
Domino Michael-aldol annulations for the stereocontrolled synthesis of bicyclo[3.3.1]nonane and bicyclo[3.2.1]octane derivatives†
Rossella Promontorio ab,
Jean-Alexandre Richard*a and
Charles M. Marson*b
ab,
Jean-Alexandre Richard*a and
Charles M. Marson*b
aOrganic Chemistry, Institute of Chemical and Engineering Sciences (ICES), Agency for Science, Technology and Research (A*STAR), 8 Biomedical Grove, Neuros, #07-01, Singapore 138665, Singapore. E-mail: jean_alexandre@ices.a-star.edu.sg
bDepartment of Chemistry, University College London, Christopher Ingold Laboratories, 20 Gordon Street, London WC1H OAJ, UK. E-mail: c.m.marson@ucl.ac.uk
First published on 22nd November 2016
Abstract
Domino Michael-aldol annulation of cycloalkane-1,3-diones with enals affords a general route to 6-hydroxybicyclo[3.3.1]nonane-2,9-diones and 2-hydroxybicyclo[3.2.1]octane-6,8-diones, notably in one-pot procedures under convenient conditions. The annulation is shown to be compatible with one or more substituents at six positions of the bicyclo[3.3.1]nonane-2,9-dione scaffold. In some cases, the relative configuration of the product can be controlled by the appropriate choice of solvent, base and temperature for the annulation. In contrast to the chair–chair conformations usually adopted, the bicyclo compounds derived from 2,4,4-trimethylcyclohexane-1,3-dione possessed boat-chair conformations. Oxidation of the annulation products gave the corresponding bicyclo triketones.
Introduction
Alicyclic frameworks often have advantageous pharmaceutical properties compared with substituted aromatic rings, principally by conferring higher aqueous solubility, lower toxicity and greater structural diversity, including stereochemistry.1,2 Polysubstituted bicyclo compounds (e.g. derivatives of bicyclo[3.3.1]nonane3 and of bicyclo[3.2.1]octane)4 have long presented challenges for organic synthesis (Fig. 1a), especially in the placement of substituents with stereocontrol, and are become increasingly important in medicinal chemistry.2,5 In particular, several bicyclo[3.3.1]nonanes of the polyprenylated acylphloroglucinol (PPAP) family (Fig. 1b) possess multiple therapeutic effects including anti-bacterial, anti-depressant, anti-viral and anti-cancer properties.6–8 | ||
| Fig. 1 (a) Representative natural products featuring bicyclo[3.3.1] and [3.2.1] scaffolds. (b) Representative polyprenylated acylphloroglucinols with biological activity. | ||
Most synthetic methodology for the construction of bicyclo[3.3.1]nonane derivatives involves a sequential process rather than a domino annulation, an excellent recent example being the alkylation of a disubstituted 1,3-dimethoxybenzene with an enantiomerically pure epibromohydrin followed by Lewis-acid ring opening of the epoxide, effecting an enantioselective desymmetrisation.9 Subsequent oxidation enabled the Shair group to complete the synthesis of a type A PPAP natural product, (+)-hyperforin.9 This approach established the utility of derivatives of dihydroresorcinols as key precursors of bicyclo[3.3.1]octane derivatives, with the possibility of later oxidation should a phloroglucinol-derived bicyclo[3.3.1]octane be required.
Seeking to develop a domino annulation, we had regard to the putative biosynthesis of hyperforin7,10 and related natural products involving the annulation of a substituted phloroglucinol (Scheme 1, eqn I) by alkylative dearomatisation with a prenyl unit, then a second electrophilic attack completing the annulation. Although to the best of our knowledge a biomimetic synthesis involving both steps is not known, a biomimetic cationic cyclisation (the second step) induced by formic acid afforded a bicyclo system that was converted into (−)-clusianone.11 Regarding domino alkylation-conjugate addition sequences, Porco and coworkers used annulating dielectrophiles comprising various allylic alcohol and 2-alkenal derivatives that contain a leaving group at the 2-position;12 bicyclo[3.3.1]nonane derivatives of the PPAP type can be obtained, stereocontrol often being possible at the central carbon atom of the annulating unit. Porco also achieved domino conjugate addition–alkylation sequences that proceed through a bicyclo[3.3.1]nonane scaffold but which result in adamantanone derivatives.13 Additionally, the Porco group has developed powerful palladium-catalysed alkylative dearomatisation-annulation domino reactions of 2-acylphloroglucinol derivatives with bis-Boc-protected methylenepropane-1,3-diol.14 A related palladium-catalysed Tsuji–Trost approach had already been demonstrated, as in the reaction of a dihydroresorcinol derivative with the dicarbonate of methylenepropane-1,3-diol (Scheme 1, eqn (II)).15 Lastly, a succinct domino approach to the synthesis of substituted bicyclo[3.3.1]nonanes involves annulation by diacylation of a substituted cyclohexanone using malonyl chloride,16–18 the Effenberger cyclisation (Scheme 1, eqn (III)). However, the reaction usually proceeds in modest yield and is largely limited to 2-unsubstituted malonyl derivatives and to the annulation of a six-membered ring.
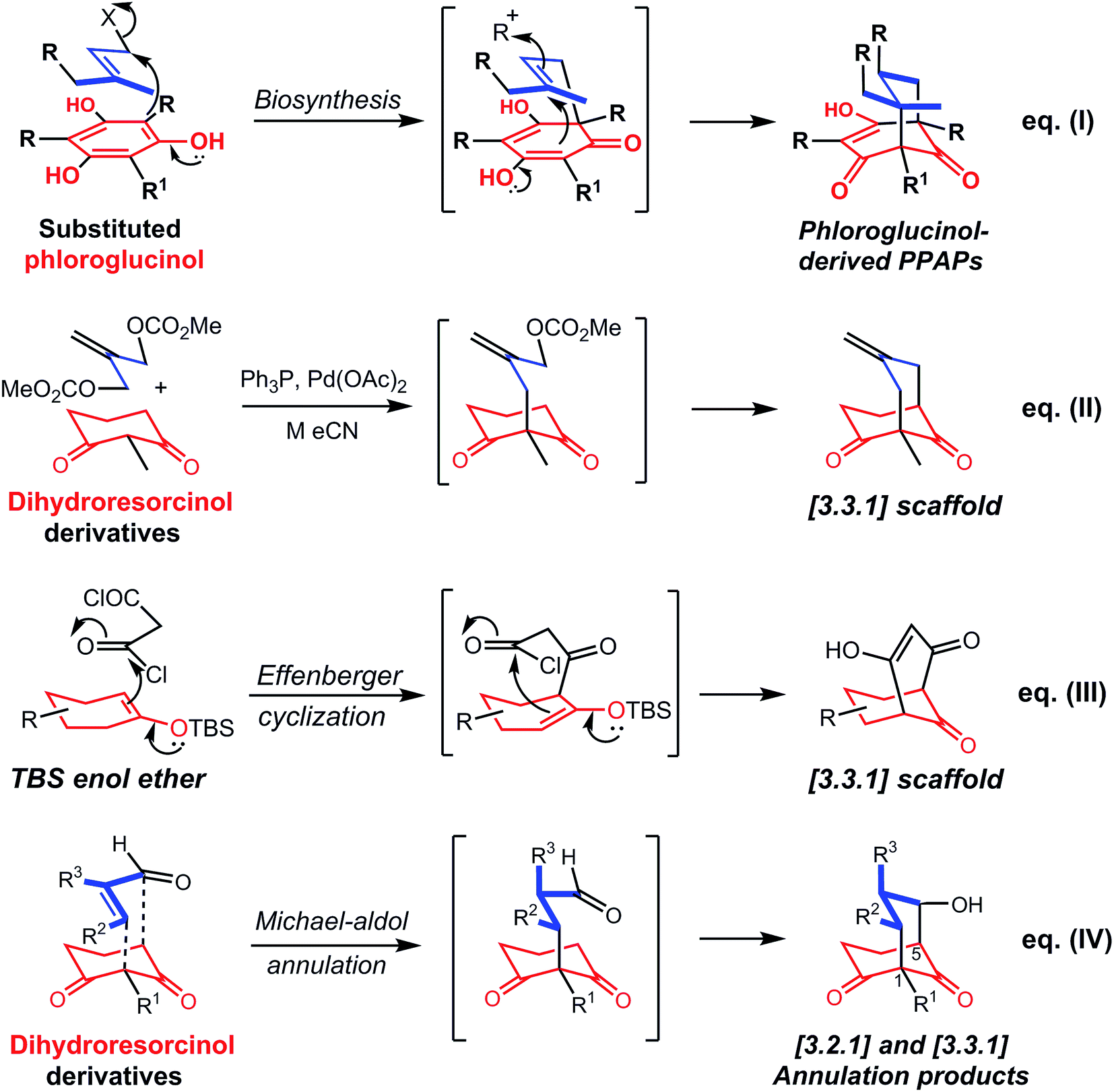 | ||
| Scheme 1 Biosynthesis of PPAPs, and selected domino annulation strategies for bicyclo systems. | ||
Given the emerging potential of bicyclo scaffolds in medicinal chemistry,1,2,5 and the limitations of current domino annulations that afford bicyclo systems, a succinct synthetic method was sought for that could generate the maximum number of stereocentres with stereocontrol, with flexibility both in the incorporation of substituents and in the size of the ring undergoing annulation. Having regard to the above criteria of diversity, and inspired by the biosynthetic annulation of phloroglucinol derivatives, we examined the feasibility of domino Michael-aldol annulations of 2-substituted cyclohexane-1,3-diones with enals, a one-pot process that can create stereocentres at any of the three carbon atoms in the annulating unit (Scheme 1, eqn (IV)). Here we report the efficacy of this domino annulation, examine its scope, and show that it can afford highly substituted bicyclo[3.2.1]octanes or bicyclo[3.3.1]nonanes, depending on the size of the cycloalkanedione used.
Results and discussion
Michael-aldol annulations have furnished polysubstituted cyclohexanone derivatives, in some cases with high enantioselectivity.19 However, with few exceptions,20 a cyclohexanone ring lacking an electron-withdrawing group at the α-position has seldom been shown to react with an enone or enal to give a bicyclo ketol. Bicyclo formation has mainly been achieved by reacting α-alkoxycarbonyl- or α-acyl-cycloalkanones with either aldehydes21–23 or ketones.24 One example of an acid-catalysed annulation of a substituted 2-acylcyclohexanone was described by Nicolaou,21 and afforded a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers of ketol 1 (Scheme 2, eqn (V)). The relative configurations were not assigned but are presumably as in Scheme 2, given that oxidation afforded the corresponding trione in 81% yield, the 3-methyl group being assigned as exo to the bridgehead carbonyl group. Michael-aldol annulation of β-keto esters has been achieved using N-heterocyclic carbene catalysts, but not usually with stereocontrol (e.g. ketol 2 in eqn (VI), Scheme 2).22 Initial formation of the enamine enables the reverse mode of annulation to be achieved, but again with little diastereoselection (e.g. ketol 3 in eqn (VII), Scheme 2).23 The corresponding reaction with methacrolein proceeded with significant stereocontrol (48% of the 6-endo-hydroxy-7-exo-methyl bicyclo ketol) but was conducted over 9 days.23
:1 mixture of diastereoisomers of ketol 1 (Scheme 2, eqn (V)). The relative configurations were not assigned but are presumably as in Scheme 2, given that oxidation afforded the corresponding trione in 81% yield, the 3-methyl group being assigned as exo to the bridgehead carbonyl group. Michael-aldol annulation of β-keto esters has been achieved using N-heterocyclic carbene catalysts, but not usually with stereocontrol (e.g. ketol 2 in eqn (VI), Scheme 2).22 Initial formation of the enamine enables the reverse mode of annulation to be achieved, but again with little diastereoselection (e.g. ketol 3 in eqn (VII), Scheme 2).23 The corresponding reaction with methacrolein proceeded with significant stereocontrol (48% of the 6-endo-hydroxy-7-exo-methyl bicyclo ketol) but was conducted over 9 days.23
 | ||
| Scheme 2 Michael-aldol annulations giving bicyclo ketols. | ||
Given the limitations in scope and/or stereocontrol using α-alkoxycarbonyl- or α-acyl-cycloalkanones in such annulation reactions, investigation of cycloalkane-1,3-diones appeared to be a potentially useful alternative to the construction of functionalised bicyclo[3.n.1]alkane scaffolds. However, to the best of our knowledge, the only such annulations involving a cyclohexane-1,3-dione derivative were reported by Dauben, in which enones were reacted at very high pressure to give ketols (Scheme 2, eqn (VII)).24 Accordingly, a pilot study of the reaction of 2-methylcyclohexane-1,3-dione (4) with acrolein was made (Table 1). Given the literature precedents for the use of secondary and tertiary organic bases (e.g. eqn (VI) and (VII), Scheme 2), including piperidine,25 a selection of bases was first studied (Table 1, entries 1–9). No reaction was observed using NaOMe or pyrrolidine (entries 1 and 2) whereas triethylamine, DIPEA, imidazole or pyridine afforded exclusively the Michael adduct 5 in 62–83% yield (Table 1, entries 3–6). In contrast to pyridine, DMAP provided the desired 6-hydroxybicyclo[3.3.1]nonane-2,9-diones 6 in excellent yield and appreciable diastereoselectivity (entry 7). The strong base DBU provided a 1:1 mixture of the Michael adduct 5 and the bicyclo ketols 6 (1:1 carbinol epimers, entry 8), whereas the weaker base 1,4-diazabicyclo[2.2.2]octane (DABCO) gave complete conversion into the bicyclo ketols 6 (1:1 carbinol epimers, entry 9). Although 10 mol% DABCO afforded a mixture of products 5 and 6 (entry 10), 20 mol% DABCO provided exclusively the bicyclo products 6 (100% conversion, 65% isolated yield of 1:1 epimers, entry 11). Under the same conditions, other solvents, including more polar solvents, did not improve the yield of bicyclo compounds 6 (entries 12–15). However, at 95 °C for 16 h DABCO (20 mol%) achieved complete conversion into ketols 6 (66:34 epimeric ratio, entry 16). Under the same conditions but heating for longer (48 h) quantitative conversion into a 90:10 ratio of epimers was achieved (entry 17); optimisation of the d.r. (entries 16–18) showed that DABCO (1 equiv.) enabled full conversion solely to the exo-ketol 6 (entry 18). Given the literature precedent for bicyclo compound formation using acidic reagents,18 the use of p-TsOH, TFA, and TfOH were examined but were found to be ineffective (entries 20, 21 and 23), except for 20% TFA in MeCN at 95 °C which afforded 36% of the ketol 6 (entry 22). Having demonstrated the benefit of heat to the selectivity of the reaction, neutral conditions were then examined; only the exo-ketol 6 was detected, with good to quantitative conversion (entries 24 and 25).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base/acid | Solvent | T (°C) | Time (h) | 5 (%) | exo-6:endo-6 (%) |
| a Percentage of conversion was determined from the H NMR spectra of the crude products.b Diastereoisomeric ratios (d.r.) were determined from integration values in the H NMR spectra of the products after work-up. | ||||||
| 1 | NaOMe | MeOH | 25 | 16 | — | — |
| 2 | Pyrrolidine (1 eq.) | MeCN | 25 | 16 | — | — |
| 3 | Et3N (1 eq.) | MeCN | 25 | 16 | 62 | — |
| 4 | (i-Pr)2NEt (1 eq.) | MeCN | 25 | 16 | 62 | — |
| 5 | Imidazole (1 eq.) | MeCN | 25 | 16 | 83 | — |
| 6 | Pyridine (1 eq.) | MeCN | 25 | 32 | 83 | — |
| 7 | DMAP (1 eq.) | MeCN | 25 | 16 | Traces | 70:30 (95) |
| 8 | DBU (1 eq.) | MeCN | 25 | 32 | 50 | 50:50 (50) |
| 9 | DABCO (1 eq.) | MeCN | 25 | 16 | — | 50:50 (100) |
| 10 | DABCO (0.1 eq.) | MeCN | 25 | 16 | 50 | 50:50 (50) |
| 11 | DABCO (0.2 eq.) | MeCN | 25 | 16 | — | 50:50 (100) |
| 12 | DABCO (0.2 eq.) | EtOH | 25 | 16 | 62 | Traces |
| 13 | DABCO (0.2 eq.) | DMF | 25 | 16 | 95 | — |
| 14 | DABCO (0.2 eq.) | DMSO | 25 | 16 | 50 | 50:50 (50) |
| 15 | DABCO (0.2 eq.) | THF | 25 | 16 | 7 | 68:32 (93) |
| 16 | DABCO (0.2 eq.) | MeCN | 95 | 16 | — | 66:34 (100) |
| 17 | DABCO (0.2 eq.) | MeCN | 95 | 48 | — | 90:10 (100) |
| 18 | DABCO (1 eq.) | MeCN | 95 | 48 | — | 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0 (100) :0 (100) |
| 19 | DABCO (0.2 eq.) | PhMe | 115 | 16 | 7 | 86:14 (93) |
| 20 | p-TsOH | CH2Cl2 | 25 | 16 | 62 | — |
| 21 | TFA | MeCN | 25 | 16 | 80 | — |
| 22 | TFA | MeCN | 95 | 16 | — | 100:0 (36) |
| 23 | TfOH | CH2Cl2 | −78 to 25 | 16 | — | — |
| 24 | — | MeCN | 95 | 72 | — | 100:0 (70) |
| 25 | — | DMF | 135 | 24 | — | 100:0 (100) |

Although DMF at 130 °C was optimal for ketols 6 and 13 (Table 2, entries 1 and 8) in terms of yield and 6-exo-diastereoselectivity, DMF was found to be unsatisfactory for enals other than acrolein. All reactions were initially run using 20 mol% of (DABCO) but under those conditions only bicyclo ketols 7 and 10 were obtained in satisfactory yields and diastereoselectivity (Table 2, entries 2 and 5). In all other cases, DABCO (1 equiv.) in MeCN gave the best yields and diastereoselectivities, and most reactions were complete within 16 h. For bicyclo[3.3.1]nonane-6-hydroxy-2,9-diones lacking substitution at the 7- and 8-positions the exo-ketols were obtained, either predominantly (entries 2 and 4) or exclusively (entries 1 and 3); that preference was also observed in the bicyclo[3.2.1]octane series (entry 8). In contrast, the 6-endo-ketols predominated in bicyclo compounds that contained an equatorial substituent on the carbon atom (in the bridging unit) adjacent to the alcohol (entry 5) or on the carbon atom remote from the alcohol (entries 9, 11 and 12). However, where a 2-prenyl group was present and also either a 7- or 8-substituent, the exo-ketols predominated (entries 6 and 7).
| a Reactions performed in the presence of DABCO (0.2 equiv. or 1.0 equiv.) at 95 °C.b endo and exo refer to the orientation of the hydroxy group.c Reaction with acrolein.d Reaction performed in DMF at 135 °C.e Sequential: the unpurified Michael adduct was isolated and then cyclised.f Reaction with methacrolein.g Reaction with crotonaldehyde.h 13% of an additional isomer was detected by 1H NMR spectroscopy.i Reaction with cinnamaldehyde.j >95% conversion by 1H NMR spectroscopy; low isolated yield attributed to partial decomposition of 16 and 17 during purification. |
|---|
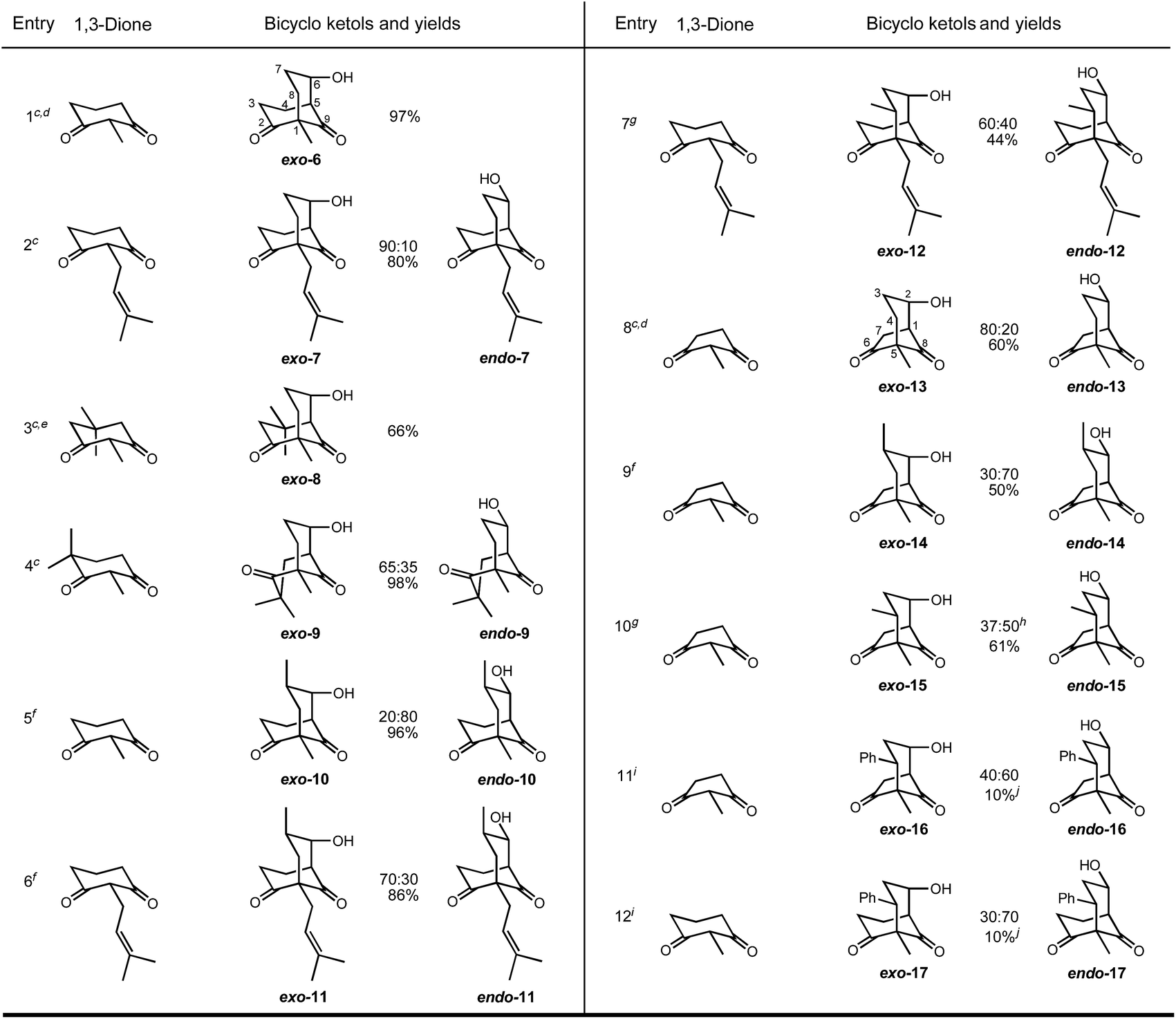 |
Assignment of the 6-exo-ketols 6 was indicated by the presence of small coupling constants (<5 Hz) for the 6-CH(OH) hydrogen atom, in contrast to that the 6-endo-ketols (e.g. trans-diaxial J5,6 = 11.5 Hz for endo-6, and 10.5 Hz for endo-10). The isolation of endo-ketol 10, together with its different NMR data from the exo-ketol 10 (isolated in 6% yield from a reaction in DMF at 95 °C) confirmed the assignments in entry 5 to be a mixture of exo- and endo-diastereoisomers, and excluded the possibility of equilibrating conformers as an explanation of the results. The situation is similar for the various optimisation runs in Table 2 which can only be explained by increasing predominance of exo-ketol 6 at higher temperatures and/or longer reaction times. The NMR data for all the bicyclo ketols comprise a pattern of chemical shifts and coupling constants consistent with the structural assignments given in Table 2. Additional support for the structures assigned by NMR spectroscopy is found in the X-ray crystal structure of the 3,5-dinitrobenzoyl derivative of exo-ketol 6 which shows that the C–O bond in the 6-CH(OH) moiety is axial; the relatively small couplings of 5.2 Hz and 1.6 Hz for the equatorial CH–OCOAr hydrogen atom in this ester parallel the small coupling constants observed for exo- versus endo-epimers (Fig. 2).
 | ||
| Fig. 2 X-ray structure (ORTEP) of the 3,5-dinitrobenzoate ester of exo-ketol 6.23 | ||
The annulation methodology was also found to be effective using gem-dimethyl-substituted cyclohexane-1,3-diones (Table 2, entries 3 and 4). In the case of 2,4,4-trimethylcyclohexane-1,3-dione, reaction with methacrolein and crotonaldehyde afforded the bicyclo ketols 18 (65%) and 19 (49%) respectively (Scheme 3); simplification of the mixtures of diastereoisomers 18 and 19 was achieved by oxidation with pyridinium chlorochromate (PCC), giving the triketones 20 and 21 respectively. Similarly, oxidation of ketols 6, 9, 10, with PCC afforded the respective triketones 22–24 (Scheme 3).
 | ||
| Scheme 3 Bicyclo[3.3.1]nonane-2,6,9-triones prepared by the oxidation of ketols with pyridinium chlorochromate. | ||
Molecular models of the gem-dimethyl-substituted trione 20 indicated that the usual chair–chair conformation adopted by most bicyclo[3.3.1]nonanes would suffer severe non-bonding interactions. That inference of an alternative conformation was confirmed by a single-crystal X-ray determination of the trione 20 which established the unusual boat-chair conformation (Fig. 3a). Compared to an sp3 carbon atom, the bridgehead carbonyl group is more able to accommodate a boat structure, and without significant flagpole interactions, for the ring containing the gem-dimethyl group.
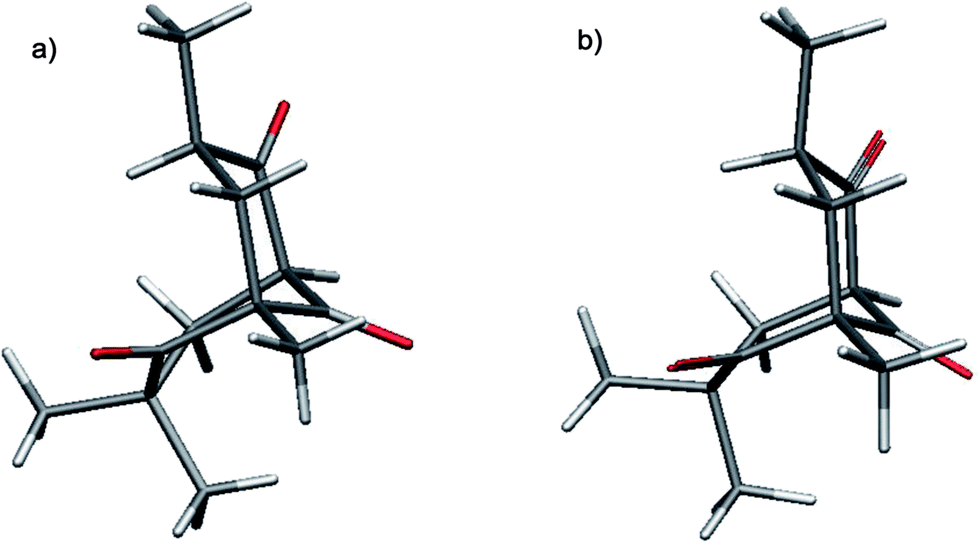 | ||
| Fig. 3 (a) X-ray structure of the trione 20 (ref. 26) and (b) lowest energy conformer obtained by OPLS3-GB/SA conformational search. | ||
OPLS3-GB/SA conformational energy searches (Table 3)27 and quantum mechanics calculations (Tables S1–S3, ESI†) support a boat-chair conformation for the trione 20. In addition, conformational searches on the other bicyclo compounds 21, 22, exo-9 and endo-9 that possess the same location of gem-dimethyl-substitution as in 20 all identified the boat-chair conformation as the lowest in energy (Table 3). Other less favourable chair-boat, twistboat–twistboat and boat–boat conformers could be detected but never the usual chair–chair conformation. The presence of a gem-dimethyl group excludes the chair–chair conformation from being adopted owing to the severe non-bonding interactions of the axial methyl group with the 7-methylene unit that would arise. In contrast, where such a non-bonding interaction is absent, as is the case for triones 23 and 24, the usual chair–chair conformation for saturated, substituted bicyclo[3.3.1]nonanes is preferred.
| Compound | 7-exo-20 | 8-endo-21 | 22 | exo-9 | endo-9 |
|---|---|---|---|---|---|
| a Energies quoted are relative to the boat–chair conformation.b The first-named conformer refers to the ring containing the gem-dimethyl group.c Not found during the conformational search. | |||||
| Boat–chairb | 0 | 0 | 0 | 0 | 0 |
| Chair–boat | 16.2 | —c | 10.0 | 28.4 | 35.6 |
| Twistboat–twistboat | 23.9 | 27.7 | 19.2 | 33.1 | 34.2 |
| Boat–boat | 27.4 | —c | 21.0 | —c | —c |
| Twistboat–twistboat 2 | —c | —c | —c | —c | 41.3 |
| Chair–chair | —c | —c | —c | —c | —c |
Trends in the mode of cyclisation are apparent. For the bicyclo[3.3.1]nonane-2,9-diones, the exo-ketol 6 is generally preferred over the endo-ketol. However, the preference for the exo-ketol can be overcome by substitution in some locations on the framework, especially for the bicyclo[3.2.1]octane-6,8-diones, as shown in Table 2. Regarding the effect of substitution on the aldehydic chain, an α-methyl group derived from methacrolein adopts the equatorial position prior to cyclisation, leading to a significant preference for the endo-alcohol, as seen by comparing entry 1 with entry 5 (Table 2), and entry 8 with entry 9. An α-substituent disfavours the formation of the exo-ketol because of three significant and adjacent developing synclinal interactions, compared with only two synclinal interactions for the endo-ketol (Scheme 4). However, a β-methyl or β-phenyl group (entries 7 and 10–12) exerts a much weaker effect than an α-methyl substituent, although both diminish the strong preference for the exo-isomer that is observed in cases where no α- or β-substituent is present (Table 2, entries 1, 2 and 8).
 | ||
| Scheme 4 Modes of cyclisation in the domino Michael-aldol annulation. | ||
Substituents both on the cycloalkane-1,3-dione ring and on the enal can have a profound effect on the conformation on the Michael adduct, and hence on its mode of cyclisation. Thus, only exo-ketol 8 (Table 2, entry 3) was detected, the developing 1,3-diaxial interaction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group with the equatorial methyl group preventing the endo-mode of cyclisation (Scheme 4). In contrast, the location of the gem-dimethyl substituents in entry 4 excludes a chair–chair conformation; consequently, the dione ring adopts a boat conformation, which having smaller non-bonding interactions with the aldehydic carbonyl group leads to a significant amount of the endo-isomer, the preference for the exo-isomer (as shown for 6 and 8) being eroded. The generally lower selectivities for the cyclopentane-1,3-dione series compared to the cyclohexane-1,3-dione series are also consistent with the flatter and less encumbering cyclopentane-1.3-dione ring that leads to the development of smaller non-bonding interactions during cyclisation.
O group with the equatorial methyl group preventing the endo-mode of cyclisation (Scheme 4). In contrast, the location of the gem-dimethyl substituents in entry 4 excludes a chair–chair conformation; consequently, the dione ring adopts a boat conformation, which having smaller non-bonding interactions with the aldehydic carbonyl group leads to a significant amount of the endo-isomer, the preference for the exo-isomer (as shown for 6 and 8) being eroded. The generally lower selectivities for the cyclopentane-1,3-dione series compared to the cyclohexane-1,3-dione series are also consistent with the flatter and less encumbering cyclopentane-1.3-dione ring that leads to the development of smaller non-bonding interactions during cyclisation.
The formation and conformations of the bicyclo ketols herein studied have implications for the potential of bicyclo compounds in medicinal chemistry. The domino Michael-aldol annulation has been shown to be effective with substituents at many locations of the bicyclo framework. Varying degrees of control of the configuration of the hydroxy group in the ketols have been achieved through optimisation of reaction conditions or through the conformational effects exerted by substituents. The bicyclo compounds derived from cycloalkane-1,3-diones often possess well-defined configurations and conformations that can contain multiple substituents with specific directionality that overall achieves a wide coverage of chemical space for a relatively compact structure. Additionally, these non-aromatic alicyclic scaffolds satisfy two important criteria for drug-likeness: high levels of saturation and suitable logP values.5
Conclusions
The present study has demonstrated the considerable scope of the domino Michael-aldol annulation in obtaining access to 6-hydroxybicyclo[3.3.1]nonane-2,9-diones and 2-hydroxybicyclo[3.2.1]octane-6,8-diones from cycloalkane-1,3-diones and enals, notably in one-pot procedures under convenient conditions. In some cases, the relative configuration of the annulation product can be controlled by the appropriate choice of solvent, base and temperature. This study has shown that the annulation is compatible with one or more substituents at six positions of the bicyclo[3.3.1]nonane-2,9-dione scaffold. The bicyclo compounds provide structural diversity suitable for use in medicinal chemistry programmes and with potential for use as precursors in natural product synthesis. Oxidation of the annulation products was achieved to give a variety of stable bicyclo triones.Experimental section
General
All moisture-sensitive reactions were performed under an atmosphere of argon and using glassware pre-dried in an oven (100 °C). Thin-layer chromatography was performed on Merck 0.2 mm aluminium-backed silica gel 60 F254 plates and visualised by UV (254 nm) or by staining with potassium permanganate with subsequent heating. Flash column chromatography was performed using Merck 0.040–0.063 mm, 230–400 mesh silica gel. Evaporation refers to the removal of solvent under reduced pressure. Melting points were determined using a Büchi B-540 apparatus. Infrared (IR) spectra were recorded on a Perkin–Elmer Spectrum One FT-IR spectrometer; absorptions are quoted in wavenumbers. 1H and 13C NMR spectra were recorded on a Bruker DRX-400 (400 MHz) spectrometer and calibrated using residual undeuterated solvent as an internal reference; chemical shifts are in parts per million (δ) and coupling constants (J) are given in Hertz (Hz). The following abbreviations were used in signal assignments: singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), and multiplet (m). Equivocal assignments are denoted by an asterisk. High-resolution mass spectra (HRMS) were obtained using either an Agilent ESI TOF (time of flight) mass spectrometer at 3500 V emitter voltage, or using a VG7070H mass spectrometer with Finigan Incos II data system at University College London.The following compounds were prepared according to the literature: 2-(3-methylbut-2-en-1-yl)cyclohexane-1,3-dione;28 2,5,5-trimethyl-1,3-cyclohexanedione.29
:7, ethyl acetate:petroleum ether) of the residue gave 2,4,4-trimethylcyclohexane-1,3-dione (2.84 g, 52%) as a white solid, stable for several weeks when stored at −20 °C; IR (film): 3005, 2988, 1711, 1458 cm−1; 1H NMR (400 MHz, CD3OD) δ 2.48 (2H, t, J = 6.5 Hz, COCH2), 1.79 (2H, t, J = 6.5 Hz, C(CH3)2CH2), 1.63 (3H, s, 2-CH3), 1.08 (6H, s, C(CH3)2); 13C NMR (100 MHz, CD3OD) δ 110.0, 40.1, 35.8, 28.0, 25.4, 7.7; HRMS (ESI-TOF) [M + H]+ C9H15O2 calcd 155.1067, found 155.1065.:1 mixture of epimers at position-6. For endo-ketol 6: 1H NMR (400 MHz, CDCl3) δ 4.10 (1H, dt, J = 11.5, 5.0 Hz, 6-CH), 3.10 (1H, m, 5-CH), 2.50–2.45 (2H, m, 3-CH2), 2.29–2.14 (4H, m, 4-CHeq, 7-CHeq and 8-CH2), 1.65–1.56 (1H, m, 4-CHax), 1.47–1.38 (1H, m, 7-CHax), 1.14 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) δ 210.2 (9-CO), 209.3 (2-CO), 73.2 (6-CH), 61.8 (1-C), 52.5 (5-CH), 38.8 (3-CH2), 35.6 (8-CH2), 27.8 (7-CH2), 16.3 (4-CH2), 15.0 (CH3).:2 dichloromethane:ethyl acetate) of the residue gave exo-5-methyl-6,9-dioxobicyclo[3.3.1]nonan-2-yl 3,5-dinitrobenzoate (30 mg, 8%) IR (film): 2936, 1731, 1704, 1629, 1545, 1050 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.24 (1H, t, J = 2.1 Hz, 4-aryl), 9.05 (2H, d, J = 2.1 Hz, 2,6-aryl), 5.62 (1H, m, 2-CH), 3.21 (1H, m, 1-CH), 2.69 (1H, ddd, J = 16.5, 7.5, 4.5 Hz, 7-CHeq), 2.47 (1H, dt, J = 16.5, 9.5 Hz, 7-CHax), 2.39–2.27 (2H, m, 3-CHeq and 8-CHeq), 2.15–2.02 (3H, m, 3-CHax and 4-CH2), 1.86 (1H, m, 8-CHax), 1.26 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) δ 210.8 (9-CO), 209.4 (6-CO), 161.6 (COOAr), 148.9 (3,5-aryl), 133.6 (1-aryl), 129.5 (2-aryl), 122.9 (4-aryl), 81.1 (2-CH), 63.2 (5-C), 48.4 (1-CH), 38.1 (7-CH2),* 37.7 (4-CH2),* 24.4 (3-CH2), 18.9 (8-CH2), 16.8 (5-CCH3); m/z (EI+, %) 395 (20), 394 (M+, 100), 364 (17); HRMS [M + H]+ C17H17N2O8 calcd 377.0979, found 377.0981.:10 mixture of exo-7:endo-7. IR (film): 3416, 2917, 1701, 1450 cm−1; exo-7: 1H NMR (600 MHz, CDCl3) δ 5.05 (1H, tsept., J = 5.0, 1.0 Hz, 1-CCH2CH), 4.34 (1H, m, 6-CH), 2.92 (1H, dm, J = 6.8 Hz, 5-CH), 2.57 (1H, m, 3-CHeq), 2.40–2.30 (2H, m, 1-CCH2), 2.25–2.05 (5H, m, 3-CHax, 4-CH2, 8-CH2), 1.80 (1H, m, 7-CHeq), 1.70 (1H, m, 7-CHax), 1.61 (3H, s, C(CH3)CH3), 1.59 (3H, s, C(CH3)CH3); 13C NMR (100 MHz, CDCl3) δ 212.0 (9-CO), 212.3 (2-CO), 134.7 (1-CCH2CHC), 118.7 (1-CCH2CH), 77.2 (6-CH), 66.5 (1-C), 52.5 (5-CH), 40.4 (3-CH2), 36.3 (8-CH2), 31.0 (1-CCH2), 26.1 (7-CH2), 25.8 (1-C-cis-CH3), 18.8 (4-CH2), 17.9 (1-C-trans-CH3); endo-7: 1H NMR (400 MHz, CDCl3) δ 5.05 (1H, m, 1-CCH2CH), 4.06 (1H, dt, J = 11.5, 4.9 Hz, 6-CH), 3.11 (1H, m, 5-CH), 2.55 (1H, m, 3-CHeq), 2.40–2.30 (2H, m, 1-CCH2), 2.26–2.15 (3H, m, 3-CHax, 4-CH2), 2.11–2.05 (2H, m, 8-CH2), 1.80 (1H, m, 7-CHeq), 1.70 (1H, m, 7-CHax), 1.58 (6H, m, C(CH3)CH3); 13C NMR (150 MHz, CDCl3) δ 212.3 (9-CO), 210.3 (2-CO), 135.0 (1-CCH2CHC), 118.6 (1-CCH2CH), 73.7 (6-CH), 65.2 (1-C), 52.8 (5-CH), 40.6 (3-CH2), 34.2 (8-CH2), 30.6 (1-CCH2), 27.2 (7-CH2), 26.1 (1-C-cis-CH3), 18.8 (4-CH2), 14.6 (1-C-trans-CH3). HRMS (ESI-TOF) [M + H]+ C14H21O3 calcd 237.1485, found 237.1481.:35 mixture of exo-9:endo-9, IR (film): 3417, 2936, 1729, 1697, 1469 cm−1. Column chromatography (silica gel, 4:6 ethyl acetate:dichloromethane) afforded exo-9: 1H NMR (400 MHz, CDCl3) δ 4.26 (1H, dq, J = 4.5, 2.4 Hz, 6-CH), 2.95 (1H, m, 5-CH), 2.20 (1H, m, 8-CHeq), 2.12–2.00 (2H, m, 4-CHeq and 8-CHax), 1.70–1.65 (2H, m, 7-CH2), 1.48 (1H, dm, J = 14.4 Hz, 4-CHax), 1.19 (3H, s, 1-CCH3),* 1.15 (3H, s, 3-C(CH3)CH3),* 0.97 (3H, s, 3-C(CH3)CH3); 13C NMR (100 MHz, CDCl3) δ 216.2 (2-CO), 213.7 (9-CO), 78.1 (6-CH), 60.4 (1-C), 51.9 (5-CH), 45.8 (3-C), 39.4 (8-CH2), 35.5 (4-CH2), 26.1 (3-CCH3eq), 25.6 (7-CH2), 24.6 (3-CCH3ax), 18.7 (1-CCH3) and endo-9: 1H NMR (400 MHz, CDCl3) δ 4.02 (1H, dt, J = 11.0, 4.5 Hz, 6-CH), 3.15 (1H, m, 5-CH), 2.17 (1H, dm, 8-CHeq), 2.05 (1H, dd = J 14.7, 2.0 Hz, 4-CHeq), 1.80–1.70 (2H, m, 4-CHax and 7-CHeq), 1.41–1.26 (2H, m, 7-CHax and 8-CHax), 1.22 (3H, 1-CCH3),* 1.20 (3H, 3-C(CH3)CH3),* 0.97 (3H, s, 3-C(CH3)CH3); 13C NMR (100 MHz, CDCl3) δ 216.1 (2-CO), 211.9 (9-CO), 74.3 (6-CH), 58.9 (1-C), 52.5 (5-CH), 45.7 (3-C), 37.0 (8-CH2), 30.9 (4-CH2), 27.1 (7-CH2), 26.0 (3-CCH3eq), 24.1 (3-CCH3ax), 18.3 (1-CCH3). HRMS (ESI-TOF) [M + H]+ C12H19O3 calcd 211.1329, found 211.1333.:20 mixture of endo-10:exo-10: IR (film): 3455, 2936, 1728, 1697 cm−1. Column chromatography (silica gel, 40:60 ethyl acetate:dichloromethane) gave endo-10: 1H NMR (400 MHz, CDCl3) δ 3.62 (1H, dd, J = 10.5, 4.8 Hz, 6-CH), 3.08 (1H, ddd, J = 8.9, 4.8, 2.1 Hz, 5-CH), 2.64 (1H, m, 3-CHeq), 2.37 (1H, m, 3-CHax), 2.24 (1H, m, 4-CHeq), 2.12–2.00 (2H, m, 7-CH and 8-CHeq), 1.92 (1H, m, 8-CHax), 1.77 (1H, m, 4-CHax), 1.13 (3H, s, 1-CCH3), 1.02 (3H, d, J = 6.3 Hz, 7-CHCH3); 13C NMR (100 MHz, CDCl3) 212.0 (9-CO), 209.9 (2-CO), 78.4 (6-CH), 62.9 (1-C), 52.0 (5-CH), 44.4 (8-CH2), 38.4 (3-CH2), 32.7 (7-CH), 17.7 (4-CH2), 16.2 (1-CCH3),* 15.5 (7-CCH3)*; exo-10: 1H NMR (400 MHz, CDCl3) δ 3.96 (1H, m, 6-CH), 2.96 (1H, ddd, J = 10.2, 4.2, 2 Hz, 5-CH), 2.62 (1H, ddd, J = 16.2, 7.4, 3.4 Hz, 3-CHeq), 2.34 (1H, m, 3-CHax), 2.20 (1H, m, 4-CHeq), 2.10 (1H, m, 7-CH), 2.01 (1H, m, 8-CHeq), 1.82 (1H, dd, J = 12.9, 4.5 Hz, 8-CHax), 1.70 (1H, m, 4-CHax), 1.14 (3H, s, 1-CCH3), 1.00 (3H, d, J = 6.6 Hz, 7-CHCH3); 13C NMR (100 MHz, CDCl3) δ 212.5 (9-CO), 211.8 (2-CO), 80.4 (6-CH), 62.5 (1-C), 52.1 (5-CH), 44.5 (8-CH2), 38.4 (3-CH2), 29.9 (7-CH), 18.6 (4-CH2), 16.8 (1-CHCH3), 16.7 (7-CCH3). HRMS (ESI-TOF) [M + H]+ C11H17O3 calcd 197.1172, found 197.1170.:30 mixture of exo-11:endo-11; IR (film): 3416, 2918, 1700, 1456 cm−1; exo-11: 1H NMR (600 MHz, CDCl3) δ 5.05 (1H, m, 1-CCH2CH), 3.98 (1H, m, 6-CH), 2.92 (1H, m, 5-CH), 2.62–2.51 (1H, m, 3-CH eq), 2.39–2.27 (3H, m, 1-C-CH2 and 3-CHax), 2.24–2.13 (m, 2H, 4-CH2), 1.98 (1H, m, 7-CH), 1.97–1.74 (2H, m, 8-CH2), 1.61 (3H, s, 1-C-cis-CH3), 1.58 (3H, s, 1-C-trans-CH3), 0.97 (3H, d, J = 7.0 Hz, 7-CHCH3); 13C NMR (150 MHz, CDCl3) 212.4 (9-CO), 210.3 (2-CO), 134.8 (1-CCH2CHC), 118.6 (1-CCH2CH), 80.6 (6-CH), 65.8 (1-C), 52.2 (5-CH), 43.3 (8-CH2), 40.5 (3-CH2), 31.0 (1-C-CH2), 29.4 (7-CH), 26.0 (1-C-cis-CH3), 18.5 (4-CH2), 17.9 (1-C-trans-CH3), 16.6 (7-CHCH3); endo-11: 1H NMR (600 MHz, CDCl3) δ 5.05 (1H, m, 1-CCH2CH), 3.55 (1H, dd, J = 7.0, 4.8 Hz, 6-CH), 3.07 (1H, ddd, J = 6.52, 3.2, 1.4 Hz, 5-CH), 2.62–2.51 (2H, m, 3-CH2), 2.39–2.27 (2H, m, 1-CCH2), 2.13–2.06 (2H, m, 4-CH2), 1.98 (1H, m, 7-CH), 1.87–1.74 (2H, m, 8-CH2), 1.65 (3H, s, 1-C-cis-CH3), 1.60 (3H, s, 1-C-trans-CH3), 1.00 (3H, d, J = 7.0 Hz, 7-CHCH3); 13C NMR (150 MHz, CDCl3) δ 212.7 (9-CO), 212.5 (2-CO), 135.0 (1-CCH2CHC), 118.5 (1-CCH2CH), 78.8 (6-CH), 66.1 (1-C), 52.2 (5-CH), 43.0 (8-CH2), 40.3 (3-CH2), 32.1 (7-CH), 30.6 (1-CCH2), 26.0 (1-C-cis-CH3), 17.6 (1-C-trans-CH3), 16.6 (7-CHCH3), 15.1 (4-CH2); HRMS (ESI-TOF) [M + H]+ C15H23O3 calcd 251.1642, found 251.1639.:8 ethyl acetate:dichloromethane) of the residue gave ketol 12 (61 mg, 44%) as 60:40 mixture of exo-12:endo-12; IR (film): 3432, 2971, 2992, 1728, 1697 cm−1. Repeated column chromatography of a small fraction enabled the exo-isomer to be isolated, and hence NMR data for the endo-isomer to be deduced: exo-12; 1H NMR (400 MHz, CDCl3) δ 5.06 (1H, tsept., J = 7.1, 1.4 Hz, 1-CCH2CH), 4.27 (1H, dt, J = 3.0, 2.5 Hz, 6-CH), 2.87 (1H, dm, J = 8.7 Hz, 5-CH), 2.60–2.33 (5H, m, 1-CCH2, 8-CH and, 3-CH2), 2.15–2.03 (2H, m, 4-CH2), 1.89–1.74 (2H, m, 7-CH2), 1.64 (6H, s, 1-C-cis-CH3 and 1-C-trans-CH3), 0.99 (3H, d, J = 6.8 Hz, 8-CHCH3); 13C NMR (100 MHz, CDCl3) δ 210.6 (9-CO), 209.5 (2-CO), 134.2 (1-CCH2CHC), 119.4 (1-CCH2CH), 75.5 (6-CH), 71.7 (1-C), 52.6 (5-CH), 39.6 (3-CH2), 38.3 (8-CH), 36.3 (7-CH2), 27.1 (1-CCH2), 26.1 (1-C-cis-CH3), 19.7 (4-CH2), 18.0 (1-C-trans-CH3), 15.8 (8-CHCH3); endo-12: 1H NMR (400 MHz, CDCl3) δ 4.98 (1H, m, 1-CCH2CH), 4.10 (1H, dt, J = 11.1, 5.4 Hz, 6-CH), 2.99 (1H, app. t, J = 6.0 Hz, 5-CH), 2.58–2.43 (5H, m, 1-CCH2, 8-CH and 3-CH2), 2.15–2.03 (2H, m, 4-CH2), 1.83–1.71 (2H, m, 7-CH2), 1.64 (6H, s, 1-C-cis-CH3 and 1-C-trans-CH3), 0.96 (3H, d, J = 6.8 Hz, CHCH3); 13C NMR (100 MHz, CDCl3) 212.6 (9-CO), 211.6 (2-CO), 135.0 (1-CCH2CHC), 118.5 (1-CCH2CH), 71.3 (6-CH), 70.2 (1-C), 52.4 (5-CH), 40.2 (3-CH2), 37.5 (8-CH), 37.4 (7-CH2), 29.8 (1-CCH2), 26.0 (1-C-cis-CH3), 18.0 (4-CH2), 17.8 (1-C-trans-CH3), 15.7 (8-CHCH3). Traces of a third diastereoisomer were detected by 13C NMR spectroscopy. HRMS (ESI-TOF) [M + H]+ C15H23O3 calcd 251.1642, found 251.1640.:20 mixture of exo-13:endo-13: IR (film): 3450, 2933, 1765, 1723, 1453 cm−1; exo-13: 1H NMR (400 MHz, CDCl3) δ 4.57 (1H, m, 2-CH), 3.03 (1H, app. t, J = 5.4 Hz, 1-CH), 2.70–2.52 (2H, m, 7-CH2), 2.25 (1H, m, 4-CHeq), 1.95–1.87 (2H, m, 3-CH2), 1.81–1.77 (1H, m, 4-CHax), 1.07 (3H, s, 5-CCH3); 13C NMR (100 MHz, CDCl3) δ 214.5 (8-CO), 211.1 (6-CO), 77.5 (2-CH), 59.3 (5-CCH3), 52.7 (1-CH), 42.1 (7-CH2), 40.1 (4-CH2), 26.6 (3-CH2), 12.2 (5-CCH3); endo-13: 1H NMR (400 MHz, CDCl3) δ 4.25 (1H, m, 2-CH), 3.08 (1H, dd, J = 7.5, 3.3 Hz, 1-CH), 2.50–2.43 (2H, m, 7-CH2), 2.25 (1H, m, 4-CHeq), 1.95–1.75 (3H, m, 3-CH2 and 4-CHax), 1.06 (3H, s, 5-CCH3); 13C NMR (100 MHz, CDCl3) δ 213.5 (8-CO), 211.6 (6-CO), 73.6 (2-CH), 58.3 (5-CCH3), 54.4 (1-CH), 38.7 (7-CH2), 35.9 (4-CH2), 27.1 (3-CH2), 11.6 (5-CCH3). HRMS (ESI-TOF) [M + H]+ C9H13O3 calcd 169.0859, found 169.0861.:30 mixture of endo-14:exo-14; IR (film): 3499, 2933, 1767, 1724, 1455 cm−1; endo-14: 1H NMR (400 MHz, CDCl3) δ 3.69 (1H, dd, J = 9.6, 3.2 Hz, 2-CH), 3.03 (1H, dd, J = 7.5, 3.3 Hz, 1-CH), 2.96 (1H, d, J = 19.4 Hz, 7-CHax), 2.48 (1H, dd, J = 19.4, 7.5 Hz, 7-CHeq), 1.92–1.67 (3H, m, 3-CH and 4-CH2), 1.02 (3H, s, 5-CCH3), 1.01 (3H, d, J = 6.6 Hz, 3-CHCH3); 13C NMR (100 MHz, CDCl3) δ 213.4 (8-CO), 211.6 (6-CO), 78.9 (2-CH), 59.4 (5-CCH3), 53.7 (1-CH), 44.5 (7-CH2), 39.1 (4-CH2), 32.9 (3-CHCH3), 17.3 (3-CHCH3), 11.6 (5-CCH3) (400 MHz, CDCl3); exo-14: 1H NMR (400 MHz, CDCl3) δ 4.25 (1H, ddd, J = 5.1, 3.4, 2.3 Hz, 2-CH), 3.01 (1H, m, 1-CH), 2.60–2.58 (2H, m, 7-CH2), 2.08 (1H, m, 3-CHeq), 1.92–1.67 (2H, m, 3-CH and 4-CHax), 1.03 (3H, s, 5-CCH3), 0.97 (3H, d, J = 6.7 Hz, 3-CHCH3); 13C NMR (100 MHz, CDCl3) δ 215.1 (8-CO), 211.4 (6-CO), 79.6 (2-CH), 58.8 (5-CCH3), 52.5 (1-CH), 46.7 (7-CH2), 41.8 (4-CH2), 30.2 (3-CHCH3), 15.4 (3-CHCH3), 12.0 (5-CCH3). HRMS (ESI-TOF) [M + H]+ C10H15O3 calcd 183.1016, found 183.1014.:50 mixture of mixture of endo-15:exo-15; IR (film): 3441, 2936, 1763, 1719, 1455, 1041 cm−1; endo-15: 1H NMR (400 MHz, CDCl3) δ 4.20 (1H, ddd, J = 11.1, 5.9, 3.4 Hz, 2-CH), 3.04 (1H, dd, J = 7.0, 3.4 Hz, 1-CH), 2.88 (1H, d, J = 19.5 Hz, 7-CHax), 2.56 (1H, dd, J = 19.5, 7.0 Hz, 7-CHeq), 2.05 (1H, dt, J = 14.4, 5.5 Hz, 3-CHeq), 1.65 (1H, m, 4-CH), 1.27 (1H, m, 3-CHax), 0.98 (3H, d, J = 5.0 Hz, 4-CHCH3), 0.88 (3H, s, 5-CCH3); 13C NMR (100 MHz, CDCl3) δ 210.4 (8-CO), 209.9 (6-CO), 71.8 (2-CH), 61.3 (5-CCH3), 53.8 (1-CH), 39.7 (4-CH), 38.6 (7-CH2), 36.3 (3-CH2), 15.0 (4-CHCH3), 9.79 (5-CCH3); exo-15: 1H NMR (400 MHz, CDCl3) δ 4.48 (1H, ddd, J = 5.1, 4.1, 1.6 Hz, 2-CH), 2.98 (1H, m, 1-CH), 2.55–2.45 (2H, m, 7-CH2), 2.36 (1H, m, 4-CH), 1.82 (1H, ddt, J = 15.8, 5.6, 1.3 Hz, 3-CHeq), 1.55 (1H, ddd, J = 15.8, 13.1, 3.9 Hz, 3-CHax), 0.99 (3H, d, J = 5.0 Hz, 4-CHCH3), 0.90 (3H, s, 5-CCH3); 13C NMR (100 MHz, CDCl3) δ 215.5 (8-CO), 214.3 (6-CO), 75.8 (2-CH), 62.4 (5-CCH3), 52.1 (1-CH), 44.0 (4-CH) 42.0 (7-CH2), 35.6 (3-CH2), 15.1 (4-CHCH3), 10.0 (5-CCH3). The 1H NMR spectrum showed the presence of third diastereoisomer (13%). HRMS (ESI-TOF) [M + H]+ C10H15O3 calcd 183.1016, found 183.1012.:80 ethyl acetate:dichloromethane) gave ketol 16 (20 mg, 10%) as a 60:40 mixture of mixture of endo-16:exo-16; IR (film): 3488, 2988, 1763, 1721, 1455, 1046 cm−1; endo-16: 1H NMR (400 MHz, CDCl3) δ 7.34–7.26 (3H, m, m- and p-H), 7.03 (2H, J = 7.8, 2.4 Hz, o-H), 4.38 (1H, ddd, J = 10.4, 5.9, 3.5 Hz, 2-CH), 3.19 (1H, dd, J = 7.5, 3.5 Hz, 1-CH), 3.16 (1H, J = 19.0 Hz, 7-CHax), 2.65 (1H, J = 14.0, 4.8 Hz, 4-CH), 2.60 (1H, J = 19.0, 7.5 Hz, 7-CHeq), 2.29–2.23 (1H, m, 3-CHeq), 2.05 (1H, m, 3-CHax), 0.77 (3H, s, 5-CCH3); 13C NMR (100 MHz, CDCl3) δ 212.5 (6-CO), 210.3 (8-CO), 137.6 (ipso-phenyl), 128.7 (phenyl), 128.6 (phenyl), 128.1 (phenyl), 71.7 (2-CH), 61.7 (5-CCH3), 54.3 (1-CH), 50.6 (4-CH), 38.8 (7-CH2), 35.3 (3-CH2), 10.7 (5-CCH3); exo-16: 1H NMR (400 MHz, CDCl3) δ 7.34–7.26 (3H, m, m- and p-H), 7.07 (2H, J = 7.8, 2.4 Hz, o-H), 4.63 (1H, ddd, J = 5.2, 3.5, 1.7 Hz, 2-CH), 3.41 (1H, dd, J = 13.7, 4.8 Hz, 4-CH), 3.13 (1H, dd, J = 8.0, 4.5 Hz, 1-CH), 2.68–2.56 (2H, m, 7-CH2), 2.26 (1H, m, 3-CHeq), 2.04 (1H, m, 3-CHax), 0.79 (3H, s, 5-CCH3); 13C NMR (100 MHz, CDCl3) δ 213.1 (8-CO), 212.5 (6-CO), 138.0 (ipso-phenyl), 128.7 (phenyl), 128.6 (phenyl), 128.1 (phenyl), 75.1 (2-CH), 62.7 (5-CCH3), 54.1 (1-CH), 52.6 (4-CH), 42.0 (7-CH2), 34.8 (3-CH2), 11.0 (5-CCH3); HRMS (ESI-TOF) [M + H]+ C15H17O3 calcd 245.1172, found 245.1172.:9 ethyl acetate:dichloromethane) gave 17 (22 mg, 10%) as a yellow oil, a 70:30 mixture of endo-17:exo-17; IR (film): 3443, 2927, 1725, 1691, 1496, 1041. endo-17: 1H NMR (400 MHz, CDCl3) δ 7.32–7.26 (3H, m, 3,4,5-phenyl), 7.03–6.97 (2H, m, 2,6-phenyl), 4.34 (1H, dt, J = 11.0, 5.5 Hz, 6-CH), 3.09 (1H, app. t, J = 5.5 Hz, 5-CH), 2.75 (1H, m, 3-CHeq), 2.81–2.70 (2H, m, 3-CHax and 8-CH), 2.58 (1H, m, 4-CHeq), 2.44–2.25 (2H, m, 7-CH2), 1.82 (1H, m, 4-CHax), 0.95 (3H, s, 1-CCH3); 13C NMR (100 MHz, CDCl3) δ 208.5 (9-CH), 208.0 (2-CH), 138.2 (ipso-phenyl), 128.7 (phenyl), 128.6 (phenyl), 128.5 (phenyl), 71.2 (6-CH), 68.9 (1-C), 52.8 (5-CH), 51.2 (8-CH), 40.2 (3-CH2), 37.0 (7-CH2), 17.2 (4-CH2), 15.0 (1-CCH3); exo-17: 1H NMR (400 MHz, CDCl3) δ 7.32–7.26 (3H, m, 3,4,5-phenyl), 7.03–6.97 (2H, m, 2,6-phenyl), 4.52 (1H, dd, J = 2.5, 2.2 Hz, 6-CH), 3.45 (1H, dd, J = 13.9, 4.7 Hz, 5-CH), 2.96 (1H, m, 3-CHeq), 2.81–2.70 (2H, m, 3-CHax and 8-CH), 2.65–2.62 (1H, m, 4-CHeq), 2.44–2.45 (2H, m, 7-CH2), 2.11 (1H, m, 4-CHax), 1.00 (3H, s, 1-CCH3); 13C NMR (100 MHz, CDCl3) δ 209.2 (9-CH), 207.8 (2-CH), 138.5 (ipso-phenyl), 128.7 (phenyl), 128.6 (phenyl), 128.5 (phenyl), 75.0 (6-CH), 70.4 (1-C), 53.0 (5-CH), 52.2 (8-CH), 39.0 (3-CH2), 36.0 (7-CH2), 20.7 (4-CH2), 15.1 (1-CCH3). HRMS (ESI-TOF): m/z [M + H]+ C16H18O3 calcd 259.1329, found 259.1328.:41:14:2 and confirmed by oxidation to 20): 1H NMR (400 MHz, CDCl3) δ 3.93 and 3.78 (1H, m), 3.50 (1H, dd, J = 10.5, 4.5 Hz), 3.15–2.63 (2H, m), 2.40–0.90 (16H, m); 13C NMR (100 MHz, CDCl3) δ 216.4, 216.1, 215.2, 213.8, 213.40, 212.1, 211.7, 207.5, 83.9, 83.1, 81.2, 79.3, 77.4, 59.9, 59.8, 59.3, 53.0, 51.9, 51.7, 46.2, 45.8, 45.72, 45.4, 45.3, 44.1, 43.4, 37.1, 35.3, 33.4, 31.9, 31.5, 31.4, 29.4, 27.4, 26.3, 26.1, 26.1, 25.8, 25.5, 25.3, 24.6, 24.1, 19.2, 18.6, 18.5, 18.3, 17.2, 16.5; m/z (EI+, %) 225 (3), 224 (M+, 17), 196 (6), 138 (52), 123 (100); HRMS M+ C13H20O3 calcd 224.1407, found 224.1408.:28:26:5 and confirmed by oxidation to 21); IR (film): 3405, 2972, 2937, 1726, 1694, 1496, 1061, 1034 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.25 and 4.00 (1H, m), 3.20 and 2.90 (1H, m), 2.70–1.20 (5H, m), 1.25–0.80 (12H, m); 13C NMR (100 MHz, CDCl3) δ 216.9, 216.8, 214.9, 214.0, 213.6, 212.5, 211.5, 80.9, 76.3, 72.6, 70.6, 63.8, 63.6, 62.5, 61.9, 52.4, 52.2, 51.7, 51.2, 46.0, 45.7, 45.6, 45.4, 45.3, 42.6, 41.1, 39.8, 36.2, 36.1, 36.0, 34.6, 34.2, 31.9, 30.9, 30.7, 27.3, 27.1, 26.6, 26.5, 24.7, 24.6, 24.3, 24.1, 17.1, 17.0 (2 lines), 16.9, 16.4, 15.7, 15.6, 14.6; m/z (EI+, %) 225 (8), 224 (M+, 58), 196 (14), 178 (22), 138 (88), 123 (100); HRMS M+ C13H20O3 calcd 224.1407, found 224.1408.:15 mixture of epimers. On standing for 2 weeks, the mixture afforded trione 20 (191 mg, 94%) as pale green needles, mp 94–95 °C; IR (film): 2262, 1716, 1699, 1270, 1037 cm−1; 1H NMR (400 MHz, CD3CN) δ 3.69 (1H, dd, J = 10.8, 1.7 Hz, 5-CH), 2.51 (1H, app. septet, J = 10.8 Hz, 7-CH), 2.31 (1H, dd, J = 14.5, 10.8 Hz, 4-CHeq), 2.32–2.22 (2H, m, 8-CH2), 1.75 (1H, d (br), J = 14.5 Hz, 4-CHax), 1.25 (3H, s, 3-CCH3eq), 1.13 (3H, s, 1-CCH3), 1.00 (3H, s, 3-CCH3ax), 0.98 (3H, d, J = 6.3 Hz 7-CHCH3); 13C NMR (100 MHz, CD3CN) δ 215.6 (2-CO), 206.7 (9-CO), 205.4 (6-CO), 64.8 (5-CH), 60.6 (1-C), 45.6 (3-C), 41.7 (8-CH2), 38.9 (7-CHCH3), 36.4 (4-CH2), 26.3 (2-CCH3eq), 24.3 (2-CCH3ax), 18.7 (1-CCH3), 13.7 (7-CHCH3); m/z (EI+, %) 223 (9), 222 (M+, 66), 194 (36), 138 (100), 123 (97); HRMS M+ C13H18O3 calcd 222.1251, found 222.1251.:9 ethyl acetate:dichloromethane) to give trione 21 (15 mg, 30%) as a 70:30 mixture of 8-endo-21:8-exo-21. IR (film): 2975, 1741, 1716, 1698, 1455 cm−1; 8-endo-21: 1H NMR (400 MHz, CDCl3) δ 3.71 (1H, dt, J = 10.7, 1.5 Hz, 5-CH), 2.57 (1H, dd, J = 15.5, 6.3 Hz, 7-CHeq), 2.47 (1H, m, 8-CH), 2.35–2.21 (2H, m, 7-CHax and 4-CHeq), 1.68 (1H, m, 4-CHax), 1.31 (3H, s, 3-CCH3eq), 1.18 (3H, s, 1-CCH3), 1.08 (3H, s, 3-CCH3ax), 0.82 (3H, d, J = 7.2 Hz, 8-CHCH3); 13C NMR (100 MHz, CDCl3) δ 215.2 (2-CO), 205.0 (9-CO), 204.2 (6-CO), 64.4 (5-CH), 63.0 (1-C), 45.2 (3-C), 42.6 (7-CH2), 37.4 (8-CH), 36.9 (4-CH2), 27.0 (3-CCH3eq), 24.8 (3-CCH3ax), 16.6 (1-CCH3), 15.3 (8-CHCH3); 8-exo-21: 1H NMR (400 MHz, CDCl3) δ 3.73 (1H, dt, J = 10.8, 1.8 Hz, 5-CH), 2.41 (1H, dd, J = 4.7, 1.8 Hz, 7-CHeq), 2.32–2.05 (3H, m, 7-CHeq, 4-CHeq and 8-CH), 1.75 (1H, m, 4-CHax), 1.36 (3H, s, 3-CCH3eq), 1.22 (3H, d, J = 6.7 Hz, 8-CHCH3), 1.17 (3H, s, 1-CCH3), 1.02 (3H, s, 3-CCH3ax); 13C NMR (100 MHz, CDCl3) δ 213.6 (2-CO), 205.5 (9-CO), 203.8 (6-CO), 63.1 (5-CH), 62.0 (1-C), 45.1 (3-C), 43.8 (7-CH2), 37.2 (4-CH2), 35.8 (8-CH), 27.4 (3-CCH3eq), 24.7 (3-CCH3ax), 17.3 (1-CCH3), 15.1 (8-CHCH3); m/z (EI+, %) 223 (7), 222 (M+, 49), 194 (19), 179 (62); HRMS M+ C13H18O3 calcd 222.1251, found 222.1251.Acknowledgements
The assistance of Dr Anders Poulsen (Experimental Therapeutics Centre (ETC), A*STAR, Singapore) in performing computational calculations and of Ms Doris Tan (ICES) for high-resolution mass spectrometric measurements is gratefully acknowledged. Financial support for a studentship (to R. P.) from the EPSRC Centre for Doctoral Training in Molecular Modelling & Materials Science, University College London and from the A*STAR Graduate Academy (A*GA), Singapore is also gratefully acknowledged.References
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed
.
- C. M. Marson, Chem. Soc. Rev., 2011, 40, 5514 RSC
-
(a) J. A. Peters, Synthesis, 1979, 321 CrossRef CAS
-
(a) M.-H. Filippini and J. Rodriguez, Chem. Rev., 1999, 99, 27 CrossRef CAS PubMed
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed
- J.-A. Richard, R. H. Pouwer and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2012, 51, 4536 CrossRef CAS PubMed
- J.-A. Richard, Eur. J. Org. Chem., 2014, 273 CrossRef CAS
- J. T. Njardarson, Tetrahedron, 2011, 67, 7631 CrossRef CAS PubMed
-
(a) B. A. Sparling, D. C. Moebius and M. D. Shair, J. Am. Chem. Soc., 2013, 135, 644 CrossRef CAS PubMed
- P. Adam, D. Arigoni, A. Bacher and W. Eisenreich, J. Med. Chem., 2002, 45, 4786 CrossRef CAS PubMed
- J. H. Boyce and J. A. Porco Jr, Angew. Chem., Int. Ed., 2014, 53, 7832 CrossRef CAS PubMed
- J. Qi and J. A. Porco Jr, J. Am. Chem. Soc., 2007, 129, 12682 CrossRef CAS PubMed
-
(a) J. Qi, A. B. Beeler, Q. Zhang and J. A. Porco Jr, J. Am. Chem. Soc., 2010, 132, 13642 CrossRef CAS PubMed
- A. J. Grenning, J. H. Boyce and J. A. Porco Jr, J. Am. Chem. Soc., 2014, 136, 11799 CrossRef CAS PubMed
- F. Buono and A. Tenaglia, Synlett, 1998, 1153 CrossRef CAS
- K.-H. Schönwälder, P. Kollatt, J. J. Stezowski and F. Effenberger, Chem. Ber., 1984, 117, 3280 CrossRef
- S. J. Spessard and B. M. Stoltz, Org. Lett., 2002, 4, 1943 CrossRef CAS PubMed
- V. Rodeschini, N. M. Ahmad and N. S. Simpkins, Org. Lett., 2006, 8, 5283 CrossRef CAS PubMed
- C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed
- T. Pouplin, B. Tolon, P. Nuhant, B. Delpech and C. Marazano, Eur. J. Org. Chem., 2007, 5117 CrossRef CAS
- K. C. Nicolaou, G. E. A. Carenzi and V. Jeso, Angew. Chem., Int. Ed., 2005, 44, 3895 CrossRef CAS PubMed
- T. Boddaert, Y. Coquerel and J. Rodriguez, Chem.–Eur. J., 2011, 17, 2266 CrossRef CAS PubMed
- D. Gravel and M. Lebelle, Can. J. Chem., 1985, 63, 1874 CrossRef CAS
- W. G. Dauben and R. A. Bunce, J. Org. Chem., 1983, 48, 4642 CrossRef CAS
- T. A. Spencer, H. S. Neel, D. C. Ward and K. L. Williamson, J. Org. Chem., 1966, 31, 434 CrossRef CAS
- The crystallographic data for structures 6 and 20 have been deposited with the Cambridge Crystallographic Data centre (CCDC).
- E. Harder, W. Damm, J. Maple, C. Wu, M. Rebout, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281 CrossRef CAS PubMed
- M. R. Garnsey, D. Lim, J. M. Yost and D. M. Coltart, Org. Lett., 2010, 12, 5234 CrossRef CAS PubMed
- S. P. Schröder, N. J. Taylor, P. Jackson and V. Franckevicius, Org. Lett., 2013, 15, 3778 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1476517 and 1485262. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra23523a |
| This journal is © The Royal Society of Chemistry 2016 |