
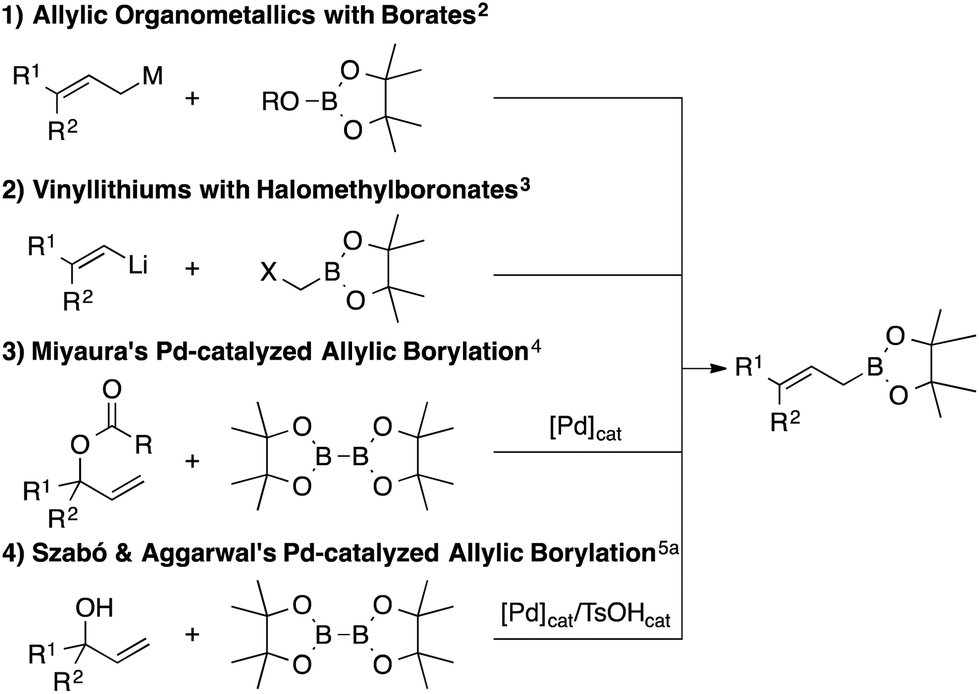
Allylic borylation of tertiary allylic alcohols: a divergent and straightforward access to allylic boronates†
Kohei
Harada‡
a,
Marina
Nogami‡
a,
Keiichi
Hirano
*a,
Daisuke
Kurauchi
a,
Hisano
Kato
a,
Kazunori
Miyamoto
a,
Tatsuo
Saito
a and
Masanobu
Uchiyama
*ab
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan. E-mail: k1hirano@mol.f.u-tokyo.ac.jp; uchiyama@mol.f.u-tokyo.ac.jp; Fax: +81 3 5841 0733; Tel: +81 3 5841 0277
bAdvanced Elements Chemistry Research Team, RIKEN, RIKEN Center for Sustainable Resource Science, and Elements Chemistry Laboratory, RIKEN, 2-1 Hirosawa, Wako-shi, Saitama, Japan. E-mail: uchi_yama@riken.jp; Fax: +81 48 467 2869; Tel: +81 48 467 2879
First published on 1st March 2016
Abstract
A facile and divergent synthetic process for converting tertiary allylic alcohols to multiply substituted allylic boronates is reported. This methodology is especially effective for tertiary alcohol substrates, providing the corresponding allylic boronates in moderate to good yields.
 Keiichi Hirano | Keiichi Hirano grew up in Nikko, Japan. He received his bachelor and master's degrees in pharmaceutical sciences from the University of Tokyo. Then, he moved to Marburg to pursue his doctoral studies supported by the DAAD fellowship and obtained his doctoral degree working with Prof. Dr. Frank Glorius at the Westfälishe Wilhelms-Universität Münster in 2009. He then carried out his postdoctoral study with Prof. Barry M. Trost at Stanford University supported by the Uehara Memorial Foundation Postdoctoral Fellowship. After another postdoctoral fellowship with Prof. Masanobu Uchiyama at the University of Tokyo funded by the JSPS postdoctoral fellowship, he was appointed as an assistant professor associated with Prof. Uchiyama in the Graduate School of Pharmaceutical Sciences at the University of Tokyo in 2012. |
 | ||
| Scheme 1 Synthesis of allylic boronates. | ||
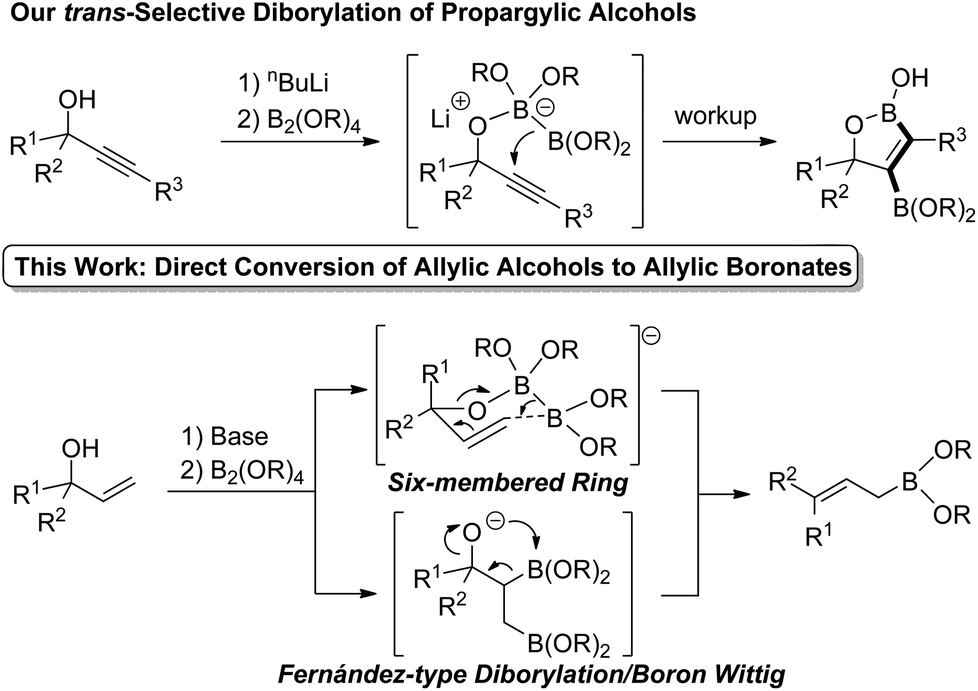
We recently reported the first trans-selective diborylation of alkynes (Scheme 2).6 This unique reactivity of electronically unactivated alkynes with a boryl anion equivalent was achieved by pseudo-intramolecular activation of diborons by propargylic alkoxide. If this approach could be applied to allylic alcohols as well, direct transformation of allylic alcohols to the corresponding allylic boronates via the Zimmerman–Traxler type six-membered cyclic transition state7 would be expected. On the other hand, Fernández-type alkoxide-mediated diborylation of alkenes8a followed by the boron-Wittig reaction9 may also result in the formation of allylic boronates. In this communication, we report a novel approach to a broad range of allylic boronates from allylic alcohols.
 | ||
| Scheme 2 Our approaches. | ||
We commenced our studies with the reaction between allylic alcohol 1a and B2pin2 as model substrates (Table 1). Compound 1a was deprotonated with nBuLi and treated with B2pin2 in THF at 140 °C to give the desired product 2a in 27% yield (entry 1). Next, we examined additives. We found that 5 equivalents of EtOH and tBuOH moderately increased the formation of 2a (entries 2 and 3). Addition of MeOH drastically improved the yield to 87% (entry 4). On the contrary, the use of water instead of MeOH significantly slowed the reaction (entry 5). Solvents were then briefly examined using MeOH as an additive. 1,4-Dioxane and 1,2-dimethoxyethane were less effective than THF (entries 6 and 7). Toluene was found to be another effective solvent (entry 8). The amount of MeOH also affected the yield. The product 2a was obtained in 90% yield by reducing the amount of MeOH to 3 equivalents (entry 9), but further reduction to 0.5 equivalents or addition of 10 equivalents was detrimental to the efficient formation of 2a (entries 10 and 11).
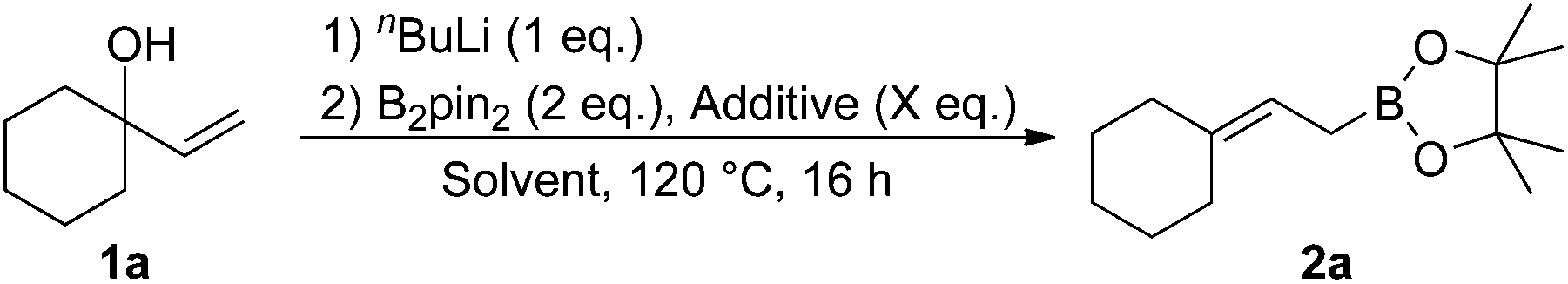
In order to further improve the yield and operational simplicity, we systematically screened inorganic bases (Table 2). tert-Butoxide bases gave lower yields than nBuLi (entries 1–3), and no significant effect of the counter cation (Li+, Na+, or K+) was observed. MeONa was found to be an appropriate base, giving 2a in 81% yield (entry 5). A combination of MeONa and 4 equivalents of MeOH as an additive gave 2a in 89% yield, and this set of reaction conditions appeared to be optimum (entry 7). Next, we examined the substrate scope (Table 3).
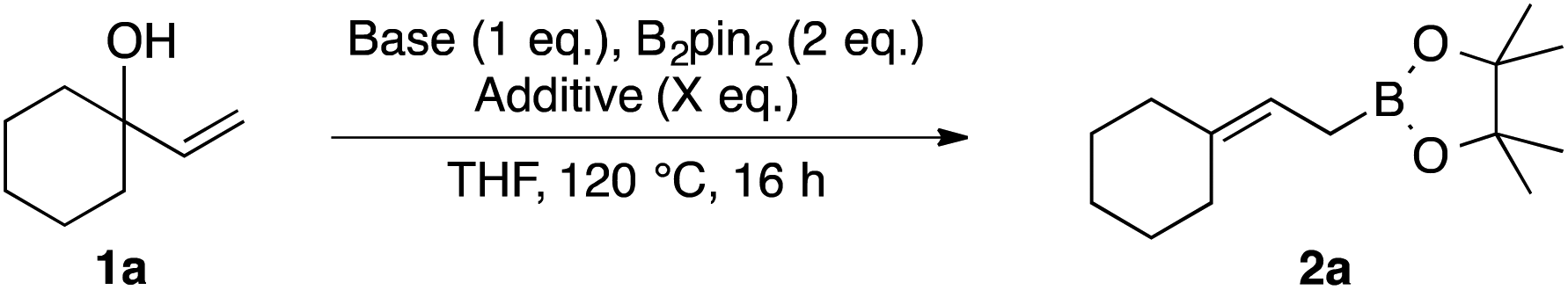
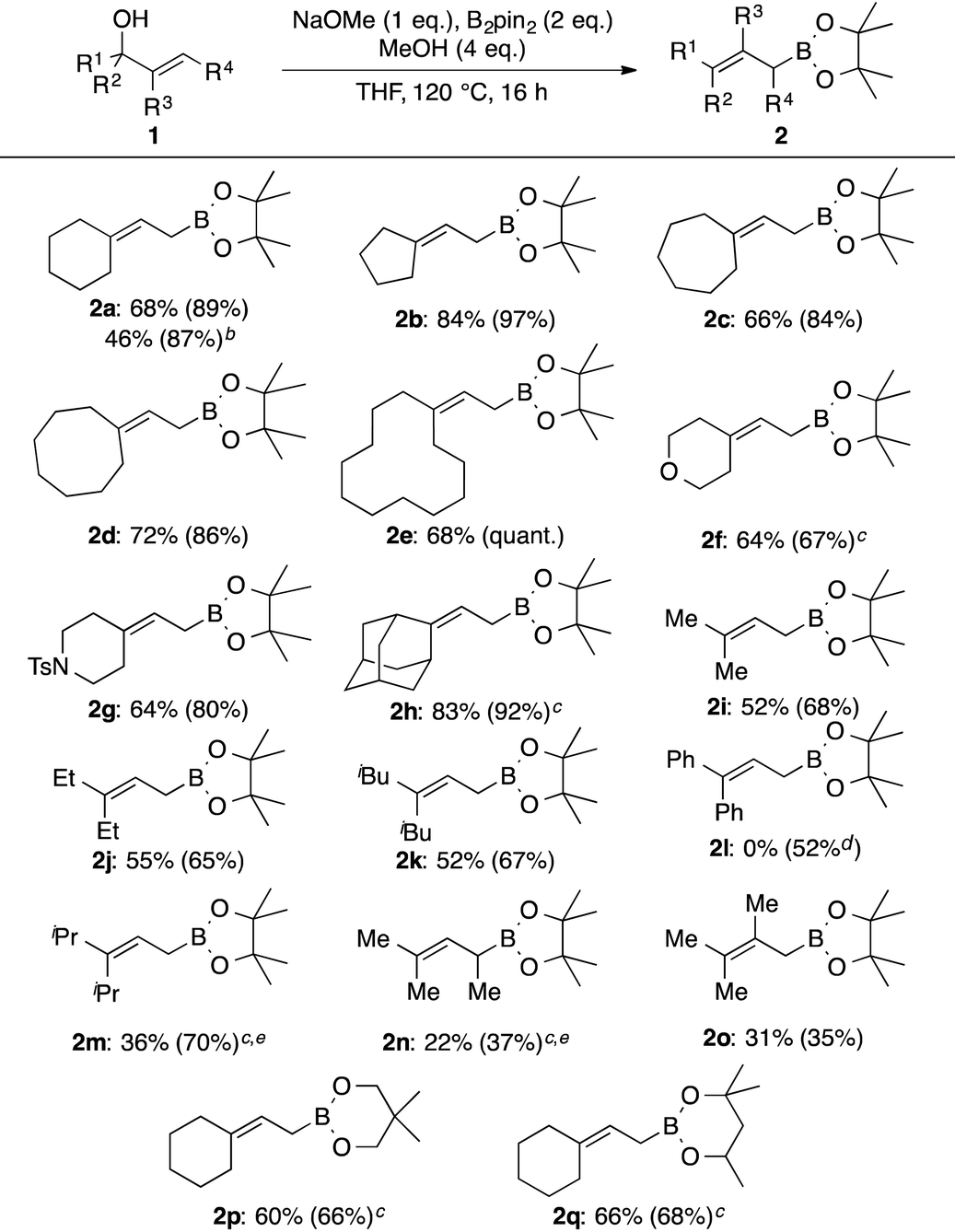
Allylic alcohols with a cyclic structure (1a–1h) were found to be generally suitable substrates for this borylation reaction. This reaction is applicable to a large-scale reaction to give the desired product 2a in high yield. The isolated yield was somewhat lower probably due to partial decomposition on silica gel during column chromatographic purification. Boronates with 5-, 6-, 7-,10 8-, and 12-membered rings were efficiently prepared in high yields (2a–2e). Heteroatoms in the ring were not deleterious to the reaction progress (2f, 2g). Even the sterically demanding adamantyl moiety did not shut down the reaction (2h). Acyclic substitution patterns at the terminal carbon of the double bond of the allylic boronates were well tolerated. Prenylboronate (2i) was obtained in 52% yield. Ethyl and isobutyl counterparts were also prepared in comparable yields to 2i (2j and 2k). On the other hand, the product with the diphenyl moiety was not obtained under these reaction conditions, and only the proto-deborylated product, 1-phenylprop-1-enylbenzene, was formed in 52% yield. This can be attributed to the lability of the C–B bond of the desired product 2l owing to the highly stable nature of the allylic anion delocalized over a double bond and benzene rings. A secondary alkyl group, such as isopropyl, decreased the reaction rate drastically and lowered the yield of the desired product 2m under the standard reaction conditions (10% NMR yield). Increasing the amount of B2pin2 to 3 equivalents at 140 °C improved the NMR yield of 2m to 70%. Allylic alcohol with an internal double bond 1n required harsher reaction conditions. Compound 2n was obtained in 27% NMR yield under the modified reaction conditions. A methyl group at the internal carbon of the double bond was tolerated (2o). Other diboron reagents participate in this borylation reaction. Reactions of 1a with bis(neopentyglycolato)diboron and bis(hexyleneglycolato)diboron gave the corresponding allylic boronates 2p and 2q in 60% and 66% yields, respectively. To demonstrate the utility of this methodology, the allylation reaction using 2f was additionally examined. In the presence of 10 mol% of Sc(OTf)3,11 4-Cl-benzaldehyde (3) was cleanly allylated to give 4 in 89% yield. Allylboronate 2h reacted reluctantly with 3 even under Aggarwal's conditions, used to activate highly substituted allylic boronates,12 presumably due to steric congestion around the reactive site.
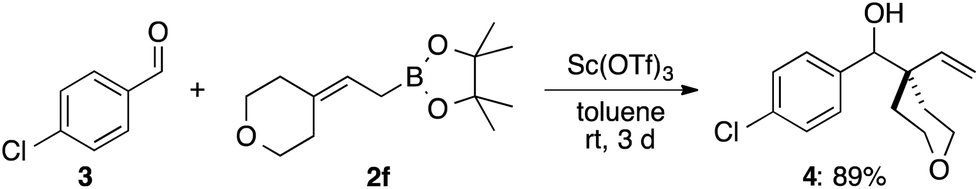
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5). If the chair-like six-membered cyclic transition state is operative, 1r should deliver the product 2r with a high degree of E-selectivity and the observed selectivity implies that this mode of action can be excluded.
:1.5). If the chair-like six-membered cyclic transition state is operative, 1r should deliver the product 2r with a high degree of E-selectivity and the observed selectivity implies that this mode of action can be excluded.
 | ||
| Scheme 3 Non-stereoselective conversion of unsymmetrical allylic alcohol 1r; isolated yield. 1H NMR yield is shown in the parentheses. | ||
In order to obtain further insight into the mechanism, DFT calculations using the model compounds (SM) were performed (Scheme 4). Sodium alkoxide and diboron form the initial complex (CP1) with a large stabilization energy (12.9 kcal mol−1). The proposed chair-like six-membered ring transition state (TS) is an extremely highly energetic species, and the energy barrier is insurmountable (+35.9 kcal mol−1).
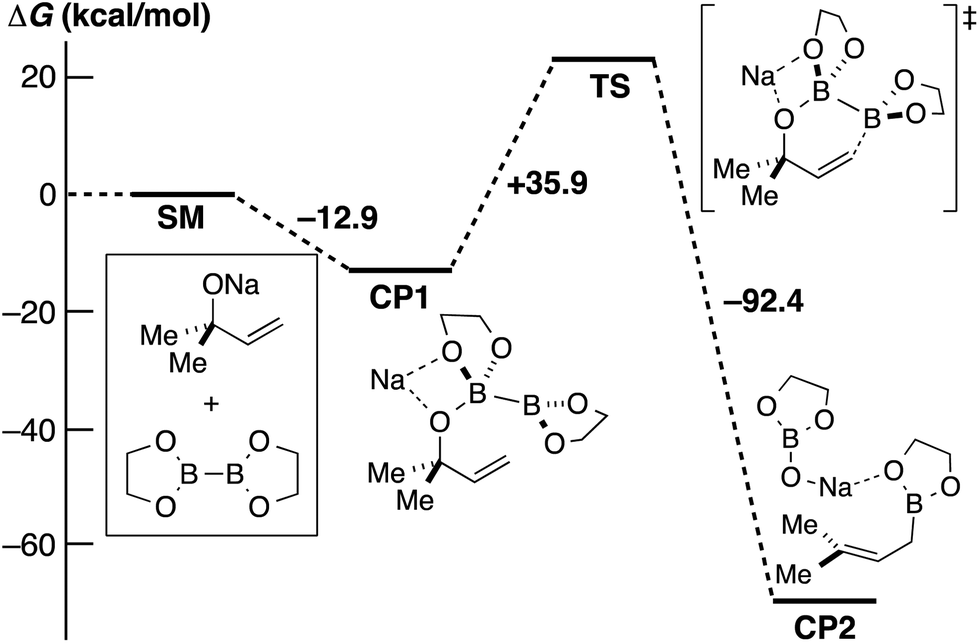 | ||
| Scheme 4 DFT calculations on our initially proposed mechanism at the level of B3LYP/6-31+G*. | ||
Fernández et al. reported an excellent combined experimental and theoretical study showing that the diborylation reaction of alkenes proceeds smoothly,8a and our computation on the following boron Wittig reaction gave a surmountable activation barrier (Scheme 5). In addition, the allylic borylation reaction using 1a at 70 °C resulted in a lower yield of 2a (52%) accompanied by the Fernández-type diborylated product 5 observed by the ESI-MS measurement, while 1a was completely consumed (Scheme 6).13 Thus, we concluded that the present allylic boronate formation was achieved via sequential processes of Fernández-type diborylation of alkene and boron Wittig reaction (Scheme 2, bottom pathway).
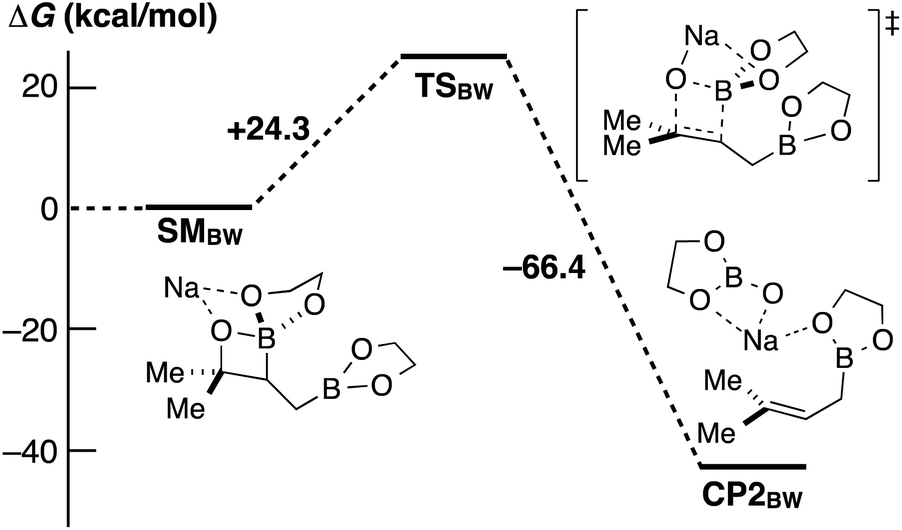 | ||
| Scheme 5 DFT calculations on the boron Wittig reaction at the level of B3LYP/6-31+G*. | ||
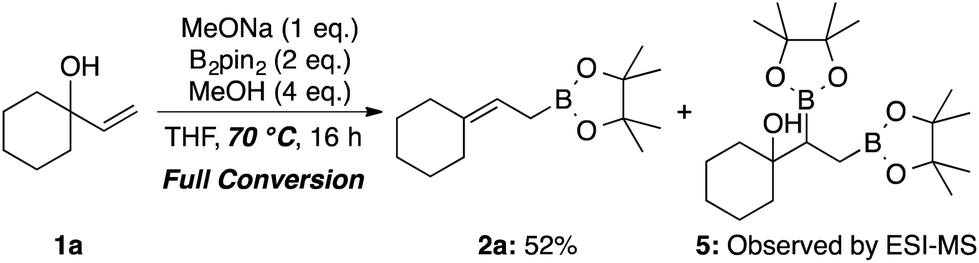 | ||
| Scheme 6 Reaction at lower temperature; 1H NMR yield is given. | ||
Additionally, we performed control experiments to elucidate whether or not the alkoxide moiety participates in pseudo-intramolecular activation of diboron (Scheme 7). The allylic methylether 6 underwent allylic boronate formation, albeit with a much lower yield compared with 2a in Table 3 (eqn (1)). Moreover, only a very low yield of the diborylated product 8 was obtained using substrate 7 without any Lewis-basic site (eqn (2)). These results indicate that some activation by the hydroxyl group operates in the case of allylic alcohols as substrates.
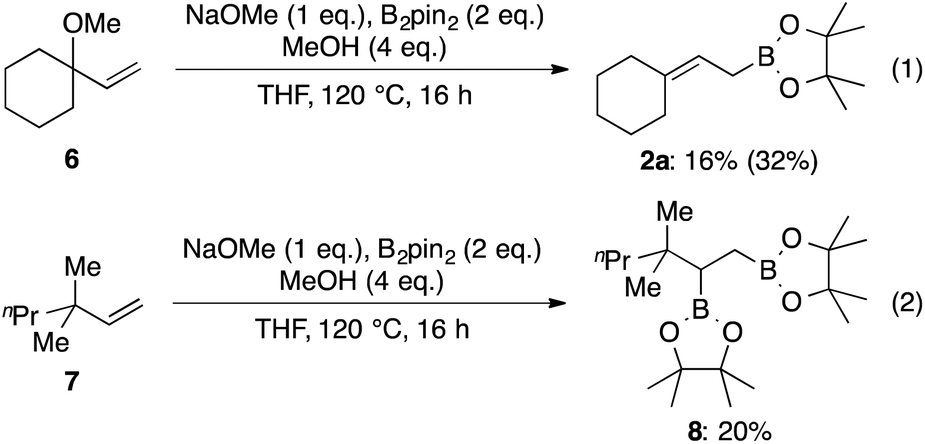 | ||
| Scheme 7 Substrates without pseudo-intramolecular diboron activation; isolated yields. 1H NMR yield is shown in parentheses. | ||
Finally, we demonstrated that allylic boronate 2a can be prepared via three sequential processes in one pot starting from vinyl bromide (Scheme 8). In situ-generated allylic alkoxide was treated with diboron to give the desired boronate 2a in 32% isolated yield.
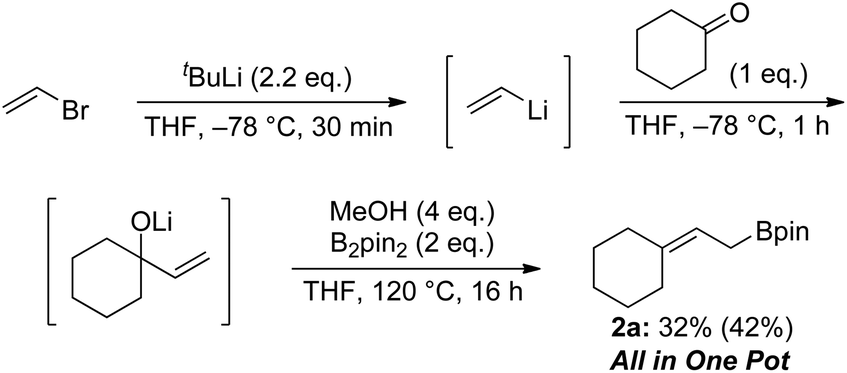 | ||
| Scheme 8 One pot synthesis of allylic boronate; isolated yield. 1H NMR yield is shown in parentheses. | ||
In summary, we have developed a novel and facile access to synthetically useful allylic boronates from simple allylic alcohols. Further mechanistic studies and application of this methodology to biologically active substances are under way in our laboratory.
Acknowledgements
This work was supported by JSPS KAKENHI (S) (no. 24229011), Takeda Science Foundation, The Asahi Glass Foundation, the Daiichi-Sankyo Foundation of Life Sciences, Mochida Memorial Foundation, Tokyo Biochemical Research Foundation, Foundation NAGASE Science Technology Development, and Sumitomo Foundation (to M. U.), JSPS Grant-in-Aid for Young Scientists (B) (no. 26860010) (to K. H.). The calculations were performed on the Riken Integrated Cluster of Clusters (RICC) and HOKUSAI GreatWave. We gratefully acknowledge the Advanced Center for Computing and Communication (RIKEN) for providing computational resources. We thank Professor Elena Fernández for her valuable suggestions.Notes and references
- (a) D. G. Hall, Boronic Acid: Preparation, Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed; (b) R. W. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 555 CrossRef; (c) Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207 CrossRef CAS; (d) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732 CrossRef CAS PubMed.
- (a) P. G. M. Wuts and S. S. Bigelow, J. Org. Chem., 1982, 47, 2498 CrossRef CAS; (b) W. R. Roush, A. E. Walts and L. K. Hoong, J. Am. Chem. Soc., 1985, 107, 8186 CrossRef CAS; (c) R. W. Hoffmann and B. Kemper, Tetrahedron Lett., 1982, 23, 845 CrossRef CAS.
- (a) P. G. M. Wuts, P. A. Thompson and G. R. Callen, J. Org. Chem., 1983, 48, 5398 CrossRef CAS; (b) R. W. Hoffmann and G. Niel, Liebigs Ann. Chem., 1991, 1195 CrossRef CAS; (c) V. Nyzam, C. Belaud and J. Villiéras, Tetrahedron Lett., 1993, 34, 6899 CrossRef CAS.
- (a) T. Ishiyama, T. Ahiko and N. Miyaura, Tetrahedron Lett., 1996, 37, 6889 CrossRef CAS; (b) G. W. Kabalka, B. Venkataiah and G. Dong, J. Org. Chem., 2004, 69, 5807 CrossRef CAS PubMed. For cross-coupling between (dialkoxyboryl)methylzinc reagents and 1-halo-1-alkenes, see: (c) T. Watanabe, N. Miyaura and A. Suzuki, J. Organomet. Chem., 1993, 444, C1 CrossRef CAS.
- (a) G. Dutheuil, N. Selander, K. J. Szabó and V. K. Aggarwal, Synthesis, 2008, 2293 CAS. For seminal work by Szabó, see: (b) V. J. Olsson, S. Sebelius, N. Selander and K. J. Szabó, J. Am. Chem. Soc., 2006, 128, 4588 CrossRef CAS PubMed; (c) N. Selander, A. Kipke, S. Sebelius and K. J. Szabó, J. Am. Chem. Soc., 2007, 129, 13723 CrossRef CAS PubMed; (d) N. Selander, S. Sebelius, C. Estay and K. J. Szabó, Eur. J. Org. Chem., 2006, 4085 CrossRef CAS.
- Y. Nagashima, K. Hirano, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2014, 136, 8532 CrossRef CAS PubMed.
- H. E. Zimmerman and M. D. Traxler, J. Am. Chem. Soc., 1957, 79, 1920 CrossRef CAS.
- (a) A. Bonet, C. Pubil-Ulldemolins, C. Bo, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2011, 50, 7158 CrossRef CAS PubMed. Morken et al. reported hydroxyl group-mediated diastereoselective diborylation mainly using homoallylic alcohols. Only one example using an allylic alcohol was given in the paper and there was no detailed information on allylic boronate formation. (b) T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264 CrossRef CAS PubMed.
- (a) D. S. Matteson, R. J. Moody and P. K. Jesthi, J. Am. Chem. Soc., 1975, 97, 5608 CrossRef CAS; (b) D. S. Matteson and P. K. Jesthi, J. Organomet. Chem., 1976, 110, 25 CrossRef CAS; (c) D. S. Matteson and R. J. Moody, J. Am. Chem. Soc., 1977, 99, 3196 CrossRef CAS; (d) D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20 CrossRef CAS; (e) A. Pelter, B. Singaram and J. W. Wilson, Tetrahedron Lett., 1983, 24, 635 CrossRef CAS; (f) A. Pelter, D. Buss, E. Colclough and B. Singaram, Tetrahedron, 1993, 49, 7077 CrossRef CAS; (g) T. Kawashima, N. Yamashita and R. Okazaki, J. Am. Chem. Soc., 1995, 117, 6142 CrossRef CAS; (h) M. Sakai, S. Saito, G. Kanai, A. Suzuki and N. Miyaura, Tetrahedron, 1996, 52, 915 CrossRef CAS; (i) K. Endo, M. Hirokami and T. Shibata, J. Org. Chem., 2010, 75, 3469 CrossRef CAS PubMed; (j) J. R. Coombs, L. Zhang and J. P. Morken, Org. Lett., 2015, 17, 1708 CrossRef CAS PubMed.
- For all the reactions, MeONa was charged into the reaction vessel in a glove box to avoid moisture absorption. We checked the moisture tolerance of the reaction using 1c by setting up the reaction outside the box, and obtained 2c in 77% yield as observed from the 1H NMR analysis of the crude mixture.
- (a) H. Lachance, X. Lu, M. Gravel and D. G. Hall, J. Am. Chem. Soc., 2003, 125, 10160 CrossRef CAS PubMed; (b) J. W. J. Kennedy and D. G. Hall, J. Am. Chem. Soc., 2002, 124, 11586 CrossRef CAS PubMed.
- J. L.-Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2015, 53, 10992 CrossRef PubMed.
- See ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00009f |
| ‡ Both authors contributed equally to this manuscript. |
| This journal is © the Partner Organisations 2016 |