
Adsorption and photocatalytic degradation behaviors of rhodamine dyes on surface-fluorinated TiO2 under visible irradiation†
Jing Guoa,
Shaojun Yuana,
Wei Jianga,
Hairong Yuea,
Zhe Cuib and
Bin Liang*a
aMulti-phase Mass Transfer & Reaction Engineering Lab, College of Chemical Engineering, Sichuan University, Chengdu 610065, China. E-mail: liangbin@scu.edu.cn; Fax: +86-28-85460556; Tel: +86-28-85460556
bCollege of Material Science and Engineering, Zhengzhou University, Zhengzhou, 450000, China
First published on 3rd December 2015
Abstract
Surface-fluorinated TiO2 (F-TiO2) particles was synthesized by a simple fluorosilanization method to serve as a visible-light photocatalyst for rapid degradation of Rhodamine B (RhB) dyes. The fluroalkylsilane (FAS-17) was covalently immobilized on the TiO2 particles via robust Si–O bonds. The changes in surface properties of the F-TiO2 particles were characterized by X-ray photoelectron spectroscopy (XPS), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), scanning electron microscope (SEM) and zeta potential measurements. The optical property of the F-TiO2 particles was determined by UV-visible spectroscopy. Upon the fluorination modification, the zeta potential of TiO2 particles switched from positive to negative, whilst the optical property of bulk TiO2 particles remained almost unchanged. Photodegradation experimental results demonstrated that zwitterionic RhB dyes were more favorably adsorbed on F-TiO2 rather than on the Ti(IV) sites of the pristine TiO2, and that the photodegradation reaction of RhB on F-TiO2 proceeded much faster than that on the pristine TiO2. However, the adsorption and photodegradation rate of anionic methyl orange (MO) dyes did not show an obvious change on F-TiO2. These results suggested that the molecular structure of dyes played a key role in visible-light photodegradation reactions on F-TiO2 particles. Positive-charged diethylamine groups of RhB molecular structures were postulated to promote the adsorption of RhB dyes on the surface of F-TiO2 particles, thus leading to easy electron injection and induction of rapid photodegradation under visible-light illumination.
1. Introduction
Industrial development is pervasively connected with a copious amount of various toxic pollutants, which are hazardous to the environment, human health, and difficult to degrade by natural means. Conventional physicochemical and biological treatment methods, such as coagulation, adsorption, filtration, and anaerobic granular sludge, have been the main-stream techniques for the purification of organic compound-contaminated water over the past decades. However, these approaches are less effective to cleanse the anthropogenic organic pollutants at very low concentrations.1 Photocatalysis, as an environmentally-benign approach, has therefore been attractive to treat persistent low-concentration organic pollutants in recent years.2TiO2 is the most extensively-studied photocatalyst in the decontamination of organic pollutants.3 Under light illumination beyond its band-gap limits, TiO2 shows strong oxidation ability to oxidize almost all organic compounds into CO2. However, one obstacle toward its practical application is that TiO2 can only be activated to induce photocatalytic reaction, by ultraviolet (UV) light, which accounts for only about 4% of the incoming sunlight. In view of practical applications, it is difficult to find an alternative photocatalyst as versatile, stable, abundant, environmentally-benign and economical as TiO2. Consequently, modification of TiO2 particles has been usually performed to achieve visible-light photosensitivity for effective photodegradation of organic pollutants under solar irradiation.
Various strategies have been proposed to modify TiO2-based catalysts for enhanced visible light performance.4–7 The doping (or incorporation) of metallic nano-materials, such as Cu2O, WO3, Ag and Au, into TiO2 lattice has been found effective to improve the photocatalytic activities of TiO2 in the visible light region. TiO2 doped with nonmetallic elements, such as N,8 S,9 C,10 I,11 Br12 and Cl,13 has also been proven to shift the optical absorption edge of TiO2 toward lower energy. A facile method to synthesize heterogeneous crystalline TiO2–halloysite nanotubes with anatase/rutile mixed phase has been described recently. The as-synthesized catalyst was found to have higher photocatalytic degradation of rhodamine B (RhB) and gentian violet than the commercial titania P25.14 TiO2 hybridized with a conducting polymer such as polyaniline has also verified to enhance photocatalytic activity towards the decomposition of organic molecules when irradiated with visible light.15 In a whole, the above doping approaches are aimed to alter the absorption band of bulk TiO2 to visible region by either narrowing the band-gap of TiO2 or introducing new absorption band. Most of them focus mainly on the improvement of the visible response of TiO2 by changing band-gap and other thermodynamic properties, but the kinetic properties are seldom investigated. Heterogeneous photocatalytic reactions primarily take place on the TiO2 surface, hence surface features of TiO2 are critical to the photocatalytic efficiencies. However, surface modification of TiO2 does not shift the band energy to the visible light regions, it usually alters the adsorption, surface acidity, surface charge, and surface functional groups of TiO2 surfaces, thus leading the change of the photocatalytic activities and mechanisms.16
In recent years, photosensitization of dyes has been reported to be one of the most effective ways to extend the photoresponse of TiO2 into the visible region.17,18 In such a system, the dye pollutants rather than the TiO2 photocatalyst are subject to the visible light excitation. The dye molecules are excited by visible-light illumination to transfer electrons into the conduction band of TiO2, thus resulting in the formation of cationic radicals of the dyes. The injected electrons then react with O2 molecules adsorbed on the surface of TiO2 to generate a series of active oxygen radicals, such as O2−˙, HO˙, and H2O2 species. The subsequent radical chain reactions lead to the degradation and mineralization of the dye pollutants.19–21 Because the photosensitization of dyes under visible irradiation can be initiated by the interfacial electron injection from the excited dye molecule into the TiO2 catalyst, more adsorption of dyes may enhance interfacial interactions between dyes and TiO2 to improve the photosensitization ability. Zhao et al. reported that rapid and more adsorption of dyes on the TiO2 particles caused the substantial enhancement of the extrinsic adsorption bands, thus leading to the increase in the degradation rate.22
Among various strategies to modify TiO2 particles, surface fluorination of TiO2 has been widely investigated.23,24 The fluorine-doped or surface fluorinated TiO2 particles exhibited the enhanced photocatalytic activity under the UV and visible light illumination. Wang et al. reported that surface fluorination not only changed the adsorption modes of dyes on the fluorinated TiO2, but also caused the dramatic change in the photocatalytic degradation kinetics and mechanisms of the adsorbed dyes.21 Surface fluorinated TiO2 has been also reported to be more effective than pure TiO2 for the photocatalytic oxidation of acid orange 7 and phenol,23 as they could enhance both photocatalytic reactions and photoelectrochemical behaviors. In the previous studies, surface fluorinations were also achieved by adding NaF into the dispersions under acidic conditions (pH 3–4), thus resulting in the replacement of surface hydroxyl groups by fluoride anion (F−),23,25 or the dissolution of anatase TiO2 to etch the surface of rutile TiO2 by HF.21,26 The NaF-modified TiO2 was found to significantly enhance the photocatalytic activities to dyes under UV light irradiation, but its photocatalytic ability was lower than the pristine TiO2 under visible light irradiation.23 However, to the best of our knowledge, few researches have been documented for the surface fluorination of TiO2 particles by a simple fluorosilanization process.
Herein, the aim of this work is to fluorinate TiO2 particles by fluorosilanization for enhanced photocatalytic degradation of dyes under visible light illumination. The fluorosilane FAS-17 was chosen due to its low surface energy, abundant fluorine atoms and big electronegativity. The surface-fluorinated TiO2 (F-TiO2) particles were characterized by X-ray photoelectron spectroscopy (XPS), X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), scanning electron microscope (SEM) and zeta potential measurements. The change in optical properties of the F-TiO2 surface was determined by UV-visible spectroscopy. The adsorption of a zwitterionic RhB dye and an anionic MO dye on the F-TiO2 surface were investigated, and the photodegradation behaviors of these two dyes were determined under visible light illumination. The postulated degradation mechanism of the RhB dye on the F-TiO2 particles was proposed to interpret the photodegradation behaviors of dyes under visible light illumination.
2. Experimental section
2.1 Materials
Powder anatase TiO2 was kindly provided by Taihai Co. (Panzhihua City, China). The chemicals, such as rhodamine B (RhB), and methyl orange (MO), rhodamine 101 inner salt (Rh 101 inner), rhodamine 6G (Rh 6G) and solvents, such as ethanol, were obtained from Aladdin Reagent Co. (Shanghai, China) and were used as received without further purifications. 1H,1H,2H,2H-Perfluorodecyltriethoxysilane (FAS-17) was purchased from Sicong Chemicals Co. (Xiamen, China). The deionized water used in the following experiments was purified using an Ultrapure reverse osmosis system (Ultrapure Technol. Co., Chengdu, China), and were used throughout the study. The molecular structures of RhB, Rh 101 inner, Rh 6G, and MO were schematically shown in ESI (Fig. S1†).2.2 Preparation of surface-fluorinated TiO2 particles
The surface fluorination of TiO2 particles with FAS-17 was performed using a typical procedure similar to those described previously.27 A 0.3 mL aliquot of FAS-17 was firstly hydrolyzed in a 5 mL ethanol solution at ambient temperature under continuous stirring for 2 h. A 0.7 g aliquot of TiO2 particles was ultrasonically dispersed in a 150 mL round flask containing 55 mL of ethanol for 30 min. Subsequently, the FAS-17 mixture was added dropwise into the round flask under continuous stirring. The fluorosilanization of TiO2 particles was allowed to proceed at 70 °C for 5 h with a reflux condenser. At the end of reaction, the surface-fluorinated TiO2 (defined as F-TiO2) particles were harvested by centrifugation at 6000 rpm and ultrasonically washed three times with copious amount of ethanol to remove the physically-adsorbed FAS-17, if any. The as-synthesized F-TiO2 particles were finally dried in a vacuum oven at 90 °C overnight.2.3 Surface characterization
The crystalline structure of TiO2 particles before and after surface fluorination was characterized by X-ray powder diffraction (XRD) (DX-2700, Dandong, China). The voltage and anode current were 40 kV and 30 mA, respectively. The monochromatic Cu Kα (1.54056 Å) and scanning mode with interval of 0.03° and set time of 0.05 s were used to collect the XRD pattern of TiO2 particles. The change in surface morphology of the F-TiO2 particles was characterized by a scanning electron microscope (SEM) (S-4800, Hitachi, Japan). The particle size distribution and zeta potential of the TiO2 particles suspensions (0.1 g L−1) were determined respectively, by dynamic laser scanning Nanoparticle and Zeta potential analyzer (ZEN3690, Malvern, UK), and the pH was adjusted between pH 2.5 and 11.0 using a 1 mol L−1 NaOH and HCl solutions. The optical properties of the pristine TiO2 and F-TiO2 particles were characterized by a UV-visible spectrophotometer with an integrating sphere (UV2100, Shimadzu, Japan). The surface composition of the F-TiO2 was determined by X-ray photoelectron spectroscopy (XPS) (XSAM800, Kratos, UK) with monochromatized Al Kα radiation at constant dwell time of 100 ms and pass energy of 40 eV. The pressure in the analysis chamber was maintained at 10−7 to 10−9 Torr during each measurement. To compensate the surface charging effect, all the core-level spectra were referred to the C 1s hydrocarbon peak with binding energy (BE) at 284.6 eV. The photoluminescence spectra (PL) were recorded via a fluorescence spectrometer (F-7000, Hitachi, Japan).The FTIR spectra of the RhB-adsorbed pristine TiO2 and F-TiO2 particles were obtained on a Fourier transform infrared spectrometer (Spectrum II L1600300, PerkinElmer) in the transmission mode. Prior to the measurement, the RhB adsorption process was as follows: the pristine TiO2 and F-TiO2 particles were dispersed separately in 16 mg L−1 solution of RhB with highly-speed stirring, and then allowed to adsorb overnight to reach the adsorption equilibrium. The RhB-adsorbed particles were subsequently harvested by centrifugation, and then dried in air at 35 °C. All the operations were carried out in dark to prevent photodegradation of the adsorbed dyes. The TiO2 particles dispersed in deionized water under the same conditions were used as the control for FTIR measurement.
2.4 Adsorption and visible light photocatalysis activities of modified F-TiO2 particles
The photocatalytic degradation of RhB and MO was investigated to determine the photocatalytic activities of F-TiO2 particles under visible light illumination. A photoreactor equipped with a 1000 W xenon lamp was used to perform the photocatalysis experiments (Institute of Electric Light Source, Kaifeng, China). Typically, the xenon lamp was positioned inside a cylindrical Pyrex vessel and surrounded by a circulating water Pyrex jacket, which served to cool the lamp for stable reaction temperature. A cutoff filter was placed outside the Pyrex jacket to secure visible-light irradiation only.To eliminate the interference caused by adsorption on the photodegradation behaviors of dyes on the pristine TiO2 and F-TiO2 particles, the adsorption profiles of zwitterionic RhB and anionic MO dyes were determined prior to the photocatalytic experiments. Typically, a 100 mg aliquot of particles was well dispersed in 10 mL of 16 mg L−1 dye aqueous solution by highly-speed stirring, and the adsorption was allowed to proceed for a predetermined time at ambient temperature. All the operations were performed in the dark to prevent the photocatalytic reactions. The dye concentration after adsorption was determined by a UV-visible spectrophotometer.
In the photocatalytic experiments, the TiO2 suspension was prepared by adding 100 mg of TiO2 particles to 10 mL of 16 mg L−1 dye (RhB or MO) aqueous solution, or alternatively adding 150 mg of TiO2 particles to 30 mL of 10 mg L−1 dye (RhB, Rh 6G, or Rh 101 inner). Prior to visible light irradiation, the suspension was stirred in the dark for 1 h to reach the adsorption/desorption equilibrium on the TiO2 particle surfaces under ambient air-equilibrated conditions. During the light irradiation process, the suspension was allowed to proceed under vigorous stirring for a predetermined time. At the end of photocatalytic reaction, the suspension was separated by centrifugation at 8000 rpm to remove the solid particles, and the supernatant was collected for further analyses. The concentration of the RhB, MO, Rh 6G and Rh 101 inner solution was determined by UV-visible spectrophotometry (TU-1810, Persee, China) at a wavelength of 553 nm, 465 nm, 526 nm and 575 nm, respectively.
3. Results and discussion
3.1 Surface properties and structure of the F-TiO2 particles
The surface fluorination of TiO2 particles was achieved by fluorosilanizing the TiO2 particles with FAS-17, as schematically illustrated in Fig. 1. The reaction involves the hydrolysis of FAS-17 to convert siloxane groups (Si–OC2H5) to silanol groups (–Si–OH) and the fluorosilanization of TiO2 particles. TiO2 particles have been widely recognized to possess abundant hydroxyl groups (–OH) on the surface.28 The ligand-exchanging reaction between the surface hydroxyl groups (–OH) of TiO2 and Si–OH of FAS-17 results in the formation of robust Si–O–Ti bonds.29,30 Meanwhile, the dehydration condensation reaction among the –Si–OH groups leads to the formation of polysiloxanes. The surface characteristics, crystalline structure and size distribution of the as-synthesized F-TiO2 particles were characterized by SEM, XPS, XRD, UV-visible and zeta potential measurements. The detailed results are discussed as follows. | ||
| Fig. 1 Schematic illustration of the fluorosilanization process of FAS-17 onto the TiO2 particles surfaces. | ||
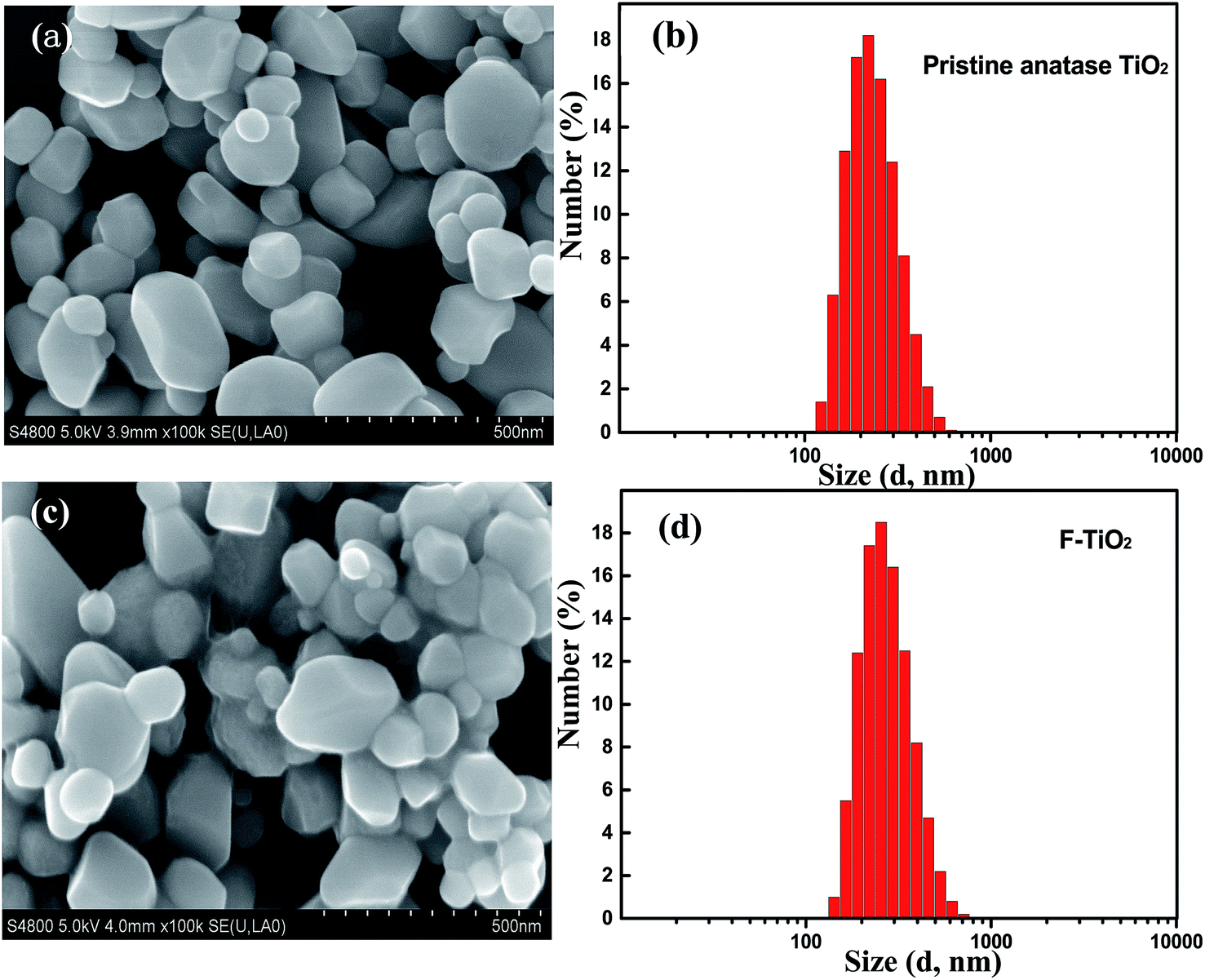 | ||
| Fig. 2 The SEM images and size distribution diagrams of (a and b) the pristine anatase TiO2 and (c and d) the F-TiO2 particles. | ||
The surface composition of the FAS-17-modified TiO2 was also characterized by XPS. Fig. 3 shows the wide scan, C 1s and F 1s core-level XPS spectra of the pristine TiO2 and F-TiO2 particles, respectively. In comparison with the wide scan XPS spectrum of the pristine TiO2 surface (Fig. 3a), four additional photoelectron lines with binding energies (BEs) at 99, 151, 685 and 832 eV, attributable to Si 2p, Si 2s, F 1s and F KLL species,34 respectively, are discernible in the wide scan spectrum of the F-TiO2 particle surface (Fig. 3a), indicative of the successfully immobilization of FAS-17 layer onto the TiO2 surface. The presence of FAS-17 layer on the TiO2 surface can also be deduced from the curve-fitted C 1s core-level spectrum, which consists of six peak components with BEs at 283.1, 284.8, 285.6, 289.4, 291.4 and 293.8 eV, attributable to C–Si, C–H, C–CFx, CF–CFx, C–F2 and C–F3 species,35 respectively (Fig. 3b), as well as the appearance of the F 1s core-level spectrum with BE at 689 eV (inset of Fig. 3b). The fluorine peak components of C–CFx, CF–CFx, C–F2 and C–F3 species are characteristics of the FAS-17 molecules.27 Thus, the surface fluorination of TiO2 particles has been successfully synthesized by covalently immobilizing of FAS-17.
 | ||
| Fig. 3 (a) Wide scan XPS spectra of the pristine TiO2 (red line) and F-TiO2 (black line) surfaces (b) the C 1s and F 1s core-level XPS spectra of the F-TiO2 particle surface. | ||
3.2 Photocatalytic activities of the modified F-TiO2 under visible light irradiation
 | ||
| Fig. 4 The adsorption profiles of (a) zwitterionic RhB and anionic MO on the pristine TiO2 and (b) F-TiO2 particles as a function of time in the dark. Adsorption conditions: initial dye concentration (C0) of 16 mg L−1, temperature: at room temperature. | ||
 | ||
| Fig. 5 (a) The photocatalytic degradation curves of RhB without and with the presence of TiO2 particles as a function of time under visible light illumination, (b) the photodegradation curve of RhB after subtracting the adsorbed amount with the presence of TiO2 and F-TiO2 particles. Reaction conditions: (a) initial dye concentration (C0) of 16 mg L−1, catalyst dosage of 10 g L−1, (b) initial dye concentration (C0) of 10 mg L−1, catalyst dosage of 5 g L−1, light intensity: 1000 W xenon lights. | ||
To investigate the role of surface fluorination in affecting the photodegradation kinetics and mechanisms of dyes, the photodegradation of anionic MO dyes were also examined, and the photocatalytic degradation behaviors of the anionic MO dyes for the pristine TiO2 and F-TiO2 particles under visible-light irradiation are show in Fig. 6. The MO dyes remains unchanged throughout under visible-light irradiation in the absence of TiO2 particles. Particularly, the MO dyes also remain relatively stable in the presence of photocatalysts, since the photodegradation ratios of the MO dyes by the pristine TiO2 and the F-TiO2 are only about 8% and about 11%, respectively, after 120 min of visible-light irradiation. These results suggest that the surface fluorination of TiO2 particles cannot improve the visible-light photocatalytic activity of the anionic MO dyes. Taking together, the surface fluorination of TiO2 particles can only lead to the selectively-enhanced photocatalytic activity to the zwitterionic RhB dyes rather than to the anionic MO dyes. This phenomenon is possibly ascribed to the enhanced pre-adsorption of RhB and the intrinsic sensitivity of RhB to visible light illumination for the F-TiO2 particles. It has been reported that the adsorption behaviors of cationic dyes on the catalyst surface play an important role in improving their photocatalytic degradation rate.22
 | ||
| Fig. 6 The photocatalytic degradation curves of MO with or without the presence of TiO2 particles as a function of time under visible light illumination. Reaction conditions: initial dye concentration (C0) of 16 mg L−1, catalyst dosage of 10 g L−1, light intensity: 1000 W xenon lights. | ||
The pre-adsorption of the zwitterionic RhB dyes on the F-TiO2 particles rapidly reaches as high as 60% prior to the visible light irradiation (Fig. 4 and 5a), while the anionic MO dyes show almost no pre-adsorption on the F-TiO2 particles. As a consequence, the enhancement in visible-light photocatalytic degradation of the zwitterionic RhB dyes is probably ascribed to, the enhanced pre-adsorption and the intrinsic visible-light sensitivity of zwitterionic RhB dyes, as compared to the anionic MO dyes. To further confirm the effect of dye types and pre-adsorption of dyes on the photocatalytic ability, other two cationic dyes such as Rh 6G and Rh 101 inner were chosen to perform the photocatalytic degradation experiments by the pristine TiO2 and F-TiO2 particles. Similar to that of the zwitterionic RhB, the photocatalytic degradation ratios of the two cationic dyes by F-TiO2 particles show a significant increase under visible light irradiation, and both reach higher than 95% (ESI, Fig. S6†). These above results are in good agreement with the previous findings that the dye structure and their anchoring groups on the photocatalyst are key factors to determine the interfacial electron transfer and the degradation ratio of the dye.19
 | ||
| Fig. 7 FTIR spectra of the (a) as-synthesized F-TiO2, (b) F-TiO2 adsorbed with RhB, and (c) F-TiO2 after degradation of the adsorbed RhB. | ||
3.3 The postulated visible-light photocatalytic mechanisms of RhB dyes on the F-TiO2 particles
Under visible light irradiation, the degradation of zwitterionic RhB dyes is substantially enhanced in the presence of F-TiO2 particles, while the degradation of anionic MO dyes shows no significant change. Therefore, it can be speculated that the enhanced photodegradation of RhB dyes is probably not caused by the intrinsic change of F-TiO2 particles, but is more possibly associated with dye sensitization. As liquid–solid heterogeneous photocatalysis is a well-known interface-based process, the contact of dye molecules with the surface of catalyst is prerequisite to efficient electron transfer. The enhanced adsorption of dyes can also eliminate the outer diffusion control in surface reaction. Therefore, the enhanced adsorption of photosensitive dyes will partly contribute to the high photocatalytic activity of F-TiO2. Take together, a postulated mechanism of the enhanced visible-light photocatalytic activity of RhB on the F-TiO2 particles has been proposed, and is schematically illustrated in Fig. 8. The positively-charged TiO2 particle surfaces show the electrostatic repulsion to resist the adsorption of RhB dyes (Fig. 8a), while the negatively-charged F-TiO2 surfaces show strong affinity to attract RhB dyes (Fig. 8b). According to the adsorption results (Fig. 4), the RhB dyes can rapidly adsorb onto the F-TiO2 surface to reach dynamic adsorption/desorption equilibrium. Thus, the pre-adsorption of RhB is an initial and important step to enhance visible-light photocatalytic degradation on the F-TiO2 particle surfaces.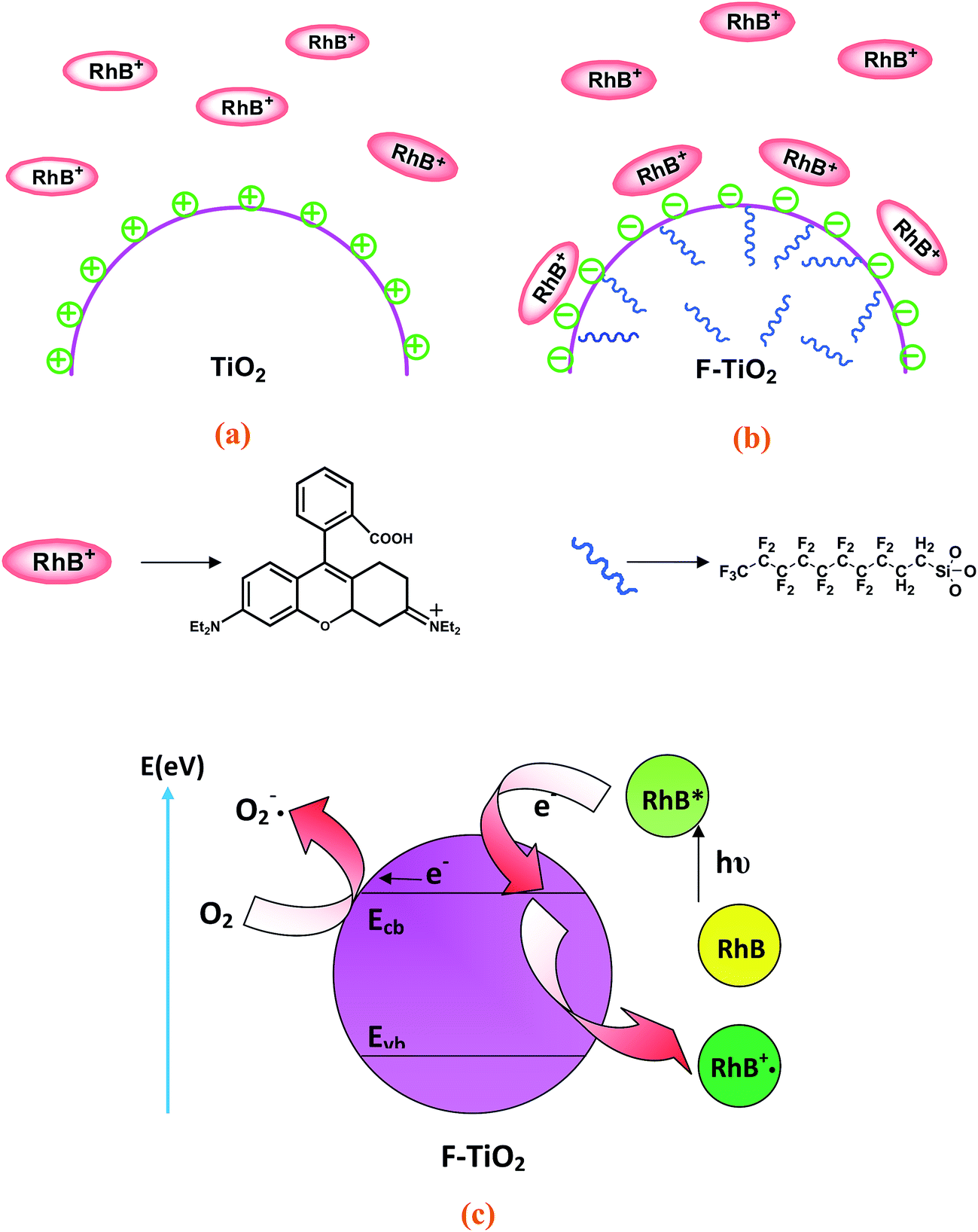 | ||
| Fig. 8 Schematic illustration of (a and b) the adsorption model of the cationic RhB dyes on the pristine TiO2 and F-TiO2 particle surface, and (c) electron-transfer processes and subsequent excitation of RhB dye on the F-TiO2 particle surfaces. | ||
Fig. 8c schematically illustrates the electron-transfer processes and subsequent excitation of RhB dye on the F-TiO2 particle surfaces. As a matter of fact, the TiO2 particles may be not excited as its absorption threshold is 385 nm under visible-light illumination, however, the adsorbed RhB on the TiO2 surface can be excited at a wavelength longer than 470 nm to produce singlet and triplet states (denoted as RhB*ads).22 The RhB*ads subsequently injects an electron into the conduction band (or to some surface states) of the TiO2 particles to convert RhB to the radical cation RhB˙+. In turn, the injected electron on the TiO2 particles, TiO2 (e−), can react with adsorbed oxidants (usually O2) to produce reactive oxygen radicals. The radical cation RhB˙+ ultimately reacts with reactive oxygen radicals and/or molecular oxygen to yield intermediate products or other radical species, for which might lead to mineralization if secondary radical processes occurred.41 As a consequence, the pre-adsorption of cationic RhB dyes and activation of the visible-light sensitive RhB dyes to inject electrons to TiO2 particles are important steps to enhance the visible-light photodegradation activities of the TiO2 particles.
To confirm the above postulated mechanism of the activation of pre-adsorbed RhB dyes to inject electron to TiO2 particles, the photoluminescence (PL) signals was obtained on a fluorescence spectrometer. The PL signals of semiconductor materials result from the recombination of photo-induced charge carriers. Generally, the lower PL intensity, the lower the recombination rate of photo-induced electron–hole pairs, and the higher the photocatalytic activity of semiconductor photocatalysts.42 Fig. 9 shows the PL emission spectra of pure TiO2, F-TiO2 and F-TiO2 adsorbed with RhB in the wavelength range 300–700 nm. The intensity of the PL spectrum of pure TiO2 is much higher than that of the F-TiO2 and F-TiO2/RhB samples. The intensity of F-TiO2/RhB is the lowest, it can be reasonably inferred that the enhanced adsorption of RhB dye should have a higher photocatalytic performance.
 | ||
| Fig. 9 The PL spectrum of the pristine anatase TiO2 (black line), the F-TiO2 (red line) and the RhB-adsorbed F-TiO2 (blue line). The adsorption experimental conditions: initial dye concentration (C0) of 100 mg L−1, catalyst dosage of 10 g L−1. | ||
4. Conclusions
With the objective to enhance the visible-light photocatalytic degradation activities of TiO2 to organic pollutants, a facile wet fluorosilanization approach was described to modify the anatase TiO2 particles. Various surface characterization techniques, including XPS, XRD, zeta potential, SEM, FTIR and UV-vis spectroscopy, were utilized to determine the surface characteristics change after the surface fluorination. The fluorination modification of TiO2 surfaces caused no significant changes in the surface morphology, crystalline phase, and optical properties, but the dramatic alternation of surface charges from positive to negative values. As a result, both the adsorption modes and degradation of the dyes were greatly altered. Adsorption experimental results revealed that the F-TiO2 particles had a high affinity to attract zwitterionic RhB dyes instead of anionic MO dyes due to the electrostatic interaction. The postulate mechanisms were proposed to interpret the electron transfer from the excited dyes to TiO2 particles rather than the intrinsic electron and holes of TiO2 particles.Acknowledgements
The authors would like to acknowledge the financial assistance of key project of National Natural Science Foundation of China (No. 21236004).References
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC
.
- K. Demeestere, J. Dewulf and H. van Langenhove, Crit. Rev. Environ. Sci. Technol., 2007, 37, 489–538 CrossRef CAS
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS
- D. Chatterjee and A. Mahata, J. Photochem. Photobiol., A, 2004, 165, 19–23 CrossRef CAS
- J. Choi, H. Park and M. R. Hoffmann, J. Phys. Chem. C, 2009, 114, 783–792 Search PubMed
- J. Lin, R. Zong, M. Zhou and Y. Zhu, Appl. Catal., B, 2009, 89, 425–431 CrossRef CAS
- B. Muktha, D. Mahanta, S. Patil and G. Madras, J. Solid State Chem., 2007, 180, 2986–2989 CrossRef CAS
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2004, 108, 10617–10620 CrossRef CAS
- T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., A, 2004, 265, 115–121 CrossRef CAS
- S. U. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed
- X. Hong, Z. Wang, W. Cai, F. Lu, J. Zhang, Y. Yang, N. Ma and Y. Liu, Chem. Mater., 2005, 17, 1548–1552 CrossRef CAS
- H. Irie, S. Washizuka, N. Yoshino and K. Hashimoto, Chem. Commun., 2003, 1298–1299 RSC
- S. Yamazaki, T. Tanimura, A. Yoshida and K. Hori, J. Phys. Chem. A, 2004, 108, 5183–5188 CrossRef CAS
- C. Li, J. Wang, S. Feng, Z. Yang and S. Ding, J. Mater. Chem. A, 2013, 1, 8045–8054 CAS
- C. Li, J. Wang, H. Guo and S. Ding, J. Colloid Interface Sci., 2015, 458, 1–13 CrossRef CAS PubMed
- X. Quan, J. Niu, S. Chen, J. Chen, Y. Zhao and F. Yang, Chemosphere, 2003, 52, 1749–1755 CrossRef CAS PubMed
- Y. Cho, W. Choi, C.-H. Lee, T. Hyeon and H.-I. Lee, Environ. Sci. Technol., 2001, 35, 966–970 CrossRef CAS PubMed
- T. Wu, T. Lin, J. Zhao, H. Hidaka and N. Serpone, Environ. Sci. Technol., 1999, 33, 1379–1387 CrossRef CAS
- D. Zhao, C. Chen, Y. Wang, W. Ma, J. Zhao, T. Rajh and L. Zang, Environ. Sci. Technol., 2007, 42, 308–314 CrossRef
- P. Chowdhury, J. Moreira, H. Gomaa and A. K. Ray, Ind. Eng. Chem. Res., 2012, 51, 4523–4532 CrossRef CAS
- Q. Wang, C. Chen, D. Zhao, W. Ma and J. Zhao, Langmuir, 2008, 24, 7338–7345 CrossRef CAS PubMed
- J. Zhao, T. Wu, K. Wu, K. Oikawa, H. Hidaka and N. Serpone, Environ. Sci. Technol., 1998, 32, 2394–2400 CrossRef CAS
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086–4093 CrossRef CAS
- J. Tang, H. Quan and J. Ye, Chem. Mater., 2007, 19, 116–122 CrossRef CAS
- J. Lee, W. Choi and J. Yoon, Environ. Sci. Technol., 2005, 39, 6800–6807 CrossRef CAS PubMed
- R. Nakamura, T. Okamura, N. Ohashi, A. Imanishi and Y. Nakato, J. Am. Chem. Soc., 2005, 127, 12975–12983 CrossRef CAS PubMed
- C. Kang, H. Lu, S. Yuan, D. Hong, K. Yan and B. Liang, Chem. Eng. J., 2012, 203, 1–8 CrossRef CAS
- L. G. Bach, M. Islam, S. Y. Seo and K. T. Lim, J. Appl. Polym. Sci., 2013, 127, 261–269 CrossRef CAS
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 2000, 16, 7268–7274 CrossRef CAS
- D. Schondelmaier, S. Cramm, R. Klingeler, J. Morenzin, C. Zilkens and W. Eberhardt, Langmuir, 2002, 18, 6242–6245 CrossRef CAS
- W.-C. Hung, S.-H. Fu, J.-J. Tseng, H. Chu and T.-H. Ko, Chemosphere, 2007, 66, 2142–2151 CrossRef CAS PubMed
- S. Sivakumar, P. K. Pillai, P. Mukundan and K. Warrier, Mater. Lett., 2002, 57, 330–335 CrossRef CAS
- S. Musić, M. Gotić, M. Ivanda, S. Popović, A. Turković, R. Trojko, A. Sekulić and K. Furić, J. Mater. Sci. Eng. B, 1997, 47, 33–40 CrossRef
- J. F. M. C. D. Wagner, J. E. Davis and W. M. Riggs, Handbook of X-ray Photoelectron Spectroscopy, Perkin-Elmer Corp., Eden Prairie, MN, USA, 1992 Search PubMed
- H. Liu, L. Pan, K. Shen, J. Lang, J. Shi, Q. Cui, H. Li and C. Liu, Fertil. Steril., 2009, 92, 1150–1152 CrossRef PubMed
- D. Li, H. Haneda, N. K. Labhsetwar, S. Hishita and N. Ohashi, Chem. Phys. Lett., 2005, 401, 579–584 CrossRef CAS
- T. Yamaki, T. Umebayashi, T. Sumita, S. Yamamoto, M. Maekawa, A. Kawasuso and H. Itoh, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 206, 254–258 CrossRef CAS
- D. Li, H. Haneda, N. Ohashi, S. Hishita and Y. Yoshikawa, Catal. Today, 2004, 93, 895–901 CrossRef
- D. Li, H. Haneda, N. K. Labhsetwar, S. Hishita and N. Ohashi, Chem. Phys. Lett., 2005, 401, 579–584 CrossRef CAS
- N. Mchedlov-Petrossyan, S. Shapovalov, S. Egorova, V. Kleshchevnikova and E. A. Cordova, Dyes Pigm., 1995, 28, 7–18 CrossRef CAS
- R. W. Matthews, Water Res., 1991, 25, 1169–1176 CrossRef CAS
- J.-G. Yu, H.-G. Yu, B. Cheng, X.-J. Zhao, J. C. Yu and W.-K. Ho, J. Phys. Chem. B, 2003, 107, 13871–13879 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Structures of all the dyes used (Fig. S1), FTIR spectra (Fig. S2), X-ray diffraction patterns (Fig. S3), the forbidden bandwidth of the pristine TiO2 and F-TiO2 particles (Fig. S4), the zeta potentials of the pristine TiO2 and F-TiO2 particles (Fig. S5), the photodegradation curves of Rh 6G and Rh 101 inner salt (Fig. S6), the forbidden bandwidth of the F-TiO2 after photodegradation (Fig. S7) and the photodegradation curves of the RhB dyes in the presence of recycled TiO2 and F-TiO2 particles (Fig. S8). See DOI: 10.1039/c5ra14379a |
| This journal is © The Royal Society of Chemistry 2016 |