxifen, long term side effects of tamoxifen have been reported. In both Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) trial and the trials of 5 years of tamoxifen versus no treatment, tamoxifen was reported to increase the incidence of developing endometrial cancer in postmenopausal women who have not had a hysterectomy before trial entry.5 This could be due to the off target effects of tamoxifen towards the organ expressing oestrogen receptors. Therefore, it is crucial to discover new strategy to treat oestrogen receptors breast cancer.
Copper is an essential component needed in appropriate amounts for multiple biological processes within the body. However, abnormal elevated levels of copper can be found in cancer tissues in comparison to normal cells.6 Based on this discovery, our group is developing copper complex to deliver anticancer agent into cancer cells.7 The increased uptake of copper-anticancer agent complex by cancer cell will increase the selectivity and activity of the anticancer agent. Our previous publication reported that S-benzyldithiocarbazate and 3-acetylcoumarin [Cu(SBCM)2] induced cell cycle arrest and apoptosis towards MDA-MB-231 triple-negative breast cancer cell.8 Nevertheless, its effect towards other breast cancer subtype has not been established. Therefore, the present study was conducted to investigate the effect of Cu(SBCM)2 towards oestrogen-receptor positive MCF-7 breast cancer cells.
2. Material and methods
2.1 Chemicals
Dimethyl sulfoxide (DMSO), ethanol, acetonitrile and pentane were purchased from Friedemann Schmidt (Frankfort, Germany). 3-Acetylcoumarin, dichlorodihydrofluorescein diacetate (DCFH-DA), α-tocopherol (vitamin E), N-acetylcysteine (NAC), hydrogen peroxide (H2O2), doxorubicin, propidium iodide, RNAse A, bromophenol blue and glycerol were purchased from Sigma-Aldrich (St Louis, MO, USA). Copper(II) acetatemonohydrate was purchased from HmbG Chemicals (Hamburg, Germany). Phenol-red-free RPMI-1640 with L-glutamine, bovine serum albumen (BSA), dulbecco Modified Eagle Medium (DMEM) F12 and Chemi-Lumi One L were purchased from Nacalai Tesque (Kyoto, Japan). Foetal bovine serum, trypsin–EDTA (1×) and penicillin–streptomycin (100×) were purchased from PAA Laboratories (Pasching, Austria). MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was purchased from PhytoTechnology Laboratories (Kansas, USA). Sodium deoxycholate, Triton-X 100 and sodium chloride (NaCl) were purchased from Merck (Darmstadt, Germany). Phenylmethanesulfonyl fluoride (PMSF) and protease inhibitor cocktails were purchased from Calbiochem (San Diego, CA, USA). Sodium dodecyl sulphate (SDS), tris-base, glycine, acrylamide, bisacrylamide, ammonium persulfate (APS), tetramethylethylenediamine (TEMED), 10% Tween-20, Bradford reagent, 2-mercaptoethanol, extra thick blotting paper and pre-stained protein marker were purchased from Bio-Rad (California, USA). Immobilon-FL polyvinylidene fluoride (PVDF) membrane with 0.45 μm pore size was purchased from Millipore (Bedford, MA, USA). Primary mouse antibodies anti-p53 (DO-1: sc-126), anti-beta-actin (sc-47778) and horseradish peroxidase-conjugated anti-mouse (sc-2005) secondary antibodies were purchased from Santa Cruz Biotechnology (CA, USA). Epidermal growth factor (EGF), hydrocortisol, insulin and horse serum were purchased from Life Technologies (Rockville, Maryland). Annexin-V-FITC kits were purchased from BD Bioscience (Singapore).2.2 Synthesis of copper complex Cu(SBCM)2
Cu(SBCM)2 was prepared and characterised as previously prescribed (Fig. 1).7 Briefly, S-benzyldithiocarbazate was reacted with 3-acetylcoumarin. The mixture was heated for 2–3 hours and allowed to stand for a few hours at 4 °C to afford SBCM-H. Cu(OAc)2·H2O was then added to the SBCM-H solution. The reaction mixture was stirred overnight and concentrated. The precipitate was filtered, washed with pentane and dried under vacuum to afford Cu(SBCM)2 (Fig. 1). Elemental analysis for C38H30CuN4S4O4: calcd. C 57.16, H 3.79, N 7.02; found C 56.53, H 3.80, N 7.02. ESI-MS: m/z = [M + K]+ calcd. 836.02, found 836.01; [M + Na]+ calcd. 820.04, found 820.03; [M + H]+ calcd. 798.06, found 798.05. IR: ν (cm−1) = 1726 (s) 1565 (m) 986 (s) 965 (s) 853 (w). The difference between the theoretical and calculated is less than 1%, indicating a high purity of our copper complex Cu(SBCM)2.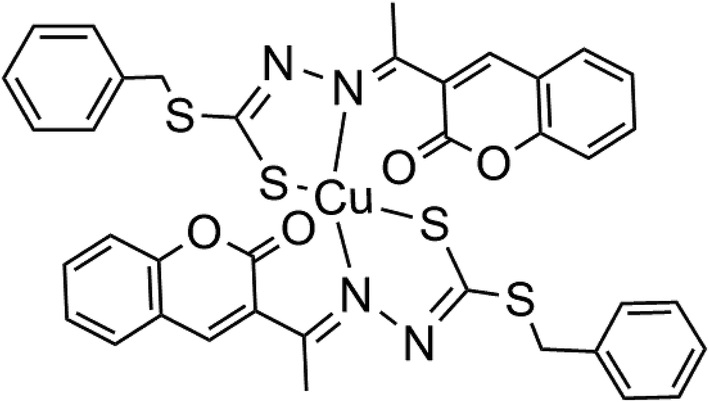 | ||
| Fig. 1 Copper complex synthesised with S-benzyldithiocarbazate and 3-acetylcoumarin [Cu(SBCM)2]. | ||
2.3 Cell culture
The human oestrogen-receptor positive MCF-7 breast cancer, triple-negative MDA-MB-231 breast cancer and non-cancerous MCF-10A cell lines were purchased from American Type and Culture Collection (ATCC, Manassas, VA, USA). MCF-7 and MDA-MB-231 breast cancer cells were grown in RPMI 1640, supplemented with 10% foetal bovine serum and 1% penicillin–streptomycin (complete growth culture media). MCF-10A cells were cultured in DMEM/F12 supplemented with 10% FBS, 20 ng mL−1 epidermal growth factor, 0.5 mg mL−1 hydrocortisone, 100 ng mL−1 cholera toxin and 10 μg mL−1 insulin.2.4 Determination of cell growth inhibition
MCF-7 cells and MCF-10A were seeded in 96-well plates with 5000 cells per well in 100 μL of complete growth culture media, followed by incubation at 37 °C (5% CO2 and 95% air) for 24 hours to allow cell attachment. The cells were then treated with Cu(SBCM)2 or tamoxifen for 72 hours. Negative control cells treated with 0.3% DMSO alone was also included. At the end of experiment, MTT (20 μL, 5 mg mL−1 in PBS) was added into each well and the plate was incubated for 3 hours. Excess MTT was then aspirated and the formazan crystals formed were dissolved by 150 μL of DMSO. The absorbance, which was proportional to cell viability, was measured at 570 nm and a reference wavelength of 630 nm by using ELx800™ Absorbance Microplate Reader (BioTek Instruments Inc., Vermont, USA). Cell viability was calculated by using formula shown below: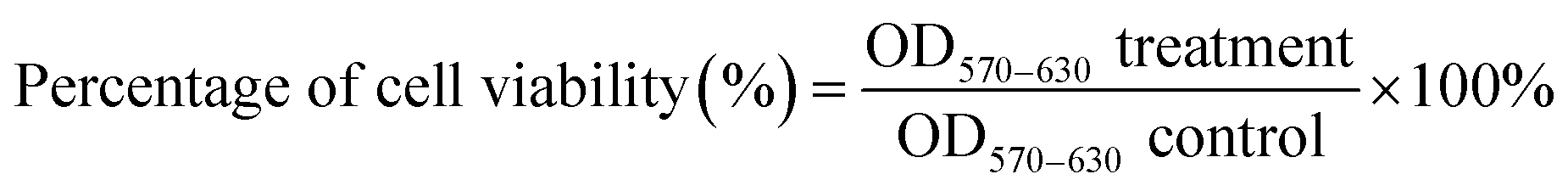
A graph of percentage of cell growth versus concentration of Cu(SBCM)2 was plotted. The concentration of Cu(SBCM)2 which inhibited 50% of cellular growth was determined. For the selective index (SI), the value was calculated by comparing the IC50 value of Cu(SBCM)2 in MCF-10A cell line against the IC50 value of Cu(SBCM)2 in MCF-7 cancer cell lines.9
2.5 Morphological study
MCF-7 cells were seeded in 6-well plates at 1.3 × 105 cells per well in 3 mL of complete growth medium, followed by incubation at 37 °C (5% CO2 and 95% air) for 24 hours and treated with Cu(SMCM)2. Control cells treated with 0.3% DMSO alone were also included. The morphological changes were observed and the images were captured under an inverted light microscope (Olympus, PA, USA) at 0, 24, 48 and 72 hours. The same spot of cells was marked and captured.2.6 Confirmation of apoptosis by Annexin V/PI flow cytometry assay
MCF-7 cells were seeded in a 25 cm2 culture flask with a population of 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells and incubated overnight. Cells were then treated with Cu(SBCM)2 at 20, 30 and 40 μM. Control cells (0.3% of DMSO as negative control and 10% DMSO as positive control) were also included in this experiment. After 48 hours, cells were washed twice with PBS (phosphate buffer saline) and were later detached by trypsinisation. Cold PBS was then used to wash cells and cells were re-suspended in 500 μL of 1× binding buffer (cell concentration 1 × 106 cells per mL). Next, 5 μL of Annexin-V-FITC and 5 μL of PI (propidium iodide) were added to cells. Cells were gently vortexed, incubated on ice for 10 minutes and analysed by NovoCyte® flow cytometry (ACEA Biosciences, San Diego, CA).
000 cells and incubated overnight. Cells were then treated with Cu(SBCM)2 at 20, 30 and 40 μM. Control cells (0.3% of DMSO as negative control and 10% DMSO as positive control) were also included in this experiment. After 48 hours, cells were washed twice with PBS (phosphate buffer saline) and were later detached by trypsinisation. Cold PBS was then used to wash cells and cells were re-suspended in 500 μL of 1× binding buffer (cell concentration 1 × 106 cells per mL). Next, 5 μL of Annexin-V-FITC and 5 μL of PI (propidium iodide) were added to cells. Cells were gently vortexed, incubated on ice for 10 minutes and analysed by NovoCyte® flow cytometry (ACEA Biosciences, San Diego, CA).
2.7 Cell cycle analysis
MCF-7 cells were seeded in 25 cm2 cell culture flasks with 300000 cells per flask, which allowed for cells attachment overnight. Cells were subsequently treated with 20, 30 and 40 μM of Cu(SBCM)2 and incubated at 37 °C for 48 hours. All attached and floating cells were washed with cold PBS, fixed gently in 70% cold ethanol and stored at −20 °C overnight. Cold PBS was used to wash cells, followed by cell staining with RNase and PI for 30 minutes to ease cell cycle detection. Cell cycle distribution was evaluated using NovoCyte® flow cytometry from ACEA Biosciences (San Diego, CA).
2.8 Western blot analysis
Western blot was performed according to the previous publication.8 Briefly, 300000 MCF-7 cells were seeded and treated with Cu(SBCM)2 for 24 and 48 hours. The cells were then lysed with RIPA buffer. The protein was separated by gel electrophoresis and transferred to PVDF membrane. The membrane was then blocked with 3% bovine serum albumen and probed overnight with mouse anti-p53 (1:2000) and anti-beta actin (1:10000). Next, the membrane was incubated with horseradish peroxidase-conjugated goat anti-mouse secondary antibodies (1:20000) for 1 hour at room temperature. Detection was performed with Chemi Lumi-One L Detection Reagent and ChemiDoc MP System.
2.9 Determination of intracellular ROS in Cu(SBCM)2-treated cells
MCF-7 and MDA-MB-231 breast cancer cells were seeded in 6-well plates at a density of 1.3 × 105 cells per well in 3 mL of complete growth culture media and incubated for 24 hours at a temperature of 37 °C under 5% CO2 to allow attachment of cells. Next, the spent media were removed and the cells were pre-treated with 10 μM DCFH-DA in complete culture media for 1 hour. Upon the removal of excess DCFH-DA, the cells were washed with phosphate-buffered saline (PBS) twice and followed by treatment with hydrogen peroxide or Cu(SBCM)2 in culture media for 30 minutes. 0.3% DMSO alone, as a negative control, was also used to treat the cells. The samples were then examined and images were captured using the Eclipse Ti2 inverted fluorescent microscope (Nikon, Shanghai, China) with excitation and emission wavelengths of 485 nm and 535 nm.2.10 Evaluation of the effects of antioxidants on Cu(SBCM)2-induced cell death in MCF-7
MCF-7 and MDA-MB-231 breast cancer cells were seeded in 96-well plates at a density of 5000 cells per well and incubated at a temperature of 37 °C under 5% CO2 and 95% air for 24 hours. Following cell attachment, cells were treated with doxorubicin or Cu(SBCM)2 or co-treated with 100 μM α-tocopherol or 100 μM N-acetylcysteine (NAC) for 48 hours. Each well was then added with 20 μL MTT (5 mg mL−1 in PBS) and followed by incubation for 3 hours. Next, the extra MTT was removed by aspiration and the purple formazan crystals formed were dissolved in 200 μL of DMSO. The cell viability, which was proportional to the amount of formazan crystals formed, was determined by measuring the absorbance at 570 nm and at background of 630 nm (reference wavelength) by employing ELx800™ Absorbance Microplate Reader (BioTek Instruments Inc., Vermont, USA). Cell viability was calculated by using formula as mentioned above and a graph of percentage of cell viability against Cu(SBCM)2 concentration were plotted from the experiments.2.11 Molecular docking studies
To explore the interactions of Cu(SBCM)2 with various biological macromolecules, we performed docking studies of Cu(SBCM)2 with DNA (PDB code 1BNA), DNA topoisomerase I (PDB code 1SC7), and human ribonucleotide reductase (PDB code 4X3V). All three targets have previously been shown to potentially account for the antitumor activity of thiosemicarbazone metal complexes.10,11The metal complex was first constructed using ChemDraw 17 and subjected to geometry optimisation using the AM1 forcefield. The target structures were downloaded from the Protein Data Bank and prepared using the Protein Preparation module implemented in Schrodinger Maestro.12 Hydrogen atoms were added, missing side chains were modelled using Prime,13 appropriate protonation states were assigned at pH 7, and hydrogen bond networks were optimised. The target structures were then subjected to restrained energy minimisation before docking. Docking calculations were performed using Glide SP,14 with the Induced Fit Protocol used for protein targets to allow for side chain flexibility.15 Default settings were used. The docking grid was centred on the major and minor groove for DNA or the co-crystallized ligand for DNA topoisomerase I and ribonucleotide reductase respectively. A total of 20 binding poses were initially generated for each target, with the final pose chosen based on the docking score and comparison with the co-crystallised ligands.
2.12 Statistical analysis
Statistical analysis was conducted using the Statistical Package for Social Science (SPSS) version 21.0. Data were expressed as mean ± standard deviation (mean ± SD). Results were analysed by one-way analysis of variance (ANOVA), followed by Dunnett's post hoc test. A difference is considered to be significant at p < 0.05.3. Results
3.1 Cu(SBCM)2 inhibited the growth of MCF-7 cells but less toxic towards MCF-10A
The growth of MCF-7 cells was not inhibited by SBCM up to 60 μM as compared to control. However, treatment with Cu(SBCM)2 significantly inhibited (p < 0.05) the growth of MCF-7 cells in a dose-dependent manner (Fig. 2A). The growth of MCF-7 cells appeared to be completely inhibited at approximately 27 μM and cell death was observed at higher concentration of Cu(SBCM)2.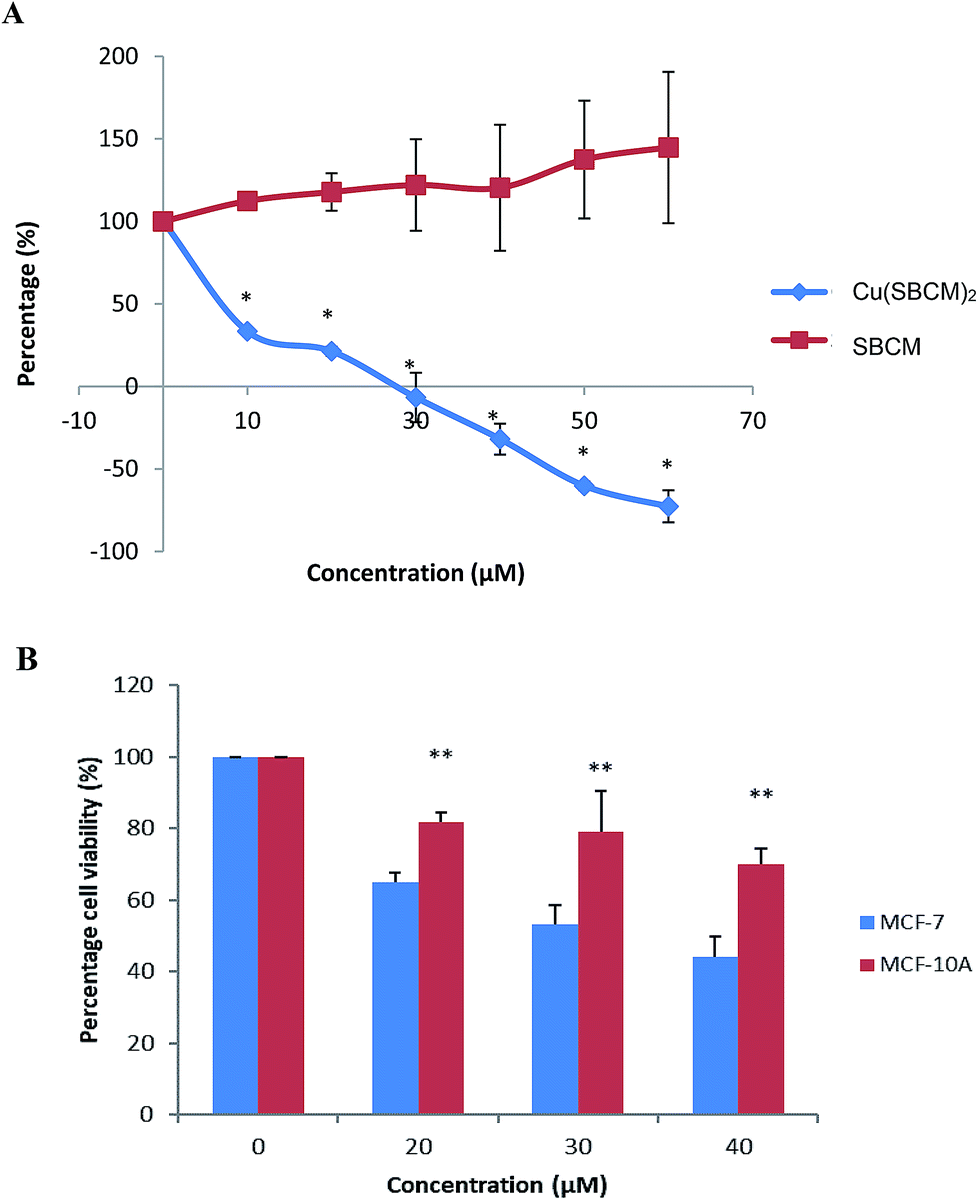 | ||
| Fig. 2 (A) Effect of SBCM and Cu(SBCM)2 and on the growth of MCF-7 breast cancer cells as determined by MTT assay. * significantly different from the control SBCM at the same concentration (p < 0.05). (B) Cell viability of MCF-7 and MCF-10A cells after 48 hours exposure with Cu(SBCM)2. ** indicates p < 0.05 when compared between MCF-7 and MCF-10A. Each data point represents the mean of three independent experiments ± SD. | ||
The viability of MCF-7 cells treated with 20, 30 and 40 μM of Cu(SBCM)2 decreased to 65.0 ± 2.5%, 53.1 ± 5.3% and 42.3 ± 5.%, respectively as compared to control. As for non-cancerous MCF-10A breast cells, similar trend was observed. Nevertheless, the viability of MCF-10A cells was 81.8 ± 2.8%, 79.0 ± 11.3% and 69.9 ± 4.5% for concentrations 20, 30 and 40 μM, respectively, in which the viabilities were significantly higher than the one in MCF-7 cancer cell lines (Fig. 2B). The IC50 value of Cu(SBCM)2 towards MCF-7 and MCF-10A were 32 and 60 μM, respectively, at 48 hours. The calculated selective index of Cu(SBCM)2 in MCF-7 is 1.9, indicating that Cu(SBCM)2 is more selective towards cancer cells than non-cancerous cells.
3.2 Cu(SBCM)2 induced apoptosis towards MCF-7 cells
The population of attached cells in the control, 10 μM and 20 μM increased from 0 to 72 hours. For the cells treated with Cu(SBCM)2 at higher concentration, increased in the cell population was noted until 24 hours and the cells detached from the substratum (Fig. 3). Cu(SBCM)2-treated MCF-7 cells demonstrated characteristics of apoptosis such as cellular shrinkage at 48 hours. Nevertheless, other features of apoptosis such as chromatin condensation and formation of apoptotic bodies were not observed (Fig. 4). To confirm our observation that Cu(SBCM)2 induced apoptosis towards MCF-7 cells, the Cu(SBCM)2-treated-MCF-7 cells were then stained with Annexin V-FITC/PI and analysed using flow cytometry. Results indicated (Fig. 5A and B) that cells underwent early and late apoptosis. The percentage of early apoptosis cells treated with Cu(SBCM)2 at 20, 30 and 40 μM was 6.70 ± 1.81%, 15.45 ± 5.72% and 30.79 ± 11.75%, respectively, in comparison to control (4.22 ± 1.23%). Similar trend was also noted for late apoptosis population. The percentage of late apoptosis cells treated with Cu(SBCM)2 at 20, 30 and 40 μM was 18.74 ± 4.26%, 24.95 ± 3.30% and 27.88 ± 1.60%, respectively, in comparison to control (10.59 ± 1.77%). | ||
| Fig. 3 The population of MCF-7 cells treated with Cu(SBCM)2 as observed under inverted light microscope at the same spot for 72 hours (100×). | ||
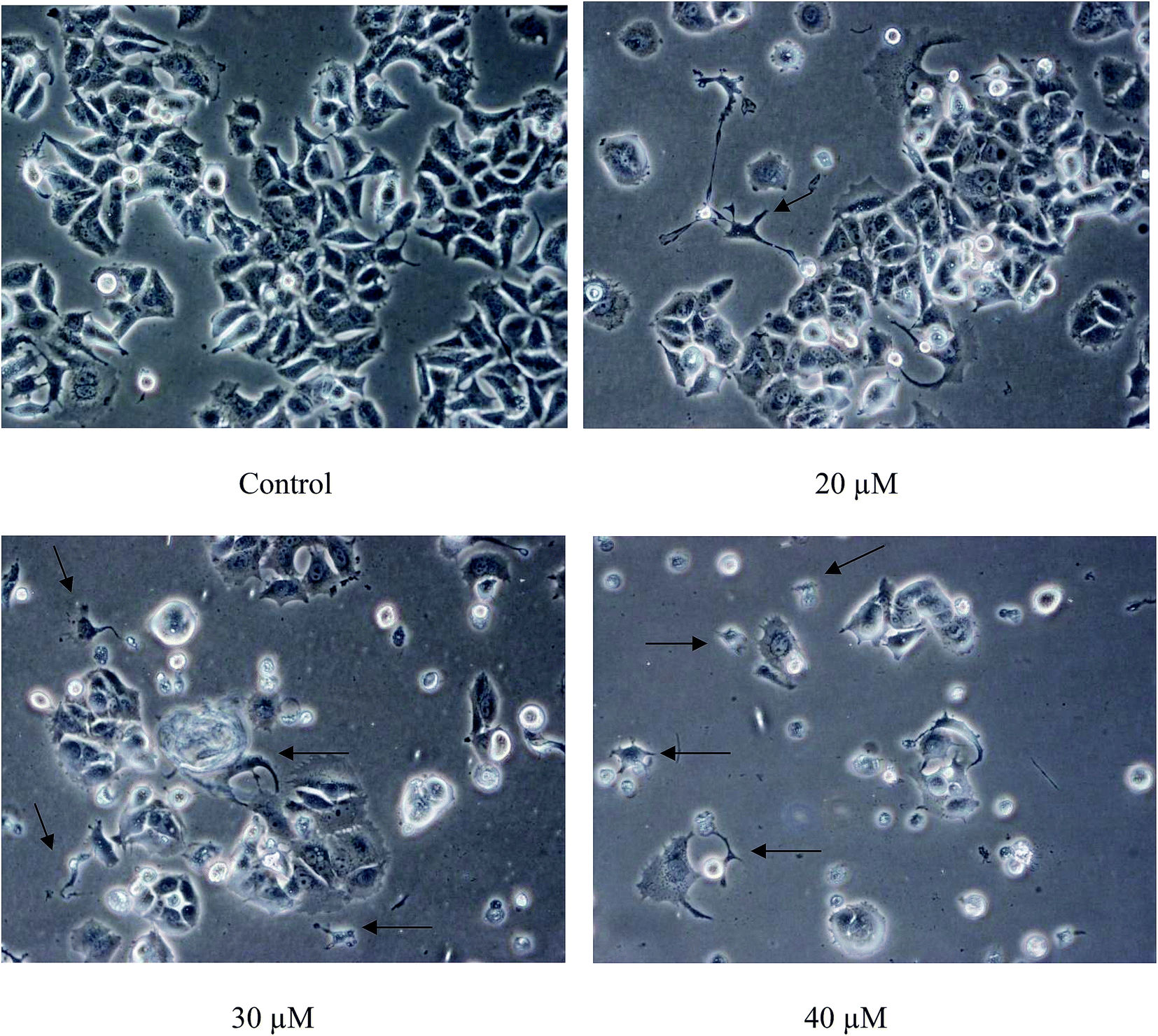 | ||
| Fig. 4 Close-up view of MCF-7 cells treated with Cu(SBCM)2 at 48 hours (200×). Cellular shrinkage was observed in Cu(SBCM)2-treated MCF-7 cells (arrows). | ||
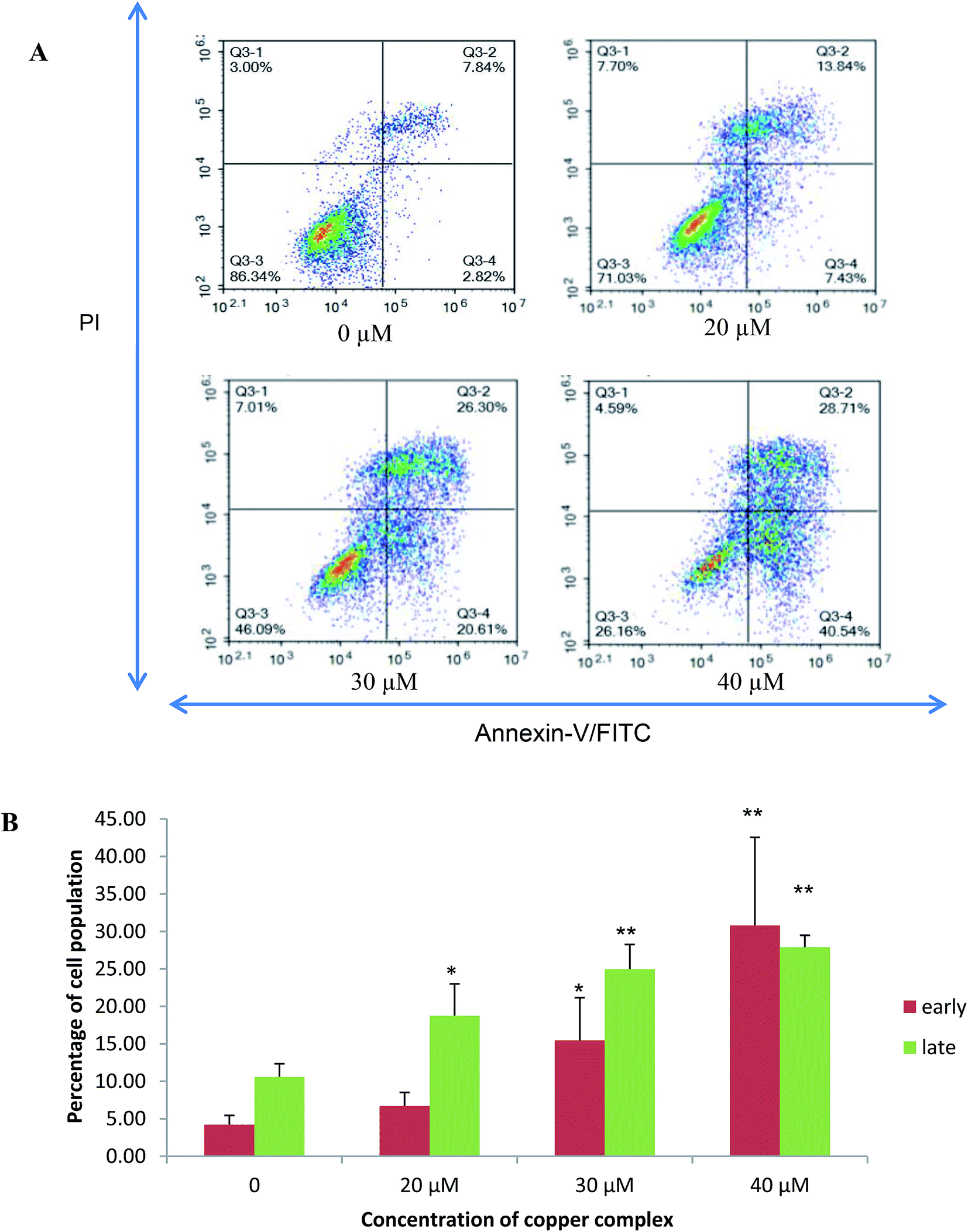 | ||
| Fig. 5 Annexin-V/-FITC/PI Flow cytometry analysis of MCF-7 breast cancer cells treated with 20, 30 and 40 μM of Cu(SBCM)2 for 48 hours. (A) These figures are from representative experiments carried out at least two independent test. The viable cells, early apoptotic and necrotic/secondary necrotic cells was represented by the lower left quadrant (Annexin-V−/PI−), lower right (Annexin-V+/PI−) and upper (Annexin-V+/PI+) quadrant, respectively. (B) Percentage of early and late apoptotic cells after Cu(SBCM)2 administration in MCF-7 cells. Data were mean ± SD of two independent experiments. (* indicates p < 0.05 when compared with control, ** indicates p < 0.001 when compared with control). | ||
3.3 Cu(SBCM)2 induced G2/M cell cycle arrest in MCF-7 breast cancer cells
MCF-7 cells treated with 20, 30 and 40 μM of Cu(SBCM)2 resulted in a drop in S phase population from 26.73 ± 3.36% to 21.94 ± 2.31%, 24.36 ± 6.87% and 22.62 ± 4.40%, respectively (Fig. 6). On the other hand, there was increments in G2/M phase in comparison to control in which the percentage was increased from 14.60 ± 1.12% to 23.57 ± 4.61%, 19.47 ± 2.75% and 18.77 ± 1.47% at 20, 30 and 40 μM of Cu(SBCM)2, respectively. | ||
| Fig. 6 Flow cytometry analysis of MCF-7 breast cancer cells treated with 20, 30 and 40 μM of Cu(SBCM)2 for 48 hours. (A) Representative figures of cell cycle distribution (G0/G1, S, and G2/M) showing accumulation of Cu(SBCM)2-treated cells in G2/M stage. Results shown are one of the two independent experiments conducted. (B) Bar chart showing accumulation of MCF-7 breast cancer cells in each cell cycle phase after Cu(SBCM)2 administration. Data were mean ± SD of two independent experiments. (* indicates p < 0.05 when compared with control). | ||
3.4 Cu(SBCM)2 up-regulated p53 protein in MCF-7 cells
In the present study, treatment of MCF-7 cells with 20, 30 and 40 μM of Cu(SBCM)2 for 24 hours significantly up-regulated the expression of wild-type p53 by 3.7, 1.6 and 1.7 folds, respectively. Similar trend was also observed when the cells were treated for 48 hours (Fig. 7).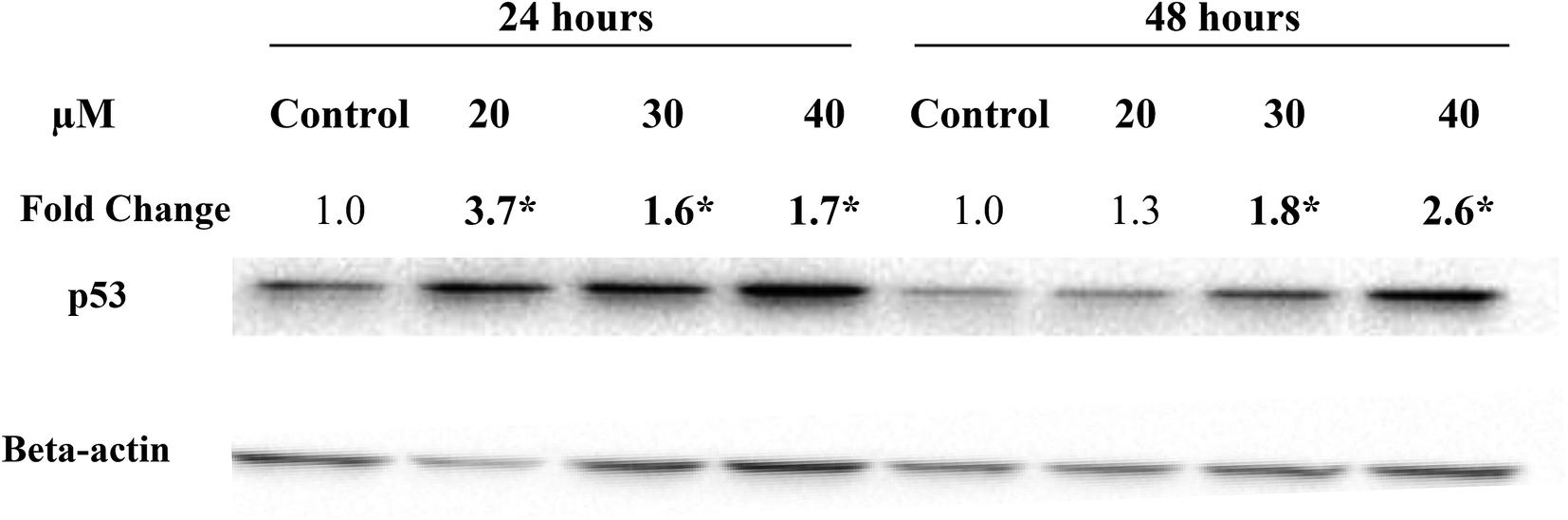 | ||
| Fig. 7 Expression levels of wild-type p53 protein in MCF-7 cells following treatment with Cu(SBCM)2. Fold change was normalised against beta-actin and compared to the control. Each data point represents the mean of three independent experiments ± SD. * significantly different from the control (p < 0.05). | ||
3.5 Effects of Cu(SBCM)2 on the intracellular ROS levels
Green fluorescence cell indicates the presence of intracellular ROS and the intensity increases with the levels of intracellular ROS. From Fig. 8, green fluorescence can be clearly seen in positive control (H2O2) of both cell lines. This indicates that H2O2 was able to induce ROS formation in both cell lines and subsequently, detected by DCFH-DA. Nonetheless, when both cell lines were exposed to concentrations of 20, 30 and 40 μM of Cu(SBCM)2 for 30 minutes, there was no increased intensity in green fluorescence for both cell lines, as compared to the control.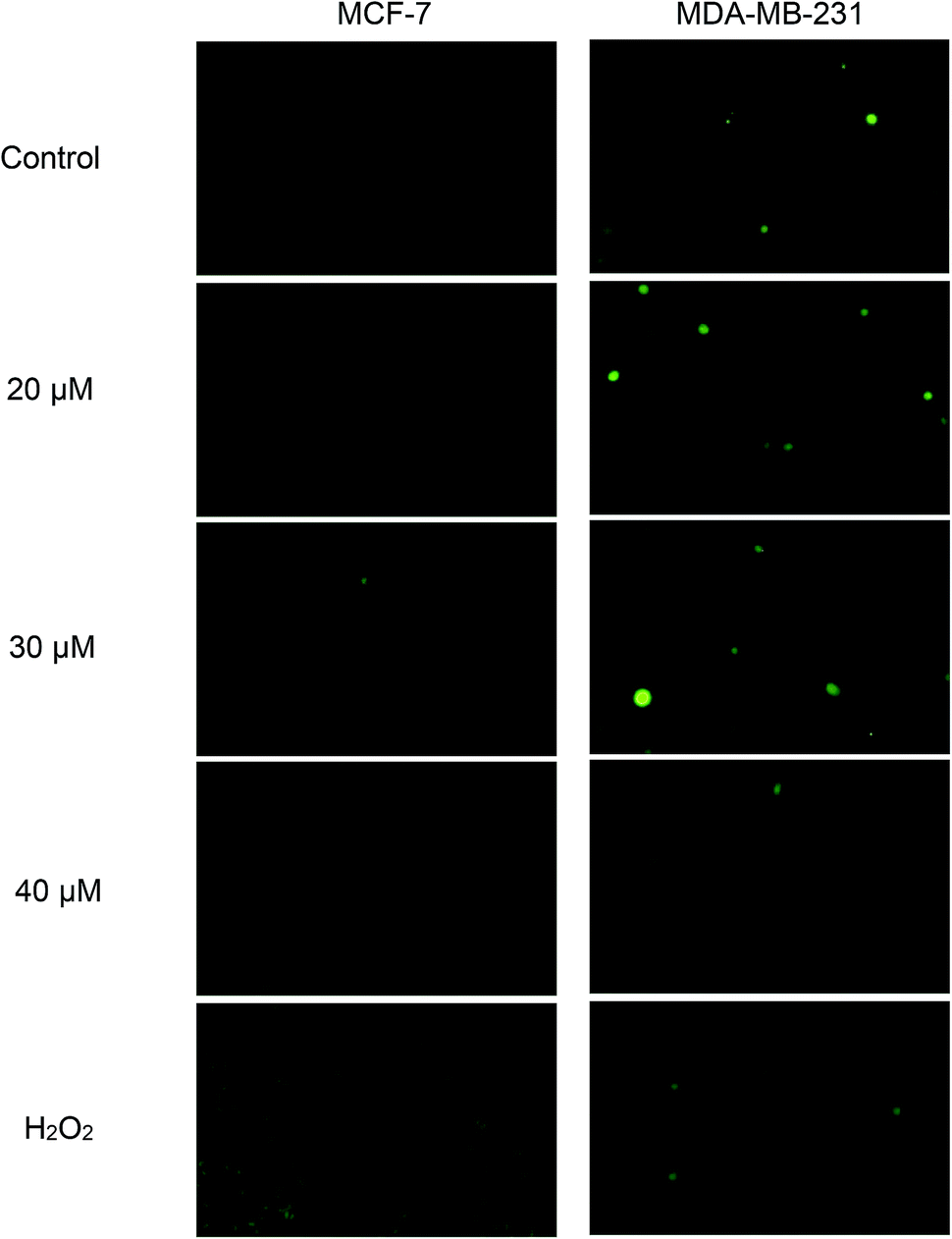 | ||
| Fig. 8 Effects of Cu(SBCM)2 on the production of ROS in breast cancer cells. After pretreatment with 10 μM DCFDA for 1 hour, cells were treated with Cu(SBCM)2 for 30 minutes and examined under fluorescence microscope. Green fluorescence indicates the presence of intracellular ROS. | ||
3.6 NAC enhanced the cytotoxicity of Cu(SBCM)2 in MCF-7 cells
Viability of MCF-7 cells after treatment with Cu(SBCM)2 alone at 20 μM, 30 μM and 40 μM for 48 hours was 55%, 53% and 42%, respectively. Co-treatment of MCF-7 cells with 100 μM NAC and Cu(SBCM)2 significantly decreased the cell viability to 29%, 33% and 24% (Fig. 9A). In contrast, co-treatment of MDA-MB-231 cells with antioxidants and Cu(SBCM)2 did not affect the cell viability as compared to Cu(SBCM)2 alone (Fig. 9B).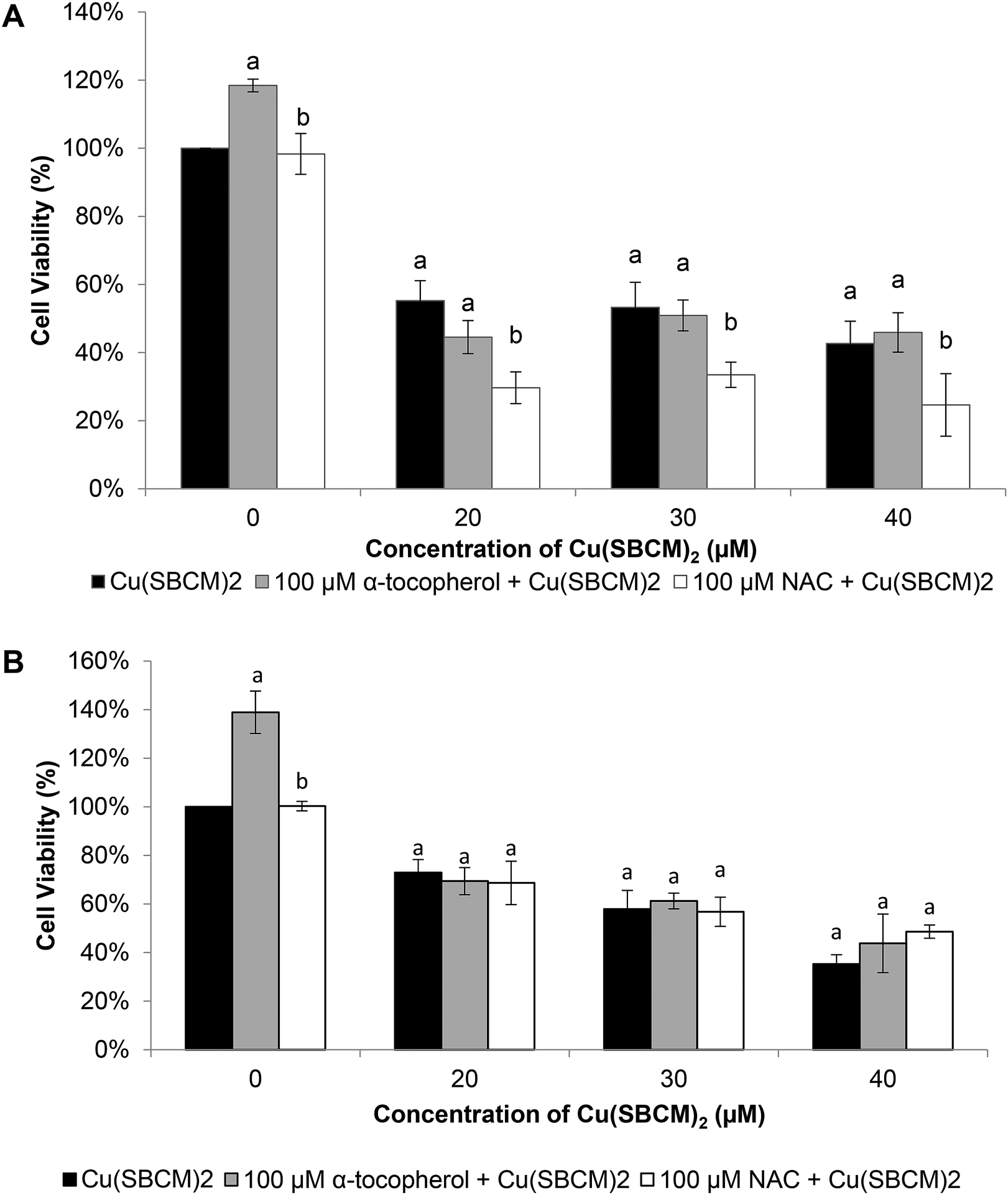 | ||
| Fig. 9 Effects of antioxidants on the cytotoxicity of Cu(SBCM)2. (A) and (B) represent the percentage of viability of MCF-7 and MDA-MB-231 cells after treatment with Cu(SBCM)2 and α-tocopherol or NAC for 48 hours. Each data point represents the mean of triplicates ± SD. a and b in the same concentration were considered significantly difference (p < 0.05). | ||
3.7 Cu(SBCM)2 has a good binding towards DNA topoisomerase I
Of the three targets investigated, Cu(SBCM)2 showed the greatest affinity for DNA topoisomerase I with a calculated binding free energy of −12.04 kcal mol−1. This was in comparison to a calculated binding free energy of −7.52 kcal mol−1 for ribonucleotide reductase and −5.22 kcal mol−1 for DNA. We then performed the molecular docking in reference to MJ238, which is a known DNA topoisomerase I inhibitor that was co-crystallised in the 1SC7 crystal structure (Fig. 10A). The predicted binding free energy of Cu(SBCM)2 towards DNA topoisomerase I (−12.04 kcal mol−1) is lower than the predicted binding free energy of MJ238 towards DNA topoisomerase I (−8.22 kcal mol−1), indicating that our Cu(SBCM)2 binds stronger to the target. Molecular docking also revealed that Cu(SBCM)2 intercalates the DNA base pair whenever the DNA is unwind (Fig. 10B). The key interaction in Cu(SBCM)2 binding to DNA topoisomerase I is demonstrated in Fig. 10C.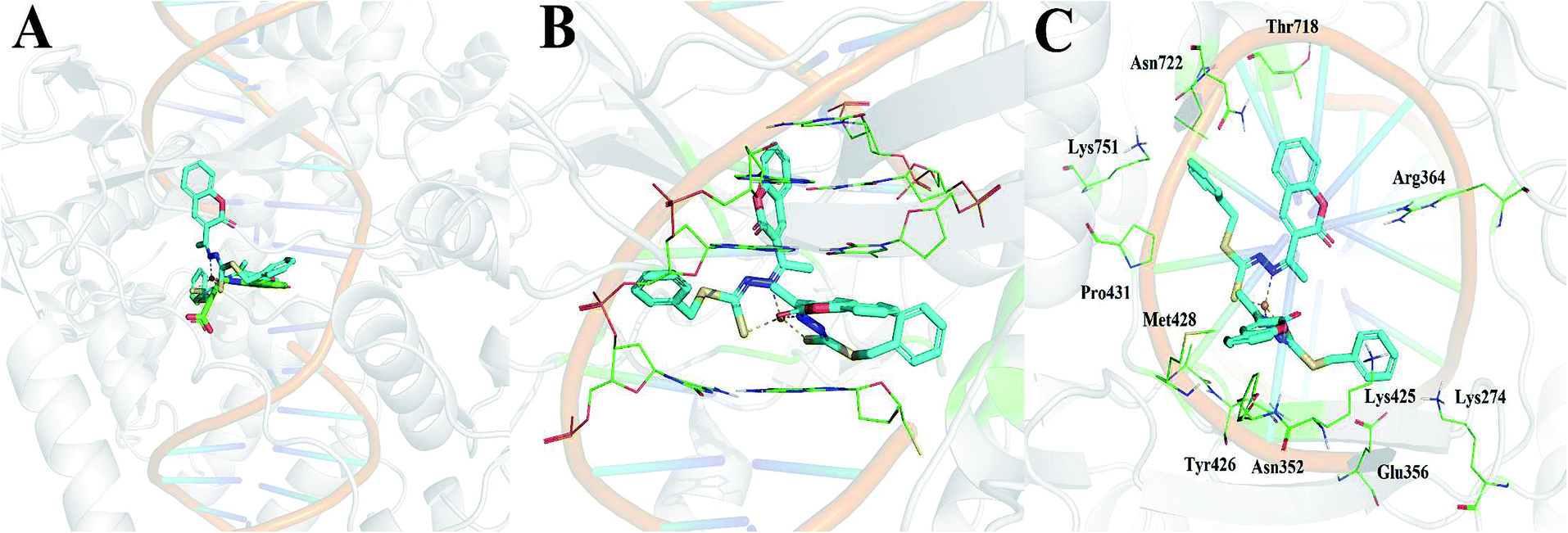 | ||
| Fig. 10 (A) Binding of Cu(SBCM)2 (cyan) in comparison to the co-crystallised DNA topoisomerase inhibitor MJ238 (green). (B) Intercalation of Cu(SBCM)2 with base pairs. (C) Key interactions in Cu(SBCM)2 binding to DNA topoisomerase I. | ||
4. Discussion
In the present study, SBCM-H Schiff base did not inhibit the growth of MCF-7 breast cancer cells. However, the conjugation of SBCM-H with copper [Cu(SBCM)2] enhanced the inhibitory properties of the Schiff base towards MCF-7 breast cancer cells, probably due to the increased uptake of Schiff base by the cells (Fig. 2A). Apart from that, Cu(SBCM)2 was also evaluated towards non-cancerous MCF-10A breast cells. It was noted that Cu(SBCM)2 demonstrated stronger inhibition towards MCF-7 than MCF-10A (Fig. 2B), suggesting that Cu(SBCM)2 is more toxic towards cancer cells but less toxic towards normal cells. This warrants the safety profile of Cu(SBCM)2 for future drug development.Observation of cells under an inverted light microscope revealed that Cu(SBCM)2 inhibited the growth (Fig. 3) and apoptosis (Fig. 4) towards MCF-7 breast cancer cells. Cells treated with Cu(SBCM)2 showed cellular shrinkage (Fig. 4) postulating that Cu(SBCM)2 induced apoptosis in MCF-7 cells, which was then further confirmed by Annexin-V/PI flow cytometry analysis (Fig. 5). Some other features of apoptosis such as membrane blebbing, chromatin condensation, nuclear fragmentation and formation of apoptotic bodies were not seen in MCF-7 cells under the inverted light microscope after treatment of Cu(SBCM)2 due to caspase-3 deficiency in the MCF-7 cells. Caspase-3 is important for typical biochemical and morphological changes of cells undergoing apoptosis such as DNA nuclear fragmentation.16 Since Cu(SBCM)2 induced apoptosis in the caspase-3-deficient-MCF-7 cells, it is suggested that the induction of apoptosis via caspase-3-independent pathways. Although the exact mechanism of apoptosis in MCF-7 breast cancer cells is yet to be determined, but there are many different mechanisms of apoptosis that could possibly be induced by Cu(SBCM)2 towards MCF-7 breast cancer cells.17
Apart from apoptosis, Cu(SBCM)2 was also found to induce G2/M phase cell cycle arrest towards MCF-7 breast cancer cells (Fig. 6). The induction of apoptosis and cell cycle arrest are very likely due to the increased expression of wild-type p53 by Cu(SBCM)2 in MCF-7 cells (Fig. 7). Apoptosis may be induced by either p53-dependent or p53-independent pathways.18 In the present study, the wild-type p53 of MCF-7 cells was significantly up-regulated by 20, 30 and 40 μM of Cu(SBCM)2 suggesting the involvement of p53 pathway in Cu(SBCM)2 induced apoptosis. In response to a stress signal, p53 up-regulation may result in extensive apoptosis.19 The significantly increased expression levels of wild-type p53 in Cu(SBCM)2-treated-MCF-7 cells indicating that Cu(SBCM)2 is a stressor that induces p53 accumulation which in turn induces apoptosis in the MCF-7 cells.
Cu(SBCM)2 exhibited a high redox potential which might be related to its cytotoxic effects.7 The high redox potential demonstrated by Cu(SBCM)2 suggests that in the Cu(SBCM)2 complex, Cu(II) is easily reduced to Cu(I). Cu(I) has been recognised to take part in Fenton-like reactions that generate ROS which can subsequently cause damage to functional biomolecules, such as protein, lipids, DNA and RNA in the cells.20 This proposes that the cytotoxicity of Cu(SBCM)2 could be due to the production of intracellular ROS as copper complex of other variants have found that they induced cell death by induction of high oxidative stress.21–25 Unexpectedly, based on the DCFH-DA assay results (Fig. 8), Cu(SBCM)2 did not increase the ROS production in both MCF-7 and MDA-MB-231 cells. However, further exploration is required as DCFH-DA is not sensitive to all types of reactive species. Hence, there is a possibility that the cytotoxic effect of Cu(SBCM)2 towards both cell lines was induced by reactive species that cannot be detected by DCFH-DA such as H2O2, reactive nitrogen species, O2˙−, and lipid peroxides.26,27 This requires further investigation and confirmation.
To assess the role of ROS in the cytotoxicity of Cu(SBCM)2, we co-treated the cancer cells with antioxidants. Surprisingly, instead of blocking the reduction in cell viability, NAC synergise the inhibition effect of Cu(SBCM)2 towards MCF-7 cells but had no influence on the viability of MDA-MB-231 treated with Cu(SBCM)2 (Fig. 9). Breast cancer is a heterogeneous disease and each breast cancer subtype is responsive towards different anticancer therapy. Different response towards the co-treatment with Cu(SBCM)2 and α-tocopherol or NAC between MCF-7 and MDA-MB-231 breast cancer cell lines suggesting a new approach for the treatment of oestrogen receptor positive breast cancer. However, the mechanism of the synergistic anticancer effects between the antioxidants and Cu(SBCM)2 is yet to be established. Similar results was reported in which co-incubation of NAC enhanced the killing effects of their copper complexes towards cancer cells and they proposed that thiol-induced redox cycling of Cu(II) complex happens outside the cells. Upon the reduction of Cu(II) to Cu(I) by NAC, the Cu(I) complex forms H2O2 readily by dismutation of O2− in the extracellular fluid. H2O2 then diffuses into the cells where more ROS formation is induced.28
Our molecular docking studies indicated that DNA topoisomerase I may be the most likely target that accounts for the antitumor activity shown by Cu(SBCM)2. The predicted binding mode of Cu(SBCM)2 to DNA topoisomerase I was found to be remarkably similar to the co-crystallised indenoisoquinoline MJ238 (Fig. 10A),29 whereby the ligand intercalates at the site of DNA cleavage between the +1 and −1 base pairs through π–π stacking with the coumarin moiety (Fig. 10B). While the predicted binding mode of Cu(SBCM)2 lacks the bidentate interaction of MJ238 with the two nitrogens on the Arg364 side chain, it is stabilised by an additional π–π stacking interaction between the phenyl ring and the cystosine base (Fig. 10C). While experimental validation will be needed to confirm these findings, they provide a useful starting point to further explore potential structure–activity relationships.
5. Conclusion
Cu(SBCM)2 induced cell cycle arrest and apoptosis in caspase-3 deficient MCF-7 breast cancer cells possibly via the activation of wild-type p53 with less toxic towards non-cancerous MCF-10A cells. Antioxidants seems enhanced the cytotoxicity of Cu(SBCM)2 towards MCF-7 cells. DNA topoisomerase I may be the most likely target that accounts for the antitumor activity shown by Cu(SBCM)2. Based on our previously published and current data, Cu(SBCM)2 has a great potential to be developed to target the breast cancer cells.Author contributions
JBF, YST, CWH and LSY contributed to the design of the study and interpretation of data. LSY allowed us to use laboratory facilities for present study. MLL synthesised and characterized the copper complex. For data acquisition, JBF, JHL, YZL and LSN contributed to the MTT assay, morphological study and Western blot analysis. LSN, PXT, CYB, SWL and LCC contributed to Flow cytometry and ROS study. JSEL performed the molecular docking study. This manuscript is prepared and critically revised by JBF, YST, CWH and LSY.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The present work was financially supported by MAHSA Research Grant (RP89-02/16) and Taylor's University Internal Research Grant Scheme (TRGS/ERFS/1/2018/SBS/034). Special thanks are delivered to the Laboratory of Vaccine and Immunotherapeutics, Institute of Bioscience and Virology Lab 1, Faculty of Biotechnology and Biomolecular Sciences from Universiti Putra Malaysia for their support and assistance in completing this study.References
- Y. Ichikawa, M. Ghanefar, M. Bayeva, R. Wu, A. Khechaduri, S. V. N. Prasad, R. K. Mutharasan, T. J. Naik and H. Ardehali, J. Clin. Invest., 2014, 124, 617–630 CrossRef CAS PubMed
.
- H. H. Wong, C. Barton, G. Acton, R. McLeod and S. Halford, Eur. J. Cancer, 2016, 66, 9–16 CrossRef CAS PubMed
- R. Jasmine and S. A. Kumar, Indian J. Pharmacol., 2016, 48, 99–100 CrossRef CAS PubMed
- S. Kaushik, H. Shyam, R. Sharma and A. K. Balapure, Indian J. Pharmacol., 2016, 48, 637–642 CrossRef CAS PubMed
- C. Davies, H. Pan, J. Godwin, R. Gray, R. Arriagada, V. Raina, M. Abraham, V. H. Medeiros Alencar, A. Badran, X. Bonfill, J. Bradbury, M. Clarke, R. Collins, S. R. Davis, A. Delmestri, J. F. Forbes, P. Haddad, M.-F. Hou, M. Inbar, H. Khaled, J. Kielanowska, W.-H. Kwan, B. S. Mathew, I. Mittra, B. Müller, A. Nicolucci, O. Peralta, F. Pernas, L. Petruzelka, T. Pienkowski, R. Radhika, B. Rajan, M. T. Rubach, S. Tort, G. Urrútia, M. Valentini, Y. Wang, R. Peto and Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group, Lancet, 2013, 381, 805–816 CrossRef CAS
- H. Xie and Y. J. Kang, Curr. Med. Chem., 2009, 16, 1304–1314 CrossRef CAS
- M. L. Low, G. Paulus, P. Dorlet, R. Guillot, R. Rosli, N. Delsuc, K. A. Crouse and C. Policar, BioMetals, 2015, 28, 553–566 CrossRef CAS PubMed
- J. B. Foo, M. L. Low, J. H. Lim, Y. Z. Lor, R. Z. Abidin, V. E. Dam, N. A. Rahman, C. Y. Beh, L. C. Chan, C. W. How, Y. S. Tor and L. S. Yazan, BioMetals, 2018, 31, 505–515 CrossRef CAS PubMed
- O. A. Peña-Morán, M. L. Villarreal, L. Álvarez-Berber, A. Meneses-Acosta and V. Rodríguez-López, Molecules, 2016, 21, 1013 CrossRef PubMed
- F. A. Beckford, A. Brock, A. Gonzalez-Sarrías and N. P. Seeram, J. Mol. Struct., 2016, 1121, 156–166 CrossRef CAS PubMed
- J. Haribabu, K. Jeyalakshmi, Y. Arun, N. S. P. Bhuvanesh, P. T. Perumal and R. Karvembu, RSC Adv., 2015, 5, 46031–46049 RSC
- Maestro, Schrödinger, LLC, New York, 2016 Search PubMed
- M. P. Jacobson, D. L. Pincus, C. S. Rapp, T. J. F. Day, B. Honig, D. E. Shaw and R. A. Friesner, Proteins: Struct., Funct., Bioinf., 2004, 55, 351–367 CrossRef CAS
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed
- W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner and R. Farid, J. Med. Chem., 2006, 49, 534–553 CrossRef CAS PubMed
- R. U. Jänicke, Breast Cancer Res. Treat., 2009, 117, 219–221 CrossRef PubMed
- J. Yuan and H. R. Horvitz, Cell, 2004, 116, S53–S56 CrossRef CAS PubMed
- L. M. McNamee and M. H. Brodsky, Genetics, 2009, 182, 423–435 CrossRef CAS PubMed
- Z. Wang and Y. Sun, Transl. Oncol., 2010, 3, 1–12 CrossRef PubMed
- P. J. Jansson, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2010, 53, 5759–5769 CrossRef CAS PubMed
- M. Hajrezaie, M. Paydar, S. Zorofchian Moghadamtousi, P. Hassandarvish, N. S. Gwaram, M. Zahedifard, E. Rouhollahi, H. Karimian, C. Y. Looi, H. M. Ali, N. Abdul Majid and M. A. Abdulla, Sci. World J., 2014, 2014, 1–12 CrossRef PubMed
- V. F. S. Pape, D. Türk, P. Szabó, M. Wiese, E. A. Enyedy and G. Szakács, J. Inorg. Biochem., 2015, 144, 18–30 CrossRef CAS PubMed
- A. A. Recio Despaigne, J. G. Da Silva, P. R. da Costa, R. G. Dos Santos and H. Beraldo, Molecules, 2014, 19, 17202–17220 CrossRef PubMed
- A. Sîrbu, O. Palamarciuc, M. V. Babak, J. M. Lim, K. Ohui, E. A. Enyedy, S. Shova, D. Darvasiová, P. Rapta, W. H. Ang and V. B. Arion, Dalton Trans., 2017, 46, 3833–3847 RSC
- A. Subastri, A. Suyavaran, E. Preedia Babu, S. Nithyananthan, R. Barathidasan and C. Thirunavukkarasu, J. Cell. Physiol., 2018, 233, 1775–1790 CrossRef PubMed
- B. Halliwell and M. Whiteman, Br. J. Pharmacol., 2004, 142, 231–255 CrossRef CAS PubMed
- Y. S. Tor, L. S. Yazan, J. B. Foo, A. Wibowo, N. Ismail, Y. K. Cheah, R. Abdullah, M. Ismail, I. S. Ismail and S. K. Yeap, PLoS One, 2015, 10, e0127441 CrossRef PubMed
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, J. Biol. Inorg Chem., 2012, 17, 409–423 CrossRef CAS PubMed
- B. L. Staker, M. D. Feese, M. Cushman, Y. Pommier, D. Zembower, L. Stewart and A. B. Burgin, J. Med. Chem., 2005, 48, 2336–2345 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra03130h |
| This journal is © The Royal Society of Chemistry 2019 |